Introduction
Pontocerebellar hypoplasia (PCH) refers to a group
of genetic, progressive disorders mostly affecting the
infratentorial structures with the involvement of the cerebellum,
pons and olivary nuclei, associated with atrophic changes of
supratentorial structures: The cerebral cortex and basal ganglia.
The actual classification is based on the identification of
underlying molecular defects leading to the current recognition of
17 PCH types associated with 25 different genes. Based on the
underlying molecular pathways, PCH may result from alteration in
genes targeting transfer (t)RNA processing (I), other forms of RNA
processing (II) or result from mutations in genes not related to
RNA processing (1).
PCH2D is caused by mutation of the gene encoding
O-phosphoseryl-tRNA selenocysteine (SEC):selenocysteinyl-tRNA
synthase [SEPSECS (chr.4p15.2)]. This enzyme converts SEC to
selenocystenenyl-tRNA in a process relying on the presence of the
selenocysteine tRNA (tRNASec) and pyridoxal-5-phosphate.
Sec-tRNASec acts as a selenium donor in selenoprotein biosynthesis.
Mice with neuronal selenoprotein deficiency show marked cerebellar
hypoplasia, associated with Purkinje cell death and decreased
granule cell proliferation, indicating that selenoproteins are
required for proper cerebellar development (2). The selenoproteins glutathione (GSH)
peroxidase (GPXs) and selenoprotein H (SelH) have critical roles in
the GSH-dependent and antioxidant systems, providing protection
from reactive oxygen species (ROS). SelH increases the expression
of the GSH-synthesis enzyme γ-glutamylcysteine synthetase, whereas
GPXs catalyse the breakdown of peroxides into water (3).
To date, a total of 26 patients with PCH2D have been
reported (1,4-21)
with different mutant alleles and variable clinical phenotypes,
exhibiting a certain degree of heterogeneity in terms of
manifestations and severity. Progressive pontocerebellar and
cerebral atrophy, microcephaly and mostly severe developmental
disability are prominent neurological features.
The present study reports on a patient harbouring a
novel SEPSECS mutation and presenting with postnatal
microcephaly, progressive motor and intellectual disability, visual
defect and progressive cerebral and pontocerebellar atrophy, as
well as recurrent blood lactate elevation. In light of the acute
neurological regression following vaccination with fever in the
present case, not previously reported in PCH2D, the clinical
spectrum associated with SEPSECS mutations was reviewed with
an emphasis on the causal link between selenoprotein biosynthesis
deficiency and mitochondrial disorders.
Case report
Case presentation
The pediatric patient was male and born full-term
(40th week of gestation) after an uncomplicated pregnancy, with an
uneventful neonatal period. At birth, the body weight was 3.2 Kg
(25th pc), and the length was 48 cm (25-50th pc), both in the
normal range (25-75th pc) according to the Neonatal Anthropometric
Charts for Italy (https://www.siedp.it). Head circumference (HC) was in
the low-normal range (33 cm, 10th pc). Early psychomotor
development was normal: Head control was gained at 3 months and
sitting with support was achieved at 5 months. Following
meningococcal vaccination at the age of six months, the patient
experienced high fever (39˚C) associated with a seizure episode,
followed by the acute onset of convergent strabismus and truncal
hypertonia. Neurological examination at the age of 9 months
revealed microcephaly (HC, 41 cm; <<3rd pc), internal
strabismus, severe developmental delay, poor active movements with
limb spasticity, absent handling and inadequate response to
environmental stimuli. The patient was unable to keep his head on
the trunk, sit, crawl and roll. Electroencephalogram (EEG) showed a
prevalence of abnormal background rapid rhythms without epileptic
discharges.
Brain MRI, performed at the age of 19 months, showed
posterior periventricular white matter signal changes bilaterally
and subcortical white matter hyperintensity, particularly in the
insular temporal areas. Neuroimaging of the cerebellum and the
