Introduction
Gastric cancer is a common lethal malignancy and
treatment modalities for this advanced disorder remain limited.
With the combination of high incidence and poor prognosis, gastric
cancer holds the position of the fourth most common cancer and
second most common cause of mortality in the world (1,2).
Although the incidence of gastric cancer is declining in several
Western countries, it is predicted that the number of gastric
cancer cases globally will increase until 2050 (3). The virulence of gastric cancer is
correlated with aggressive tumor biology and also indicates the
current lack of effective systemic therapy. Dissecting the
mechanisms of gastric cancer growth and progression is, therefore,
critical to identify novel targets for desperately needed adjuvant
therapies.
Obesity is a worldwide epidemic and more than one
third of all adults are currently obese in the United States alone
(4). White adipose tissue is an
active endocrine organ, secreting a series of soluble mediators
called adipocytokines that play an important role in regulating
metabolism, inflammation and immunity. The altered adipocytokine
milieu of obesity results in a generalized proinflammatory state
and is a risk factor for developing systemic diseases, including
diabetes, atherosclerosis and asthma (5). Notably, obesity has also been
considered as a risk factor for developing numerous types of
malignant tumors, including adenocarcinoma of the colon, prostate
and breast (6). A meta-analysis
demonstrated that the body mass index is closely correlated with
the risk of gastric cancer, particularly cardia gastric cancer
(7). Limited fundamental research
exists regarding the mechanisms by which obesity affects gastric
cancer biology and no in vivo animal models currently exist.
Adopting this novel in vivo model, we sought to investigate
the effect of obesity on gastric cancer growth and progression.
Materials and methods
Animals and cell culture
All experiments were carried out with approval from
the Xi’an Jiaotong University Institutional Animal Care and Use
Committee (Xi’an, China). Three- to five-week-old male C57BL/6j
mice were obtained from Shanghai SLAC Laboratory Animal, Co., Ltd.
(Shanghai, China), housed in standard conditions, divided into 2
groups and fed with a high-fat diet (35.5% fat, 36.3% carbohydrate,
20.0% protein) and a normal diet (5.4% fat, 51.0% carbohydrate,
22.9% protein) (8) for 12 weeks,
respectively. The cancer cells used in animal studies, murine
forestomach carcinoma cell line (MFC), were established in the
Chinese Academy of Medical Sciences (9) and purchased from the Type Culture
Collection of the Chinese Academy of Sciences (Shanghai, China).
Cells were cultured in RPMI-1640 medium (Cellgro, Herndon, VA, USA)
and supplemented with 10% fetal bovine serum (Valley Biomedical,
Winchester, VA, USA), 1% penicillin/streptomycin and 1% glutamine
(Cellgro).
Experimental design
After 12 weeks, 12 mice consuming the normal-fat
diet were referred to as ‘lean’, and 12/20 mice consuming the
high-fat diet and chosen by the criterion that the body weight
exceeded the mean plus 2-fold standard deviation (SD) of the lean
mice were referred to as ‘obese’. All mice had insulin and glucose
tolerance tests to confirm altered metabolism between the obese and
lean mice. Then 2.0×106 MFC cells were injected
subcutaneously into the right flank. Mice were monitored daily, and
the body weight of the mice and tumor sizes were measured every 3
days. After 2 weeks of tumor growth, both obese and lean mice were
sacrificed after anesthesia and blood drawn from the retro-orbital
venous plexus was preserved for analysis of metabolites and flow
analysis of CD3+, CD4+/−, CD8+/− T
lymphocytes. Anti-mouse CD8a-APC, anti-mouse CD3e-FITC and
anti-mouse CD4-PE monoclonal antibodies were purchased from
eBioscience (San Diego, CA, USA). The tumors were carefully
dissected and preserved in formalin for histological evaluation, as
well as assay of proliferation and apoptosis. Laparotomy was
performed to determine the existence of metastasis, which was
confirmed by histology.
Insulin and glucose tolerance tests
In order to determine the influence of obesity on
glucose regulation and insulin sensitivity, we performed the
insulin and glucose tolerance tests on obese and lean mice. The
insulin tolerance test was performed at noon by intraperitoneal
(i.p.) injection of 0.75 U/kg insulin, the i.p. glucose tolerance
test was performed after overnight fasting by administering 20%
glucose to mice (8). Blood glucose
was measured using a Glucometer Elite (Bayer, Elkhart, IN, USA) at
the indicated time points.
Tumor characteristics
Cellular proliferation was assayed by DNA
incorporation of 5-bromodeoxyuridine (5-BrdU). The mice were
injected i.p. with 120 mg/kg 5-BrdU 1 h after sacrifice (10). A monoclonal 5-BrdU antibody and
streptavidinbiotin staining system was used according to the
manufacturer’s instructions (Santa Cruz Biotechnology, Inc., Santa
Cruz, CA, USA), and the quantity of positively stained cells/10
high-power fields (original magnification, ×40) of formalin-fixed,
paraffin-embedded tumor sections was analyzed.
Terminal deoxynucleotidyl transferase mediated
deoxyuridine triphosphate nick-end labeling (TUNEL) assay (Roche
Diagnostics, Brussels, Belgium) was used to identify and quantify
apoptosis in formalin-fixed, paraffin-embedded tumor sections.
Nuclear condensation, perinuclear clearing and cell shrinkage
suggested TUNEL positivity and the number of cells/10 high-power
fields was recorded. The number and extent of adipocytes and
microvessel density were measured in 10 high-power fields of tumor
sections that were stained with hematoxylin and eosin (H&E).
All histological analyses were performed by three independent
observers who were unaware of the tumor tissue source.