brainstem were otherwise normal.
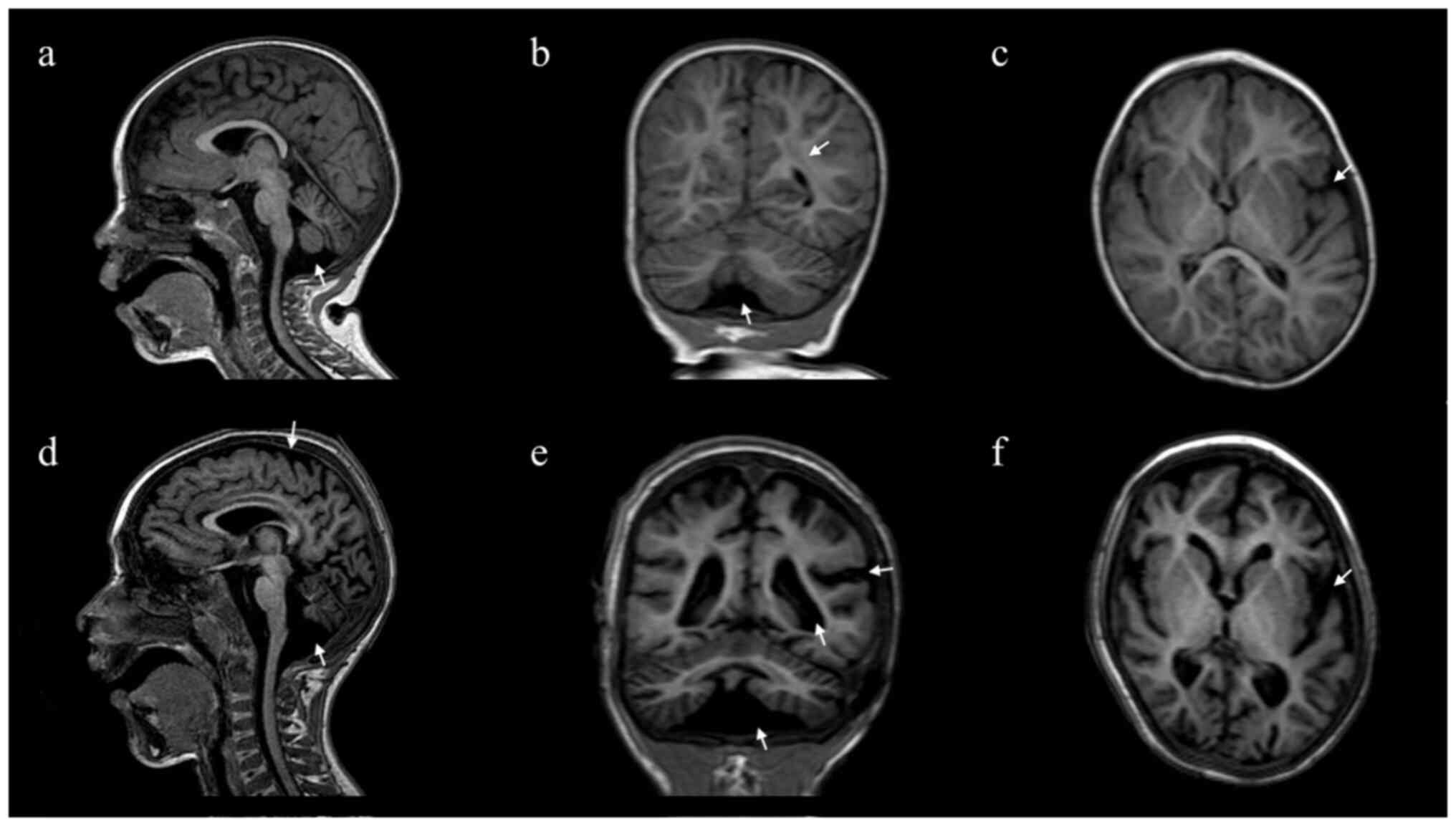
At 26 months of age, brain MRI showed slight
peritrigonal white matter hyperintensity with a normal corpus
callosum and reduction of the cerebral white matter with
enlargement of the periencephalic and peritruncal cerebrospinal
fluid (CSF) spaces associated with pontobulbar and vermian atrophy
(Fig. 1A-C). Between two and five
years of age, the patient had recurrent seizures associated with
fever. Repeated EEG recording showed hypersynchronous activity in
the central posterior regions.
The patient was first evaluated at our department
(Child and Adolescent Neurology and Psychiatric Section, University
Hospital Policlinico, Department of Clinical and Experimental
Medicine, University of Catania, Catania, Italy), at age 6
(November 2021). On physical examination, a sloping forehead, round
face, thick nasal alae, low-set ears and thin elongated fingers
were noted. Pertinent neurological findings included bilateral
convergent strabismus, microcephaly and spastic quadriplegia with
fixed fits and absent verbal language. The patient had recurrent
episodes with irritability and inconsolable crying when away from
home and daytime episodes of perioral tremor. No improvement with
gradual titration of baclofen and clonazepam per os was
observed over time.
Brain MRI showed slight asymmetric enlargement of
the lateral ventricles, an enlarged fourth ventricle, multiple
areas of altered signal in the peri/supraventricular and
subcortical white matter, as well as deepening of the
temporo-occipital sulci bilaterally. The white-matter volume was
globally reduced with enlargement of subtentorial, pontine and
bulbar cisternae, large cisterna magna and a wide pontocerebellar
angle. There were overt signs of pontobulbar, cerebellar vermis and
hemisphere atrophy with signal alteration in fluid-attenuated
inversion recovery sequences (Fig.
1D-F).
Screening methods
Extensive metabolic screening and Array Comparative
Genomic Hybridization (aCGH) were performed using the SurePrint G3
Custom CGH Microarray, 4x180K (Agilent Technologies, Inc.)
according to the manufacturer's protocol, with appropriate Agilent
reference DNAs (Euro male and Euro female). The array data
extraction and analysis were performed using CytoGenomics v.5.0.2.5
(Agilent Technologies, Inc.). The extensive metabolic screening
included quantitative analysis of urinary organic acids by gas
chromatography/mass spectrometry (MS) (22). A blood amino acid (AA) profile was
obtained from dried blood spots (DBS) by electrospray
ionization-tandem MS (ESI-MS) (25 metabolites, including 14 AAs and
11 AA ratios, were simultaneously measured). The following AAs were
determined: Alanine (Ala), arginine (Arg), citrulline (Cit),
glutamate (Glu), glutamine (Gln), glycine (Gly), leucine (Leu),
methionine (Met), ornithine, phenylalanine (Phe), proline, tyrosine
(Tyr) and valine (Val). The following AA ratios were measured:
Leu+Val/Phe+Tyr, Cit/Arg, Cit/Phe, Leu/Phe, Met/Leu, Met/Phe,
Phe/Tyr, Glu/Gln, Tyr/Leu, Tyr/Met and Val/Phe). The anion gap to
check for metabolic acidosis (22)
was determined to measure the difference between the primary
measured cations (sodium and potassium) and the primary measured
anions (chloride and bicarbonate) in serum. Determination of blood
acyl-carnitine levels included measures of short-chain,
medium-chain and long-chain acyl-carnitines from DBS by using
ESI-MS (22). Serum transferrin
glycoform analysis was performed by capillary electrophoresis and
serum lysosomal enzymes (β-hexosaminidase and β-galactosidase) were
measured fluorometrically (23).
Whole blood (3 ml) was collected from the proband
and the proband's parents for NGS analysis after informed consent
had been provided. NGS analyses were performed by Research &
Innovation Genetics Srl (Padua-Italy). DNA library preparation and
whole-exome enrichment were performed using the SureSelect All Exon
V6 kit (Agilent Technologies, Inc.). The library was sequenced
using the NextSeq 2000 Illumina Sequencer (100-bp paired-end reads;
Illumina, Inc.) according to the manufacturer's instructions.