Serum assays
Enzyme-linked immunosorbent assay (ELISA) was used
to determine serum concentration of insulin (Crystal Chem, Inc.,
Downers Grove, IL, USA) and visfatin (Linco Research, Inc., St.
Charles, MO, USA) following the manufacturer’s instructions. Serum
glucose was determined by colorimetric assay.
Statistical analysis
Values are expressed as the mean ± SD. Analysis of
the data and plotting of the figures were performed with the aid of
software (Origin version 7.5 and SPSS version 13.0). Analysis of
variance and the Tukey’s test were applied where appropriate.
P<0.05 was considered to indicate a statistically significant
result.
Results
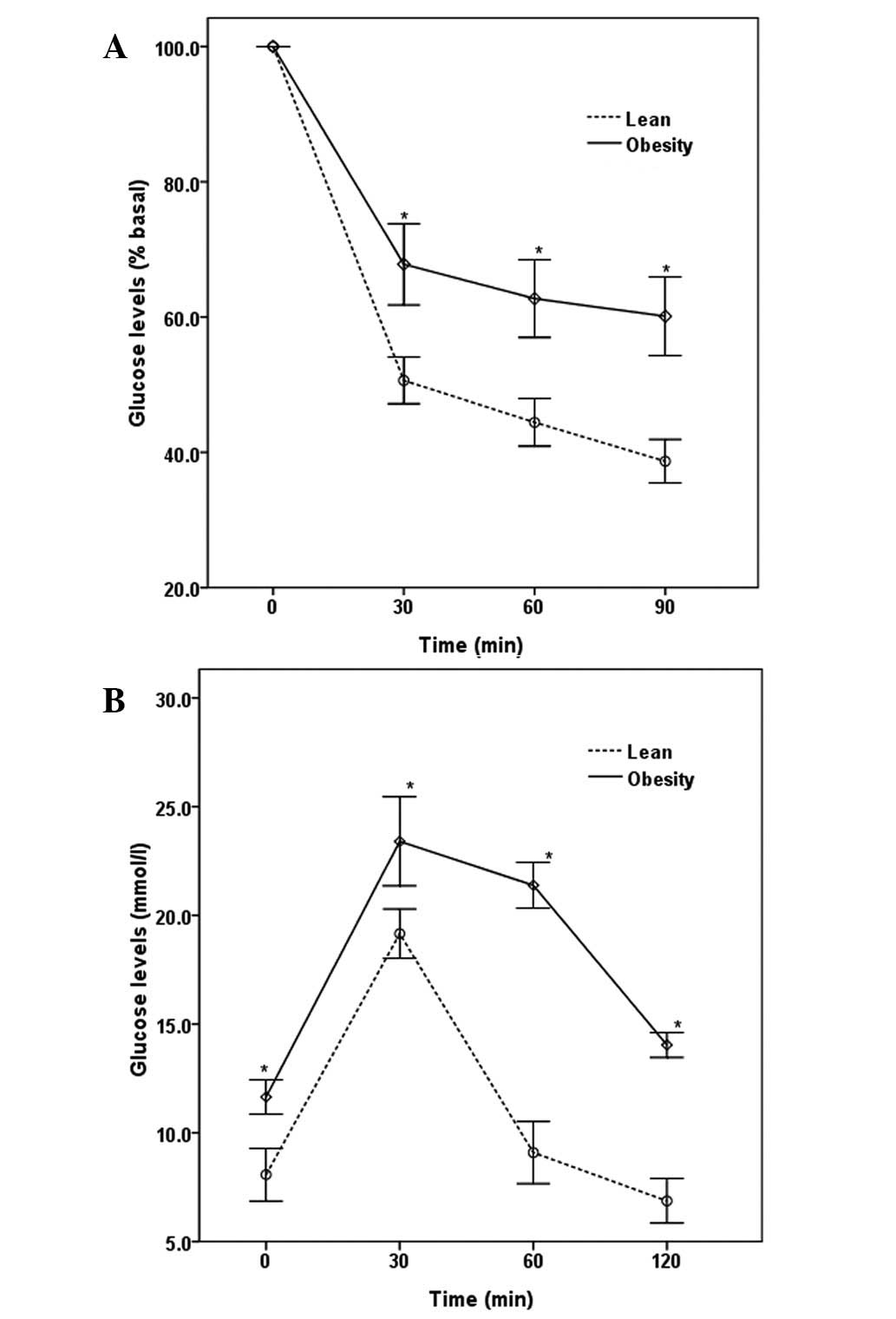
Metabolic changes in mice
The mice were maintained on a normal or high-fat
diet for 12 weeks. At the time of transplantation, the obese mice
were significantly heavier than the lean animals (35.42±2.83 vs.
28.50±1.15 g; P<0.01). Several metabolic parameters were also
altered in the obese mice, and those mice were insulin resistant
and glucose intolerant (Fig.
1).
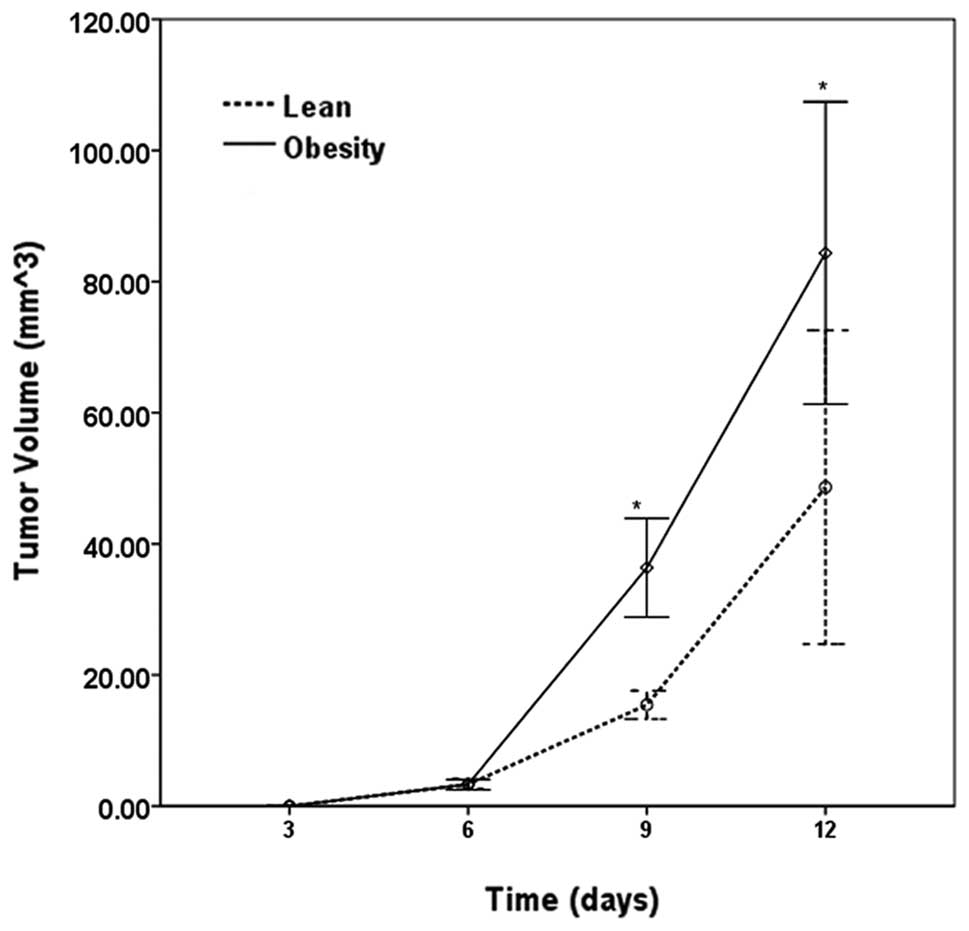
Tumor growth, metastasis and
mortality
Injected animals were maintained on a normal- or
high-fat diet for another 2 weeks. All mice were alive and
metastases were not detected during the experimental time frame.
The tumors became palpable 4 days after injection and tumor growth
was observed in 10/12 (83.3%) of the lean mice and in 100% of the
obese mice. Tumors grew larger and faster in the obese mice than
those in the lean mice within 2 weeks (Fig. 2). Both obese and lean mice were
then sacrificed after anesthesia and the tumors were preserved. The
tumor weights were as follows: lean, 77.2±14.9 mg; obese,
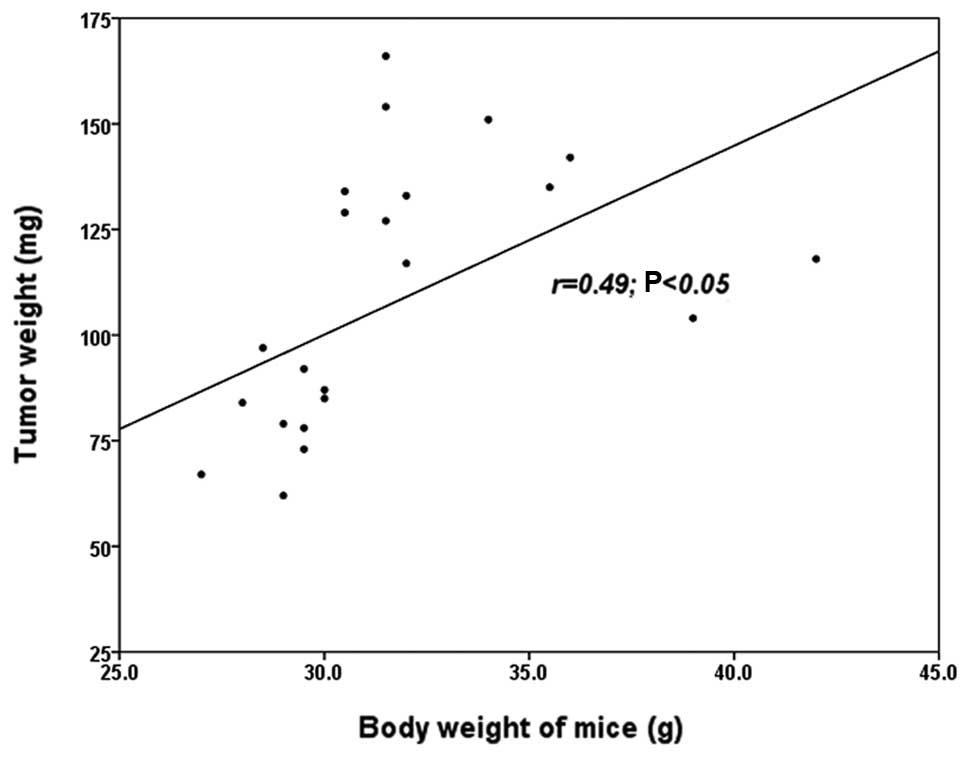
134.2±17.3 mg (P<0.05 vs. lean). Tumor weight demonstrated a
strong positive correlation with the body weight of the mice
(r=0.49, P<0.05; Fig. 3).