Bioinformatics analysis included the following: i) NGS reads were
aligned to the GRCh37 human reference genome using the
Burrows-Wheeler Alignment tool (https://bio-bwa.sourceforge.net/) with the default
parameters; ii) PCR duplicate removal using Picard (http://picard.sourceforge.net); iii) identification of
single nucleotide polymorphisms and insertions/deletions using the
Genome Analysis Toolkit (GATK 4; https://gatk.broadinstitute.org/hc/en-us); iv) variant
annotation using snpEff (http://snpeff.sourceforge.net). Whole-exome sequencing
(WES) data and read alignment analysis were checked for coverage
depth and alignment quality using the Bedtools software package
(https://github.com/arq5x/bedtools2).
Variant classification was performed in accordance with the
guidelines from the American College of Medical Genetics and
Genomics (ACMG) (24).
Phenotype-driven analysis, coupled with the employment of in
silico multigene panels specific for microcephaly and
neurodevelopmental disorders, was used to filter, select and
interpret genetic variants obtained following exome sequencing. The
PCR products containing significant variants underwent Sanger
sequencing and capillary electrophoresis using an 3100-avant
automatic sequencer (Applied Biosystems; Thermo Fisher Scientific,
Inc.) according to the manufacturer's instructions (https://tools.thermofisher.com/content/sfs/manuals/cms_041003.pdf).
Findings of the screening
analysis
Extensive metabolic screening, including urinary
organic acids and plasma amino acids, anion gap, serum transferrin
glycoform analyses and lysosomal enzymes yielded normal results.
Isolated, recurrent elevations of lactic acid levels (25 mg/dl at
the age of 12 months and 40.2 mg/dl at around the age of 2 years;
normal range: 5.7-22.0 mg/dl) were detected (Table I). Additional evidence of
mitochondrial dysfunction was found by acyl-carnitine analyses,
illustrating a decrease in blood acetyl-carnitine. This is formed
by the enzyme carnitine acetyltransferase, which transfers a
2-carbon moiety from acetyl-CoA to L-carnitine. A decrease of
acetyl-CoA, the substrate for the synthesis of acetyl-carnitine, is
found in the case of increased oxidative stress due to oxidative
damage, particularly when several key enzymes of acetyl-CoA and
energy metabolism are oxidatively modified (25).
 | Table IClinical, neuroimaging and genetic
data of reported patients with SEPSECS mutations. |
Table I
Clinical, neuroimaging and genetic
data of reported patients with SEPSECS mutations.
| First author,
year | Families | Pts | Sex | Mutation | Effect | Genotype | Brain MRI | Microcephaly | Spasticity | Walking | Epilepsy (seizure
type) | Vision | DD/ID | Language | Other | First signs | Mitochondrial
signs | (Refs.) |
|---|
| Ben-Zeev, 2003;
Agamy, 2010 | 2 | 4 | M | c.1001A>G | Missense | Homozygous | Cerebellar/cerebral
atrophy | + (secondary) | + | ND | + (generalized,
myoclonic) | Nystagmus | + | Non-verbal | - | ND | - | (4,5) |
| | | | F | c.1001A>G | Missense | Homozygous | Normal at 5 m;
progressive cerebellar/frontal parasylvian atrophy | + (secondary) | + | - | - | ND | + | Non-verbal | Chorea | ND | - | |
| | | | M | c.1001A>c.
715G>A | Missense
missense | Compound
heterozygous | Normal at 5 m;
progressive cerebellar/frontal parasylvian atrophy | + (secondary) | + | - | + (focal,
generalized) | Partial visual
pursuit | + | Non-verbal | Chorea | ND | - | |
| | | | F | c.1001A>G
c.715G>A | Missense
missense | Compound
heterozygous | w.m..changes thin
corpus callosum at 8 m; progressive cerebellar atrophy | + (secondary) | + | - | - | No visual
pursuit | + | Non-verbal | - | ND | - | |
| Makrythanasis,
2014 | 1 | 1 | F | c.1466A>7 | - | Homozygous | Cerebellar vermis
atrophy | + | + | ND | + | ND | ND | ND | - | DD | - | (6) |
| Alazamy, 2015 | 1 | 1 | - | c.1027_1120del | - | Homozygous | - | ND | + | ND | ND | Nystagmus | ND | Dysarthria | - | ND | - | (7) |
| Anttonen, 2015 | 3 | 3 | F | c.974C>G
c.1287C>A | Missense
nonsense | Compound
heterozygous | Early w.m. changes;
progressive cerebellar/cerebral atrophy | ND | + | - | + (infantile
spasms) | Central
blindness | + | Delay | High TSH | Opisthotonus
(birth) | Elevated blood and
CSF lactate | (8) |
| | | | F | c.974C>Gc.