T lymphocytes in cell immunity
When tumors grew for 2 weeks, mice were sacrificed
after anesthesia, and blood was collected to isolate lymphocytes
for flow analysis of CD3+, CD4+/−,
CD8+/− T lymphocytes. Obese mice had a significantly
lower level of CD3+, CD3+CD4+ T
cells (P<0.05), and a lower level of
CD4+/CD8+ in peripheral blood compared with
these levels in the lean animals (P<0.05). No difference between
obese and lean mice in regards to the levels of
CD3+CD8+ T lymphocytes was observed
(P>0.05; Table I).
 | Table IT lymphocytes involved in cell
immunity in the peripheral blood of the obese and lean mice. |
Table I
T lymphocytes involved in cell
immunity in the peripheral blood of the obese and lean mice.
| Group | CD3+
(%) |
CD3+CD4+ (%) |
CD3+CD8+ (%) |
CD4+/CD8+ (%) |
|---|
| Lean | 37.44±6.10 | 19.49±3.71 | 15.99±2.96 | 1.23±0.18 |
| Obese | 32.23±2.86a | 13.87±1.98a | 17.47±1.95 | 0.80±0.13a |
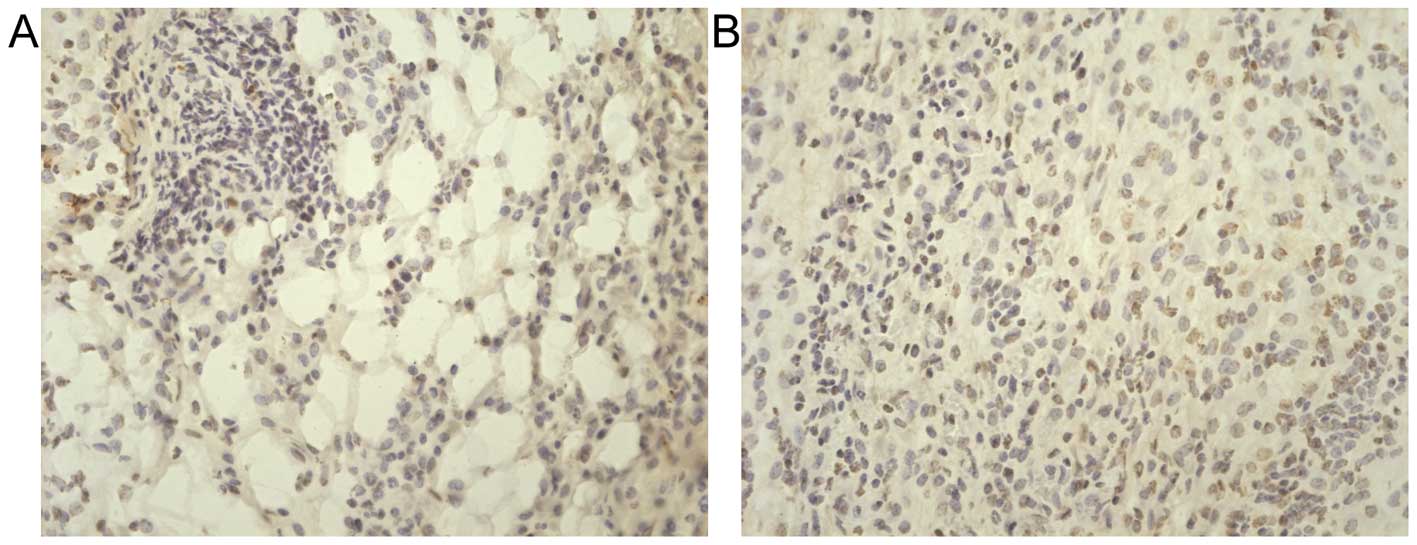
Proliferation and apoptosis
Tumor proliferation was measured by 5-BrdU uptake
and apoptosis was measured by TUNEL assay. Cellular 5-BrdU uptake
in tumors from lean and obese mice was as follows: 47.7±10.2
cells/hpf; 88.1±8.8 cells/hpf (P<0.01 vs. lean; Fig. 4). Cellular apoptosis in tumors from
lean mice and obese mice were as follows: 54.7±5.7 cells/hpf;
34.7±4.6 cells/hpf (P<0.01 vs. lean; Fig. 5). These results demonstrated that
increased tumor size in obese mice was not only correlated with
apoptotic arrest, but also was a function of more rapid tumor
proliferation.
Metabolic parameters
Table II shows the
results of serum glucose, insulin and visfatin concentrations, as
well as the homeostatic model assessment (HOMA) score, which is a
measure of insulin resistance. Obese mice were hyperglycemic,
hyperinsulinemic and insulin resistant. Serum visfatin was
increased in obese compared to that in the lean mice.
| Table IISerum metabolic parameters in
mice. |
Table II
Serum metabolic parameters in
mice.
| Group
|
|---|
| Parameters | Obese | Lean |
|---|
| Visfatin (ng/ml) | 44.3±3.6a | 39.6±3.4 |
| Insulin (mU/l) | 133.2±19.8a | 104.7±11.8 |
| Glucose
(mmol/l) | 11.9±1.6a | 7.8±1.6 |
| HOMA-IR | 4.2±0.2a | 3.8±0.2 |
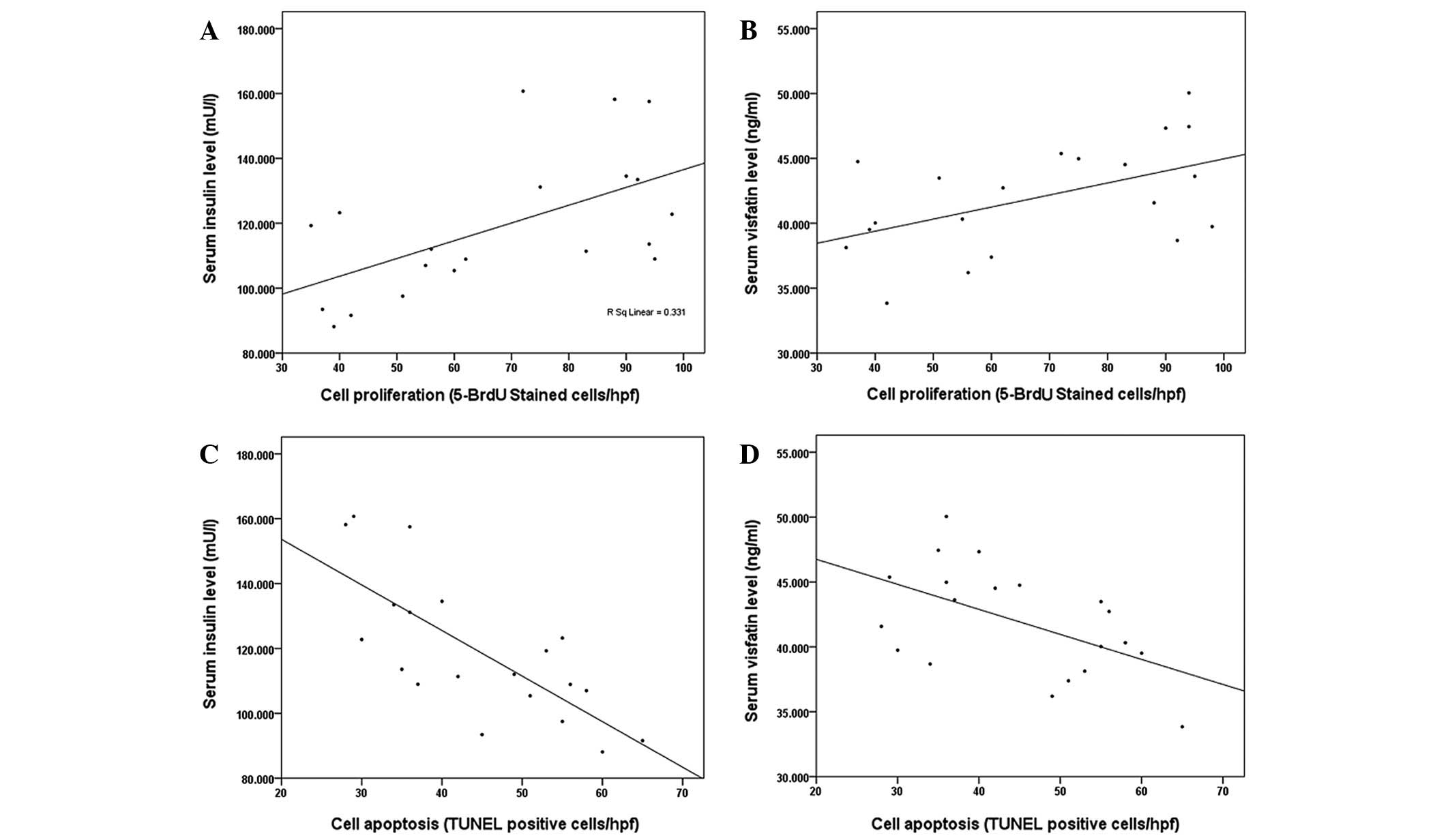
Tumor proliferation correlated significantly with
serum insulin (r=0.58, P=0.01; Fig.
6A) and visfatin concentrations (r=0.51, P=0.02; Fig. 6B), while it did not correlate with
serum glucose concentration (r=0.20, P=0.1). Tumor apoptosis showed
a strong negative correlation with circulating insulin (r=−0.74,
P<0.01; Fig. 6C) and serum
visfatin concentrations (r=−0.53, P=0.02; Fig. 6D), but not with glucose
concentration (r=−0.16, P=0.25). The level of CD3+,
CD3+CD4+ T cells did not correlate
significantly with serum glucose, insulin or visfatin
concentrations (P>0.05).
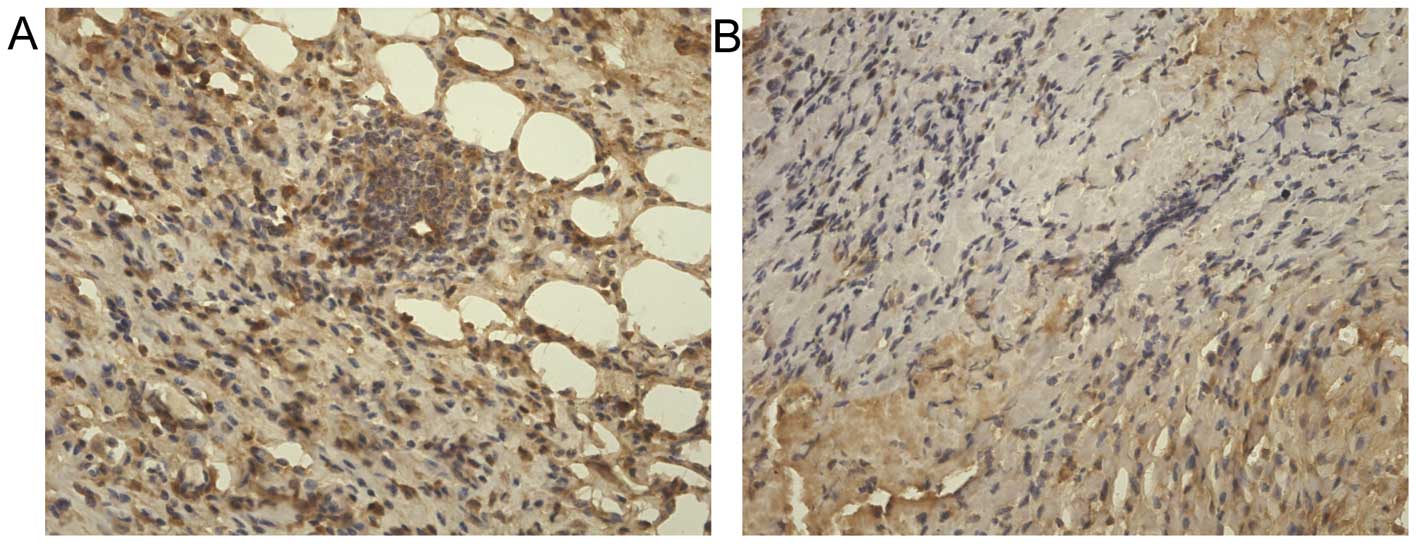
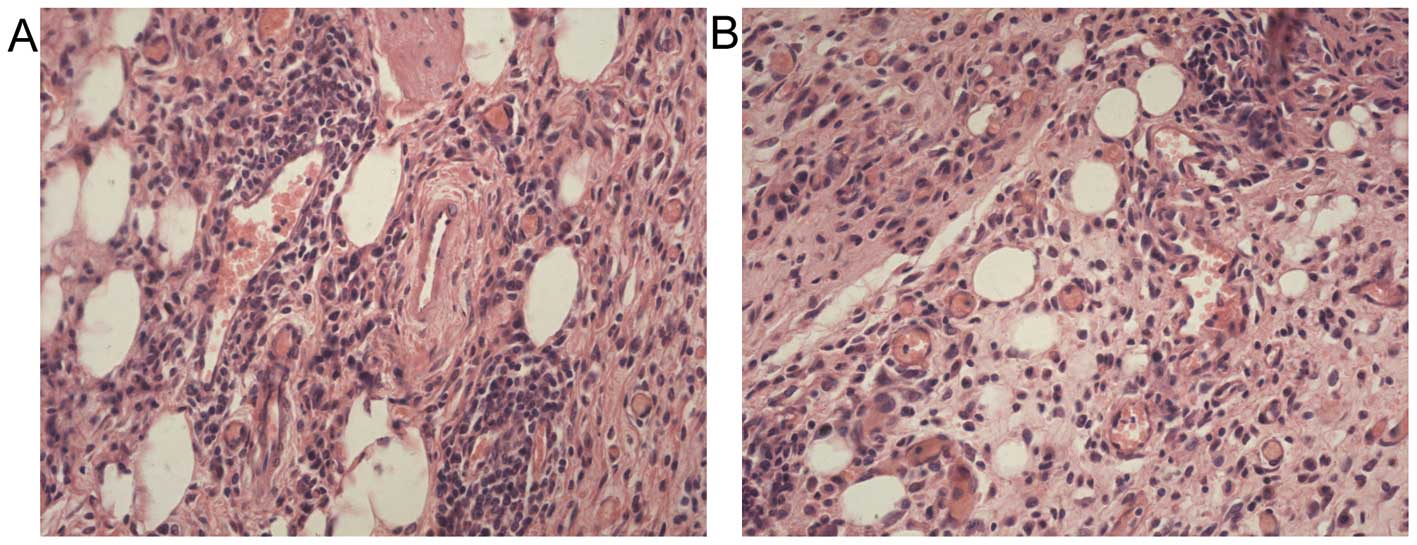
Tumor microenvironment
Microscopic analysis of tumors demonstrated an
interesting observation. In addition to the fibrosis typically
apparent in gastric cancers, a significant number of adipocytes
were present within the tumors (Fig.
7). Although the average number of adipocytes exhibited no
changes among tumors from obese and lean mice (17.2±4.2 cells/hpf
vs. 15.4±2.7 cells/hpf, P>0.05), intratumoral adipocytes present
in tumors from obese mice were significantly larger than those from
lean animals (169.9±5.7 vs. 67.3±8.2 μm2, P<0.01).
Another notable observation was that a great number of microvessels
existed within the tumors. The microvessel density in the tumors
from obese mice was greater than those from lean mice, but the
difference was not significance (9.2±1.0 vs. 7.1±1.5/hpf,
P>0.05).
Discussion
This study is the first to use a completely novel
in vivo animal model of gastric cancer growth in obesity.