1287C>A | Missense
nonsense | Compound
heterozygous | Early w.m. changes;
progressive cerebellar/cerebral atrophy | ND | + | - | + (infantile
spasms) | Central
blindeness | + | Delay | High TSH | Opisthotonus (1
m) | Elevated blood and
CSF lactate | |
| | | | M | c.974C>G
c.1287C>A | Missense
nonsense | Compound
heterozygous | Progressive
cerebellar and cerebral atrophy | + (primary) | + | - | + | Central visual
defect | + | Delay | - | Opisthotonus (2
m) | Elevated CSF
lactate | |
| Zhu, 2015 | 1 | 1 | F | c.1A>G
c.388+3A>G | Missense
splicing | Compound
heterozygous | Progressive
cerebellar atrophy | + | ND | ND | ND | ND | ND | ND | - | Hypotonia, DD (5
m) | - | (9) |
| Iwama, 2016 | 2 | 2 | F | c.356A>G
c.77delG | - | Compound
heterozygous | Normal at 2 y;
progressive cerebellar/frontoparietal atrophy | + (secondary) | + | Ataxic gait | - | Nystagmus | + | Slurred speech | - | Downward nystagmus
(3 m) | - | (10) |
| | | | F | c.356A>G
c.467G>A | - | Compound
heterozygous | Normal at 5 y;
progressive cerebellar/cerebral atrophy | + (primary) | + | Ataxic gait | - | ND | ND | Slurred speech | - | DD | - | |
| Pavlidou, 2016 | 1 | 1 | M | c.1001T>C | Missense | Homozygosity | Ventricular
widening at 18 m; progressive cerebellar/cerebral atrophy | + (secondary) | + | Quadriplegia | ND | Optic nerve
atrophy | + | Delay | High CK | Hypotonia | Myopathic features;
decreased mitochondrial respiratory chain complex IV at muscle
biopsy | (11) |
| Olson, 2017 | 1 | 1 | M | c.176CC>T | - | Homozygous | w.m changes;
cerebral/pontocerebellar atrophy | + | ND | ND | + (epileptic
encephalopathy) | ND | ND | ND | - | Hypotonia DD | - | (12) |
| Van Dijk, 2018 | 1 | 1 | F | c.1321G>A | Missense | Homozygous | Progressive
cerebellar atrophy | + | - | Broad-based
gait | ND | Horizontal
nystagmus | + | Able to say few
words | - | DD | - | (13) |
| Hengel, 2020 | 1 | 1 | - | c.181A>G | - | Homozygous | Pontocerebellar
hypoplasia | ND | ND | ND | ND | ND | + | ND | - | ND | - | (14) |
| Arrudi-Moreno
2021, | 1 | 1 | F | c.114+3A>G | Splicing
disruption | Homozygous | Normal at 14 m;
progressive pontocerebellar atrophy | + (secondary) | + | Tetraplegia | - | Convergent
strabismus | ND | Able to say few
words | - | DD | - | (15) |
| Nicita, 2022 | 1 | 1 | M | c.865C>T
c.1297T>C | Missense | Compound
heterozygous | Cerebral atrophy;
thin corpus callosum | + | - | Wide-based gait;
bradykinesia | ND | Nystagmus, optic
nerve atrophy | ND | Dysarthria | Dystonia, low
IgA | Head titubation,
ocular nystagmus (13 m) | - | (16) |
| Martinez-Martin,
2022 | 1 | 2 | M | c.1321G>A | Missense | Homozygous | Mild atrophy of
cerebellar vermis | - | Pyramidal
signs | Ataxic gait | - | Convergent
strabismus | + | Scanning
speech | - | Learning
difficulties and motor impairment (7 y) | - | (17) |
| | | | M | c.1321G>A | Missense | Homozygous | Normal | - | - | Unable to walk in
tandem | - | Nystagmus | + | Scanning
speech | - | Motor and language
impairment (5 y) | - | |
| Ramadesikan,
2022 | 1 | 2 | F | c.1A>T
c.846G>A | Loss of function;
splicing disruption | Compound
heterozygous | w.m. changes at
birth; progressive cerebral/pontocerebellar atrophy | + (secondary) | ND | - | + (febrile, later
focal clusters) | Cortical
blindness | + | Non-verbal | - | Abnormal posturing
(birth) | Decreased activity
of complex I -II at muscle biopsy | (18) |
| | | | M | c.1A>T
c.846G>A | Loss of function;
splicing disruption | Compound
heterozygous | Progressive
cerebral/cerebellar atrophy | + (secondary) | ND | - | + (febrile, later
focal clusters) | Cortical vision
imparment | + | Non-verbal | Respiratory
failure | Hypotonia | Decreased activity
of complex I-II at muscle biopsy | |
| Rong, 2022 | 1 | 1 | F | c.701+1G> A
c.194A>G | Splicing;
missense | Compound
heterozygous | Bilateral pallidum
signal changes at 15 m; progressive frontotemporal atrophy | - | ND | ND | - (15 m
seizure-like symptoms) | Esotropia | + | Delay | - | Hypotonia | Limbs myogenic
changes | (19) |
| Zhao, 2022 | 1 | 1 | - | c.628C>T
c.770T>C | - | Compound
heterozygous | Normal at 28 y;
progressive cerebellar atrophy | ND | ND | Ataxic gait | - | ND | + | Slurred speech | Dysphagia | Bradykinesiat 24
y | - | (21) |
| Ghasemi, 2024 | 2 | 2 | M M | c.208T>C | Missense | Homozygous | ND | ND | ND | Motor delay at 3
y | Febrile
seizures | Strabismus | + | Delay | - | ND | - | (1) |
| | | | | c.1274A>G | Missense | Homozygosity | Cerebellar atrophy
at 4 years | ND | + | Ataxia at 4 y | Febrile
seizures | Nystagmus | + | Delay | Neuropathy | DD | - | |
| Present study | 1 | 1 | M | c.114+3A> G
c.440 G>A | Splicing
missense | Compound
heterozygous | w.m. changes at 17
m; progressive pontocerebellar and cerebral atrophy | + (secondary) | + | Tetraplegia | + (febrile seizures
generalized epilepsy) | Convergent
strabismus | + | Non-verbal | - | Squint, hypertonia
following fever | Elevated blood
lactate (25 mg/dl at the age of 12 months and 40.2 mg/dl at around
the age of 2 years; normal range: 5.7-22.0 mg/dl) | - |
aCGH analysis was normal. Sequencing analysis of the
coding exons of the genes included in the genetic panel for the
molecular diagnosis of microcephaly and neurodevelopmental
disorders showed two variants in SEPSECS: A paternal
NM_016955.4:c.114+3A>G and maternal NM_016955.4: c.440G>A
(p.Ser147Asn) (Fig. 2A).