The fact that tumors grew larger and faster in obese mice relative
to lean animals provides powerful evidence for the direct influence
of obesity on gastric cancer growth. Rapid tumor growth was not
only a function of decreased apoptosis, but was also correlated
with increased cellular proliferation. Notably, tumor cell
proliferation was positively correlated with serum visfatin and
insulin concentrations, and tumor apoptosis showed a strong
negative correlation with circulating visfatin and serum insulin
concentrations. Tumor survival and growth in immunocompetent mice
correlated with T lymphocyte levels involved in cell immunity;
fewer CD3+, CD3+CD4+ T cells in
peripheral blood from obese than lean mice led to tumors growing
larger and faster in obese relative to lean mice.
A significant observation was the unexpected
difference in tumor microenvironment. Tumors growing in obese mice
had significantly greater adipocyte mass than tumors from lean
animals. The effect of the tumor microenvironment on cancer growth
and progression has become well understood, but the provocative
concept that adipocytes active in metabolism may correlate with
this milieu is completely novel and deserves further investigation.
This model clearly constitutes a powerful instrument with which to
further understand the mechanisms by which obesity affects gastric
cancer growth.
Obesity always accompanies hypertriglyceridemia and
hypercholesterolemia. Hypertriglyceridemia, but not
hypercholesterolemia, was found to be an independent risk factor
for lymph node metastasis in male patients of early gastric cancer,
indicating that elevated serum TG levels may provide circumstances
conducive to the development of lymph node metastasis in the early
stage of gastric cancer, at least in male patients (11,12).
The long-term survival of patients with gastric cancer is governed
by the volume of intraperitoneal adipose tissue, and obese patients
with stage 2 had a significantly lower mean survival rate than lean
patients (13,14). Carcinogenesis in obese patients is
determined by a range of important mechanisms and metabolites,
including insulin, insulin resistance, inflammatory cytokines and
visfatin (15,16), and a strong positive correlation
was observed between serum insulin concentration and gastric cancer
cell proliferation in this report. Despite these observations, the
mechanisms by which obesity affects gastric cancer growth and
progression remain completely unknown.
White adipose tissue was considered to be an inert
tissue functioning solely as an energy store for a number of years,
but has currently attracted increased attention for secreting
adipocytokines as an endocrine tissue. Adipocytokines are small,
hormonally active molecules that are structurally similar to
cytokines and are produced mainly by adipocytes. These pleiotropic
compounds exert a wide range of biological functions that include
inflammation, immunity and other metabolic effects (5). As such, the altered adipocytokine
milieu of obesity is a striking mechanistic link to potentiating
tumor growth and progression. It is worthwhile to note the role
played by visfatin in mediating insulin sensitivity, and this
prominent adipocytokine may well prove to be an important
mechanistic link in the network of factors affecting
obesity-associated tumor growth.
Visfatin was originally identified as pre-B-cell
colony-enhancing factor (PBEF), a putative cytokine isolated from
peripheral blood lymphocytes, and described as a secreted growth
factor for early B cell proliferation (17), and drew more attention after
Fukuhara et al (18)
reported it as an insulin-mimetic adipocytokine secreted by
visceral fat. The circulating visfatin concentration increases with
increasing obesity, and contributes to a general proinflammatory
state in the periphery, and is gaining more attention and is a more
widely studied adipocytokine in relation to cancer biology. To
date, however, these studies have been limited to in vitro
models. Generally, visfatin has been observed to potentiate tumor
proliferation and metastasis in a variety of cancers including
breast (19,20) and prostate (21). Common mechanistic pathways include
activation of the extracellular signal regulated kinase (ERK1/2)
pathway and phosphorylation of the signal transducer and activator
of transcription 3 (STAT3) (22).
In the current study, circulating visfatin concentration correlated
with tumor progression in malignant astrocytomas (23), gastric cancer (24), colorectal cancer (25) and this report demonstrated the
association with gastric cancer growth and visfatin, that played an
important role in increasing gastric cancer cell proliferation and
decreasing cell apoptosis.
One limitation of the murine model is that the MFC
cell line originated from 615 mice, not from C57BL/6j. MFC was
established in the Chinese Academy of Medical Sciences in the late
1980s by culturing small pieces of forestomach squamous cell
carcinoma xenograft tumor from 615 mice. The 615 mice, with a
partial gene background of C57BL/6j mice, were established in 1961
by the Blood Transfusion and Blood Institute, Chinese Academy of
Medical Sciences (9). Therefore,
C57BL/6j mice possessing a complete immune system may reject the
MFC xenograft. However, we observed that the MFC xenograft injected
subcutaneously in C57BL/6j mice grew larger in 2 weeks, then became
smaller after a longer time. Also, this report investigated the
changes in T lymphocytes in cell immunity correlating with MFC
survival in C57BL/6j mice. Fewer CD3+,
CD3+CD4+ T cells in peripheral blood from
obese than lean mice led to tumor survival and increased growth in
obese mice relative to lean mice. Another limitation of the murine
model is the small volume of serum and tissue available for
analysis. We measured the visfatin concentration which is relative
to carcinogenesis and was recently identified as an adipocytokine.
It is likely that some cytokines and adipocytokines are involved in
regulating gastric cancer growth in obesity. Further studies are
required to completely characterize the model.
In the present study, simple hematoxylin and eosin
(H&E) histology indicated a completely striking and most
provocative observation. Tumors that originated from the same cell
type grew and exhibited a discrete physiological in vivo
phenotype, and developed remarkably different microenvironments.
Specifically, tumors from obese mice had significantly greater
adipocyte mass than those tumors growing in lean mice. Adipose
stromal cells promote tumor growth by secreting adipocytokines
assisting in the formation of new blood vessels, a process
necessary for the expansion of tumor mass, indicating that the
tumor microenvironment in cancer may be modulated by white adipose
tissue-derived trophic factors in a paracrine rather than in an
endocrine manner, and stromal and vascular progenitor cells from
white adipose tissue grafts were also associated with acceleration
of cancer progression (26,27).
The complicated interaction between extracellular matrix stromal
cells and epithelial cells has attracted much recent attention, and
several peices of evidence support the concept that aberrant
signaling by extracellular matrix cells directly affects epithelial
carcinogenesis and potentiates metastasis of malignant cells
(28). As a result, we suggest
that adipocytes active in metabolism play an important role in the
process, and the potential to identify unique therapeutic targets
is clear. In conclusion, these experiments show the first in
vivo study of gastric cancer in the context of obesity. These
data support the concept that insulin resistance and the altered
adipocytokine milieu observed in obesity may lead directly to
changes in the tumor microenvironment, thereby potentiating gastric
cancer growth.
Acknowledgements
The authors thank Dr Hai-Tao Shi for
his technical assistance from the GI Medicine Department of The
Second Affiliated Hospital and the Institution of Genetic Disease
Research of Xi’an Jiaotong University (Xi’an, China). This study
was supported by a grant from the National Natural Science
Foundation of China (no. 81172357).
References
|
1
|
Parkin DM, Bray F, Ferlay J and Pisani P:
Global cancer statistics, 2002. CA Cancer J Clin. 55:74–108. 2005.