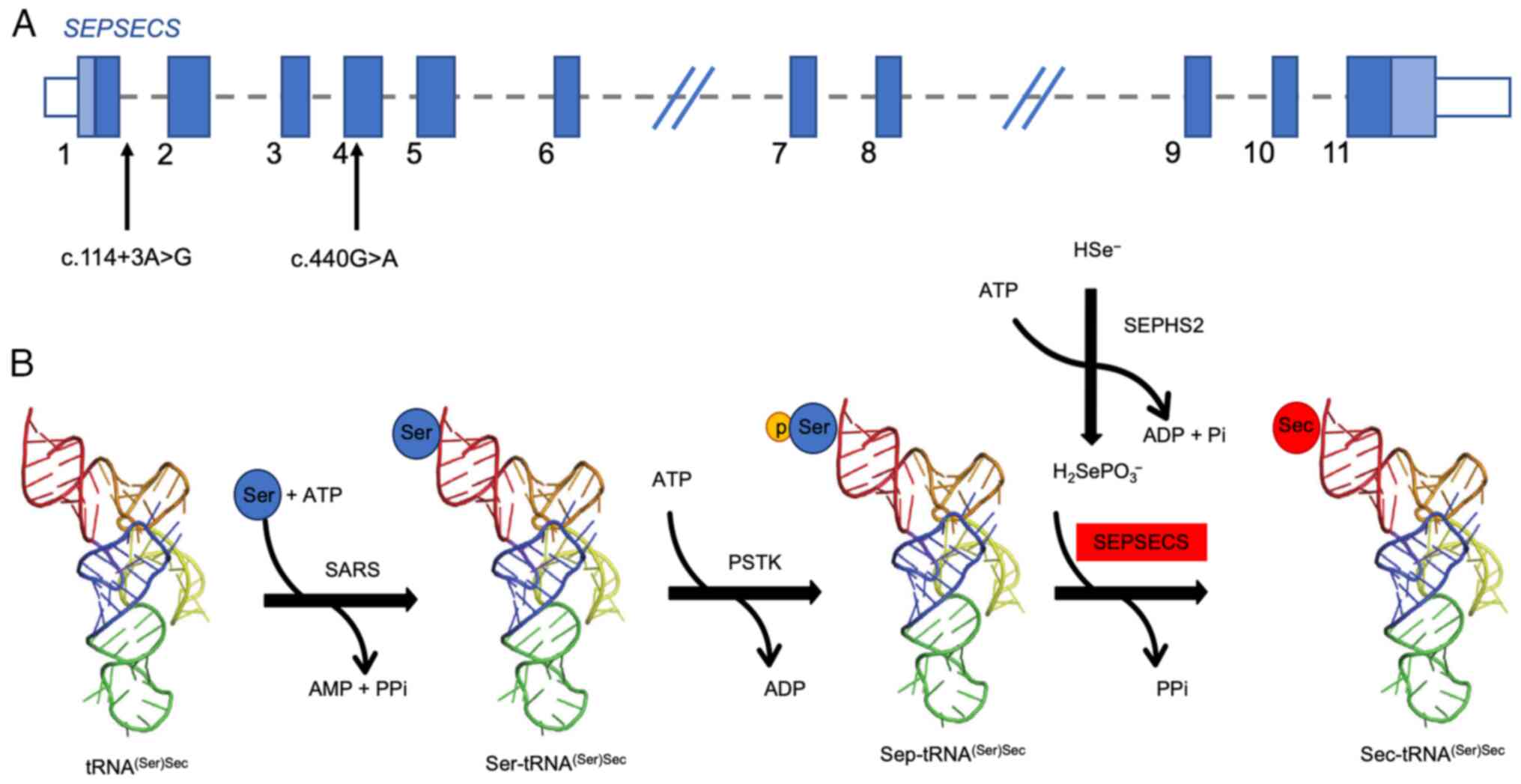 | Figure 2Location of the whole-exome sequencing
variants of the patient within the SEPSECS gene and role of
SepSecS in selenocysteine biosinthesis. (A) The two variants found
in the proband are shown in the schematic representation of the
SEPSECS gene. (B) In humans, Sec-tRNA(Ser)Sec
synthesis follows a two-step process via a phosphorylated
intermediate: tRNA(Ser)Sec is converted to
Ser-tRNA(Ser)Sec, which is phosphorylated to
Sep-tRNA(Ser)Sec.
O-phosphoseryl-tRNA:selenocysteinyl-tRNA synthase, encoded by the
SEPSECS gene, converts Sep-tRNA(Ser)Sec to
Sec-tRNA(Ser)Sec, which acts as a selenium donor in
selenoprotein biosynthesis. SepSeS, O-phosphoseryl-tRNA
selenocysteine:selenocysteinyl-tRNA synthase;
Sec-tRNA(Ser)Sec, selenocysteine tRNA (serine)
selenocysteine; Ser-tRNA(Ser)Sec, seryl tRNA (serine)
selenocysteine; Sep (O-phosphoserine) tRNA, Sec (selenocysteine)
tRNA synthase; Sec-tRNA(Ser)Sec,
selenocysteine-tRNA (serine) selenocysteine. |
The variant c.114+3A>G arises in the consensus
sequence of the splicing donor site (+3 of exon 1). Functional
studies performed on the transcript revealed that it induces exon 1
skipping in the SEPSECS gene, including the translation
initiation codon ATG, thus resulting in a possible loss of function
of the protein (20). The variant
is considered pathogenic according to the ACMG criteria (24).
The variant c.440G>A (p.Ser147Asn) has not been
reported in the medical literature, to the best of our knowledge.
It leads to the substitution of serine at codon 147 by asparagine
in a conserved protein position as measured by PhyloP scores
(https://ionreporter.thermofisher.com/) which measure
evolutionary conservation at individual alignment sites with
positive scores indicating sites that are predicted to be conserved
(PhyloP-Vertebrate=5.64/6.42; phyloP-Primate=0.56/0.65;
PhastCons=1.00/1.00; https://varsome.com/). In silico computational
analysis indicated the harmful effect probability of p.Ser147Asn
amino acid substitution on the structure/activity of the resulting
protein [PolyPhen2=0.996/1.00 (PolyPhen-2: Prediction of functional
effects of human nsSNPs; http://genetics.bwh.harvard.edu/pph2/);
SIFT=0.005/0.00; MutationTaster=1.00/1.00 (https://www.mutationtaster.org/); CADD PHRED=26;
Mutation Assessor=2.85/5.00 (http://mutationassessor.org/r3/)].
According to the ACMG criteria PP3 (multiple
computational evidence supports a deleterious effect on the gene)
and PM3 (absence or extremely low frequency of the variant from
controls in the 1000 Genomes Project, Exome Sequencing Project, or
other large population datasets), the variant can be classified as
likely pathogenic. Both variants were deposited in ClinVar
[accession nos. SCV005061756 (https://www.ncbi.nlm.nih.gov/clinvar/variation/984624/?oq=SCV005061756&m=NM_016955.4(SEPSECS):c.114%203A%3EG)
and SCV005061752 (https://www.ncbi.nlm.nih.gov/clinvar/variation/3242529/?oq=SCV005061752&m=NM_016955.4(SEPSECS):c.440G%3EA%20(p.Ser147Asn),
respectively]. No other significant variants were identified in the
whole dataset (dataset available at https://doi.org/10.5281/zenodo.13234362).
Discussion
To date, 27 patients with ascertained SEPSECS
mutations have been identified, including the present one (Table I). Most patients had an uneventful
pre- and perinatal history with the onset of neurological signs in
the first 2 years of life. A small minority of patients (10,17,21)
had a later onset of neurological symptoms, even in adulthood. In
the most severe phenotypes, patients rapidly experienced an obvious
worsening of clinical conditions, with a post-natal onset of
microcephaly, cerebellar signs and spasticity. Congenital
microcephaly was described in 2 subjects (8,10) and
a normal cranial circumference throughout the follow-up was
reported in 3 patients (17,19).
Motor function impairment was variable with most patients
experiencing spastic quadriplegia, inability to walk and decreased
motor function (12 patients) (4,8,11,15,18)
and the current study. Milder cerebellar symptoms were reported in
milder affected subjects with later disease onset (4 patients).
Choreiform movements (2 patients) (4) and extrapyramidal symptoms resembling
dystonia-parkinsonism (1 patient) (16) were infrequent. Intellectual
disability was an almost constant feature and verbal communication
appeared to be variably affected from the total lack of
communicative skills (4,18) to dysarthria and slurred speech
(21).
Visual impairment was reported in 9 patients
(4,8,11,16,18)
with strabismus and nystagmus as common early signs. Epilepsy,
reported in 7 patients (4,6,8,12,18),
may be either focal or generalized with tonic, myoclonic and
tonic-clonic seizures; 2 patients developed febrile convulsions
with later onset of non-febrile seizures (18) and an additional patient presented
with an early-onset developmental and epileptic encephalopathy with
a burst suppression EEG pattern (12).
Brain MRI was normal in the first months of life in
certain patients. In milder phenotypes (10,17,21),
no alterations were detected even for years, and in one patient,
noticeably, neuroimaging was normal until the age of 28 years
(21). In the majority of patients,
however, white matter changes, particularly in the frontal lobes,
were detected early in infancy and preceded the emergence of
progressive pontocerebellar atrophy associated with cerebral
atrophy. Neuropathological findings included laminar subtotal
necrosis of the neocortex, particularly in the parieto-occipital
regions, myelin loss and gliosis of white matter, consistent with
degeneration secondary to neuronal loss, severe degeneration and
atrophy of the brainstem and cerebellar cortex with an
olivopontocerebellar atrophy-like appearance, subtotal striatal
degeneration and thalamic atrophy (8).
Pathogenic recessive mutations in the SEPSECS
gene have been shown to alter conserved residues and to be mostly
nonsense, missense or inducing splicing disruption. Among the
reported genetic variants, some, like Ala239Thr, Thr325Ser,
Tyr334Cys and Tyr429*, are associated with severe early-onset
phenotypes, reducing protein stability and increasing misfolding,
diminishing the ability of the SepSecS tetramer to bind
tRNASec and affecting the integrity of the active site
(26). Others, such as Gly441Arg,
which was found in homozygosity in subjects with milder and
late-onset phenotypes, may have a less destructive effect on the
catalytic capacity of the enzyme (13).
The current study illustrates a severe early onset
PHC2D type caused by compound heterozygous variants c.114+3A>G
and c.440G>A (p.Ser147Asn), splicing and missense mutations,
respectively. The first variant has been previously described in
homozygosity in two unrelated patients with a severe early-onset
phenotype and classified as likely pathogenic or pathogenic
(15,20). The latter has not been reported in
the literature and dedicated databases, to the best of our
knowledge; it is rare, arises in a conserved protein position and
in silico prediction analysis indicated a probably harmful
effect.
The trace element Selenium (Se) is a vital
micronutrient incorporated into proteins, named selenoproteins, by
the amino acid SEC. Serine is added to tRNASec by seryl-tRNA
synthetase and then modified to phosphoserine by phosphoseryl-tRNA
kinase. Dietary selenium is added to phosphoserine by
selenocysteine synthetase to produce SEC (Fig. 2B). Selenoproteins have critical
roles in both the GSH-dependent and thioredoxin-dependent
antioxidant systems (3).