View Article : Google Scholar
|
|
2
|
Brenner H, Rothenbacher D and Arndt V:
Epidemiology of stomach cancer. Methods Mol Biol. 472:467–477.
2009. View Article : Google Scholar
|
|
3
|
Forman D and Burley VJ: Gastric cancer:
global pattern of the disease and an overview of environmental risk
factors. Best Pract Res Clin Gastroenterol. 20:633–649. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Flegal KM, Carroll MD, Kit BK and Ogden
CL: Prevalence of obesity and trends in the distribution of body
mass index among US adults, 1999–2010. JAMA. 307:491–497. 2012.
|
|
5
|
Tilg H and Moschen AR: Adipocytokines:
mediators linking adipose tissue, inflammation and immunity. Nat
Rev Immunol. 6:772–783. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Calle EE and Kaaks R: Overweight, obesity
and cancer: epidemiological evidence and proposed mechanisms. Nat
Rev Cancer. 4:579–591. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yang P, Zhou Y, Chen B, Wan HW, Jia GQ,
Bai HL and Wu XT: Overweight, obesity and gastric cancer risk:
results from a meta-analysis of cohort studies. Eur J Cancer.
45:2867–2873. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yakar S, Nunez NP, Pennisi P, et al:
Increased tumor growth in mice with diet-induced obesity: impact of
ovarian hormones. Endocrinology. 147:5826–5834. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Qian SS, Gao J, Wang JX, Liu Y and Dong
HY: Establishment of a mouse forestomach carcinoma cell line (MFC)
with spontaneous hematogenous metastasis and preliminary study of
its biological characteristics. Zhonghua Zhong Liu Za Zhi.
9:261–264. 1987.(In Chinese).
|
|
10
|
Zyromski NJ, Mathur A, Pitt HA, et al:
Obesity potentiates the growth and dissemination of pancreatic
cancer. Surgery. 146:258–263. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kitayama J, Tabuchi M, Tsurita G, Ishikawa
M, Otani K and Nagawa H: Adiposity and gastrointestinal malignancy.
Digestion. 79:26–32. 2009. View Article : Google Scholar
|
|
12
|
Kitayama J, Hatano K, Kaisaki S, Suzuki H,
Fujii S and Nagawa H: Hyperlipidaemia is positively correlated with
lymph node metastasis in men with early gastric cancer. Br J Surg.
91:191–198. 2004. View
Article : Google Scholar
|
|
13
|
Moriwaki Y, Kunisaki C, Kobayashi S,
Harada H, Imai S and Kasaoka C: Does body mass index (BMI)
influence morbidity and long-term survival in gastric cancer
patients after gastrectomy? Hepatogastroenterology. 50:284–288.
2003.PubMed/NCBI
|
|
14
|
Ojima T, Iwahashi M, Nakamori M, et al:
Influence of overweight on patients with gastric cancer after
undergoing curative gastrectomy: an analysis of 689 consecutive
cases managed by a single center. Arch Surg. 144:351–358. 2009.
View Article : Google Scholar
|
|
15
|
Payer J, Jackuliak P and Nagyová M:
Obesity and a risk of carcinoma. Vnitr Lek. 56:1082–1087. 2010.(In
Slovak).
|
|
16
|
Pollak M: Insulin-like growth
factor-related signaling and cancer development. Recent Results
Cancer Res. 174:49–53. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Samal B, Sun Y, Stearns G, Xie C, Suggs S
and McNiece I: Cloning and characterization of the cDNA encoding a
novel human pre-B-cell colony-enhancing factor. Mol Cell Biol.
14:1431–1437. 1994.PubMed/NCBI
|
|
18
|
Fukuhara A, Matsuda M, Nishizawa M, et al:
Visfatin: a protein secreted by visceral fat that mimics the
effects of insulin. Science. 307:426–430. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim SR, Park HJ, Bae YH, et al: Curcumin
down-regulates visfatin expression and inhibits breast cancer cell
invasion. Endocrinology. 153:554–563. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kim JG, Kim EO, Jeong BR, et al: Visfatin
stimulates proliferation of MCF-7 human breast cancer cells. Mol
Cells. 30:341–345. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Patel ST, Mistry T, Brown JE, Digby JE,
Adya R, Desai KM and Randeva HS: A novel role for the adipokine
visfatin/pre-B cell colony-enhancing factor 1 in prostate
carcinogenesis. Peptides. 31:51–57. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bi TQ and Che XM: Nampt/PBEF/visfatin and
cancer. Cancer Biol Ther. 10:119–125. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Reddy PS, Umesh S, Thota B, et al:
PBEF1/NAmPRTase/visfatin: a potential malignant
astrocytoma/glioblastoma serum marker with prognostic value. Cancer
Biol Ther. 7:663–668. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nakajima TE, Yamada Y, Hamano T, et al:
Adipocytokine levels in gastric cancer patients: resistin and
visfatin as biomarkers of gastric cancer. J Gastroenterol.
44:685–690. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nakajima TE, Yamada Y, Hamano T, et al:
Adipocytokines as new promising markers of colorectal tumors:
adiponectin for colorectal adenoma, and resistin and visfatin for
colorectal cancer. Cancer Sci. 101:1286–1291. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang Y, Daquinag A, Traktuev DO, et al:
White adipose tissue cells are recruited by experimental tumors and
promote cancer progression in mouse models. Cancer Res.
69:5259–5266. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang Y, Bellows CF and Kolonin MG:
Adipose tissue-derived progenitor cells and cancer. World J Stem
Cells. 2:103–113. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Comoglio PM and Trusolino L: Cancer: the
matrix is now in control. Nat Med. 11:1156–1159. 2005. View Article : Google Scholar : PubMed/NCBI
|