In the present patient, it appears that acute
neurological regression may have been triggered by fever related to
vaccination, which is a phenomenon observed in patients with
primary mitochondrial diseases (27). Similarities between mitochondrial
disorders and selenoprotein biosynthesis defects have been
emphasized owing to the consolidated role of selenoproteins in
maintaining cellular redox potential and detoxifying
H2O2 (3). In
this regard, recurrent elevation of lactate levels in blood or CSF
was observed in 3 out of four studied patients with PCH2D (8) and in the present one. Furthermore, the
patient of the present study had a depletion of acetyl-L-carnitine
in blood, indicating a deficiency in the regulation of energy
metabolism and oxidative stress, since the acetyl moiety is used
for producing energy and acts as an antioxidant protecting against
oxidative stress (28). Myopathic
features and decreased mitochondrial respiratory chain complex I,
II and IV were found in three studied patients (11,18).
Increased brain protein carbonylation as a sign of oxidative stress
was described post-mortem in patients with SEPSECS
deficiency, as well as moderate liver degeneration resembling
mitochondrial encephalopathy (8).
In sum, the present study was the first report of acute
neurological regression following a catabolic stressor as the
presenting sign of PCH2D. In light of the current understanding of
the potential relationship between selenoprotein defects and
mitochondrial dysfunction (3,8,11,18),
it is important to highlight that patients with SEPSECS
mutations may produce elevated levels of toxic metabolites and ROS
during a catabolic state induced by physiological stressors such as
fasting, fever, illness, trauma or surgery. Therefore, it may be
advisable to consider preventive measures to avoid catabolic
stressors in afflicted patients.
Further studies are necessary to characterize the
role of SEPSECS pathogenic variants in triggering oxidative
damage contributing to a severe neurological presentation of
PCHD2.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
Original data generated using WES have been
deposited in the public database Zenodo (https://doi.org/10.5281/zenodo.13234362, accession no.
10.5281/zenodo.13234362). Detected variants were submitted to
ClinVar (https://submit.ncbi.nlm.nih.gov/subs/variation_clinvar;
accession nos. SCV005061756 and SCV005061752, respectively).
Additional data generated in the present study are included in the
figures and tables of this article.
Authors' contributions
RB and FP conceptualized the study. RB, FP, VM, FC
and GR performed clinical procedures and acquired data. MF
processed the data generated by WES and deposited them in public
databases. RB, FP and MF checked and confirmed the authenticity of
the raw data. FP and RB were major contributors in writing the
manuscript. RR, MDC, MF and RB reviewed and edited the manuscript.
All authors have read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
All procedures performed in this study are part of
the routine clinical care of patients with neurodevelopmental
disorders, performed in accordance with the ethical standards of
the institutional research committee at the University Hospital
Policlinico ‘G. Rodolico-San Marco’ Catania (Catania, Italy) and
with the 1964 Helsinki declaration and its later amendments.
Written informed consent for all medical procedures was obtained
from the patient's parents.
Patient consent for publication
Written consent for the publication of case data,
medical images, genetic information and data reported within this
article, was obtained from the patient's parents.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ghasemi MR, Tehrani Fateh S, Moeinafshar
A, Sadeghi H, Karimzadeh P, Mirfakhraie R, Rezaei M, Hashemi-Gorji
F, Rezvani Kashani M, Fazeli Bavandpour F, et al: Broadening the
phenotype and genotype spectrum of novel mutations in
pontocerebellar hypoplasia with a comprehensive molecular
literature review. BMC Med Genomics. 17(51)2024.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Wirth EK, Bharathi BS, Hatfield D, Conrad
M, Brielmeier M and Schweizer U: Cerebellar hypoplasia in mice
lacking selenoprotein biosynthesis in neurons. Biol Trace Elem Res.
158:203–210. 2014.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Bellinger FP, Raman AV, Reeves MA and
Berry MJ: Regulation and function of selenoproteins in human
disease. Biochem J. 422:11–22. 2009.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Ben-Zeev B, Hoffman C, Lev D, Watemberg N,
Malinger G, Brand N and Lerman-Sagie T: Progressive
cerebellocerebral atrophy: A new syndrome with microcephaly, mental
retardation, and spastic quadriplegia. J Med Genet.
40(e96)2003.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Agamy O, Ben Zeev B, Lev D, Marcus B, Fine
D, Su D, Narkis G, Ofir R, Hoffmann C, Leshinsky-Silver E, et al:
Mutations disrupting selenocysteine formation cause progressive
cerebello-cerebral atrophy. Am J Hum Genet. 87:538–544.
2010.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Makrythanasis P, Nelis M, Santoni FA,
Guipponi M, Vannier A, Béna F, Gimelli S, Stathaki E, Temtamy S,
Mégarbané A, et al: Diagnostic exome sequencing to elucidate the
genetic basis of likely recessive disorders in consanguineous
families. Hum Mutat. 35:1203–1210. 2014.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Alazami AM, Patel N, Shamseldin HE, Anazi
S, Al-Dosari MS, Alzahrani F, Hijazi H, Alshammari M, Aldahmesh MA,
Salih MA, et al: Accelerating novel candidate gene discovery in
neurogenetic disorders via whole-exome sequencing of prescreened
multiplex consanguineous families. Cell Rep. 10:148–161.
2015.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Anttonen AK, Hilander T, Linnankivi T,
Isohanni P, French RL, Liu Y, Simonović M, Söll D, Somer M,
Muth-Pawlak D, et al: Selenoprotein biosynthesis defect causes
progressive encephalopathy with elevated lactate. Neurology.
85:306–315. 2015.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Zhu X, Petrovski S, Xie P, Ruzzo EK, Lu
YF, McSweeney KM, Ben-Zeev B, Nissenkorn A, Anikster Y, Oz-Levi D,
et al: Whole-exome sequencing in undiagnosed genetic diseases:
Interpreting 119 trios. Genet Med. 17:774–781. 2015.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Iwama K, Sasaki M, Hirabayashi S, Ohba C,
Iwabuchi E, Miyatake S, Nakashima M, Miyake N, Ito S, Saitsu H and
Matsumoto N: Milder progressive cerebellar atrophy caused by
biallelic SEPSECS mutations. J Hum Genet. 61:527–531.
2016.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Pavlidou E, Salpietro V, Phadke R,
Hargreaves IP, Batten L, McElreavy K, Pitt M, Mankad K, Wilson C,
Cutrupi MC, et al: Pontocerebellar hypoplasia type 2D and optic
nerve atrophy further expand the spectrum associated with
selenoprotein biosynthesis deficiency. Eur J Paediatr Neurol.
20:483–438. 2016.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Olson HE, Kelly M, LaCoursiere CM, Pinsky
R, Tambunan D, Shain C, Ramgopal S, Takeoka M, Libenson MH, Julich
K, et al: Genetics and genotype-phenotype correlations in early
onset epileptic encephalopathy with burst suppression. Ann Neurol.
81:419–429. 2017.PubMed/NCBI View Article : Google Scholar
|
|
13
|
van Dijk T, Vermeij JD, van Koningsbruggen
S, Lakeman P, Baas F and Poll-The BT: A SEPSECS mutation in a
23-year-old woman with microcephaly and progressive cerebellar
ataxia. J Inherit Metab Dis. 41:897–898. 2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Hengel H, Buchert R, Sturm M, Haack TB,
Schelling Y, Mahajnah M, Sharkia R, Azem A, Balousha G, Ghanem Z,
et al: First-line exome sequencing in Palestinian and Israeli Arabs
with neurological disorders is efficient and facilitates disease
gene discovery. Eur J Hum Genet. 28:1034–1043. 2020.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Arrudi-Moreno M, Fernández-Gómez A and
Peña-Segura JL: A new mutation in the SEPSECS gene related to
pontocerebellar hypoplasia type 2D. Med Clin (Barc). 156:94–95.
2021.PubMed/NCBI View Article : Google Scholar : (In English,
Spanish).
|
|
16
|
Nicita F, Travaglini L, Bombelli F, Tosi
M, Pro S, Bertini E and D'Amico A: Novel SEPSECS pathogenic
variants featuring unusual phenotype of complex movement disorder
with thin corpus callosum: A case report. Neurol Genet.
8(e661)2021.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Martínez-Martín Á, García-García J,
Díaz-Maroto Cicuéndez I, Quintanilla-Mata ML and Segura T: Bringing
light into the darkness: Autosomal recessive cerebellar ataxia due
to a recessive mutation in the SEPSECS gene. Neurologia (Engl Ed).
37:709–710. 2022.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Ramadesikan S, Hickey S, De Los Reyes E,
Patel AD, Franklin SJ, Brennan P, Crist E, Lee K, White P, McBride
KL, et al: Biallelic SEPSECS variants in two siblings with
pontocerebellar hypoplasia type 2D underscore the relevance of
splice-disrupting synonymous variants in disease. Cold Spring Harb
Mol Case Stud. 8(a006165)2022.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Rong T, Yao R, Deng Y, Lin Q, Wang G, Wang
J, Jiang F and Jiang Y: Case Report: A relatively mild phenotype
produced by novel mutations in the SEPSECS Gene. Front Pediatr.
9(805575)2022.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Schlüter A, Rodríguez-Palmero A, Verdura
E, Vélez-Santamaría V, Ruiz M, Fourcade S, Planas-Serra L, Martínez
JJ, Guilera C, Girós M, et al: Diagnosis of Genetic White Matter
Disorders by Singleton Whole-Exome and Genome Sequencing Using
Interactome-Driven Prioritization. Neurology. 98:e912–e923.
2022.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Zhao R, Zhang L and Lu H: Analysis of the
Clinical Features and Imaging Findings of Pontocerebellar
Hypoplasia Type 2D Caused by Mutations in SEPSECS Gene. Cerebellum.
22:938–946. 2023.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Barone R, Alaimo S, Messina M, Pulvirenti
A and Bastin J: MIMIC-Autism Group. Ferro A, Frye RE and Rizzo R: A
Subset of Patients With Autism Spectrum Disorders Show a
Distinctive Metabolic Profile by Dried Blood Spot Analyses. Front
Psychiatry. 9(636)2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Barone R, Carchon H, Jansen E, Pavone L,
Fiumara A, Bosshard NU, Gitzelmann R and Jaeken J: Lysosomal enzyme
activities in serum and leukocytes from patients with
carbohydrate-deficient glycoprotein syndrome type IA
(phosphomannomutase deficiency). J Inherit Metab Dis. 21:167–172.
1998.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al: ACMG
Laboratory Quality Assurance Committee. Standards and guidelines
for the interpretation of sequence variants: A joint consensus
recommendation of the American College of Medical Genetics and
Genomics and the Association for Molecular Pathology. Genet Med.
17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Mailloux RJ, Jin X and Willmore WG: Redox
regulation of mitochondrial function with emphasis on cysteine
oxidation reactions. Redox Biol. 2:123–139. 2013.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Puppala AK, French RL, Matthies D, Baxa U,
Subramaniam S and Simonović M: Structural basis for early-onset
neurological disorders caused by mutations in human selenocysteine
synthase. Sci Rep. 6(32563)2016.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Muraresku CC, McCormick EM and Falk MJ:
Mitochondrial Disease: Advances in clinical diagnosis, management,
therapeutic development, and preventative strategies. Curr Genet
Med Rep. 6:62–72. 2018.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Ferreira GC and McKenna MC: L-Carnitine
and Acetyl-L-carnitine Roles and Neuroprotection in Developing
Brain. Neurochem Res. 42:1661–1675. 2017.PubMed/NCBI View Article : Google Scholar
|