Introduction
Advances in medicine over the past few decades
significantly lowered cardiovascular disease-linked mortality by up
to 75% and increased the survival rate of patients with cardiac
disease, but at the same time this has led to a great increase in
the number of people surviving with injured heart (1). However, the increasing incidence of
obesity, and associated hypertension and diabetes coupled with
unhealthy lifestyles is causing a significant increase in the
number of surviving individuals with heart disease, adding burden
on the society in terms of health, economy and productivity
(2,3). Several diseases and metabolic
disturbances can be contributed to heart failure (HF) and these
include myocardial infarction (MI), hypertension, valvular disease,
genetic disorders, diabetes and obesity. HF occurs because of the
compromised ability of myocardium to exert systolic contraction
with enough force to pump blood; with characteristic reduced
ejection fraction or it can be due to lowered diastolic filling,
but with the preservation of ejection fraction. While acute HF is
the sudden appearance of HF symptoms such as congestion and
difficulty to breath (4), chronic HF
is marked by the inability of heart to function optimally over an
extended period of time (4,5).
Pathological remodeling of the myocardium is an
integral part of the HF syndromes (6), which involves altered gene expression
and disturbed signaling pathways and altered contractile response
of myocardium. Of particular significance are the changes in the
functionality of proteins that play a central part in intracellular
Ca2+ handling as well as ion channels involved in
Ca2+ transport. Cardiac muscle contraction is dependent
on the maintenance of Ca2+ homeostasis, which is
essential for excitation-contraction (E-C) coupling of
cardiomyocyte. Thus, electrical depolarization of the cardiomyocyte
membrane swiftly moves to the center of the cell via the network of
transverse tubules (t-tubules), which terminate close to
sarcoplasmic reticulum (SR), with a 12 nm gap. Membrane
depolarization triggers the rapid diffusion of extracellular
Ca2+ to the SR, through these gaps, facilitated by the
L-type Ca2+ channels (LTCC). This influx of
Ca2+ leads to Ca2+-induced calcium release
(CICR) from the SR through the type 2-ryanodine receptor (RyR2).
This elevated calcium promotes cross-bridge cycling by relieving
actin from troponin C-dependent inhibition, thereby causing
cardiomyocyte contraction. Then, Ca2+ is taken back into
the SR lumen by the SR Ca2+ ATPase 2a (7). Considering the significance of
Ca2+ in heart muscle contraction, it is appreciated that
disturbances in the Ca2+ handling machinery in
cardiomyocyte can potentially lead to HF. Thus HF is characterized
by disturbed Ca2+ leak from SR, mediated by RyR2, even
though the precise mechanisms are not clear (8). Other players such as SERCA are also
deranged in HF. Besides membrane depolarization, intracellular
Ca2+ is important in many other cell processes including
oxidative stress, mitochondrial function, apoptosis and
autophagy.
Ca2+/calmodulin-dependent protein
kinase II
Several studies have indicated that a
Ca2+-regulated protein kinase,
Ca2+/calmodulin-dependent protein kinase II (CaMKII)
plays a critical role in E-C coupling, contractility of
cardiomyocyte (9,10), mitochondrial function and
cardiomyocyte survival (11,12). Expression and activity of CaMKII have
been found to be elevated in various conditions of stressed
myocardium and in different heart diseases in both animal models as
well as heart patients (9–14). The activation of CaMKII can be either
at the level of this enzyme protein itself or at an upstream
signaling event involving catecholamines (15) or renin-angiotensin-aldosterone
systems (16). Abnormally elevated
CaMKII activity can cause dysfunction of several downstream events
whose components are regulated by CaMKII, such as E-C coupling,
structural remodeling, and transcriptional activation of certain
inflammatory proteins and apoptosis (17).
Structure/function features of CaMKII
CaMKII is a serine/threonine kinase with a broad
range of protein substrates and wide tissue distribution. There are
4 isoforms of CaMKII (α, β, δ and γ), coded for by 4 separate
genes, with heart expressing predominantly the δ-isoform, with some
γ-isoform as well. Alternate splicing of mRNA adds further
complexity to the CaMKII isoforms and their function and regulation
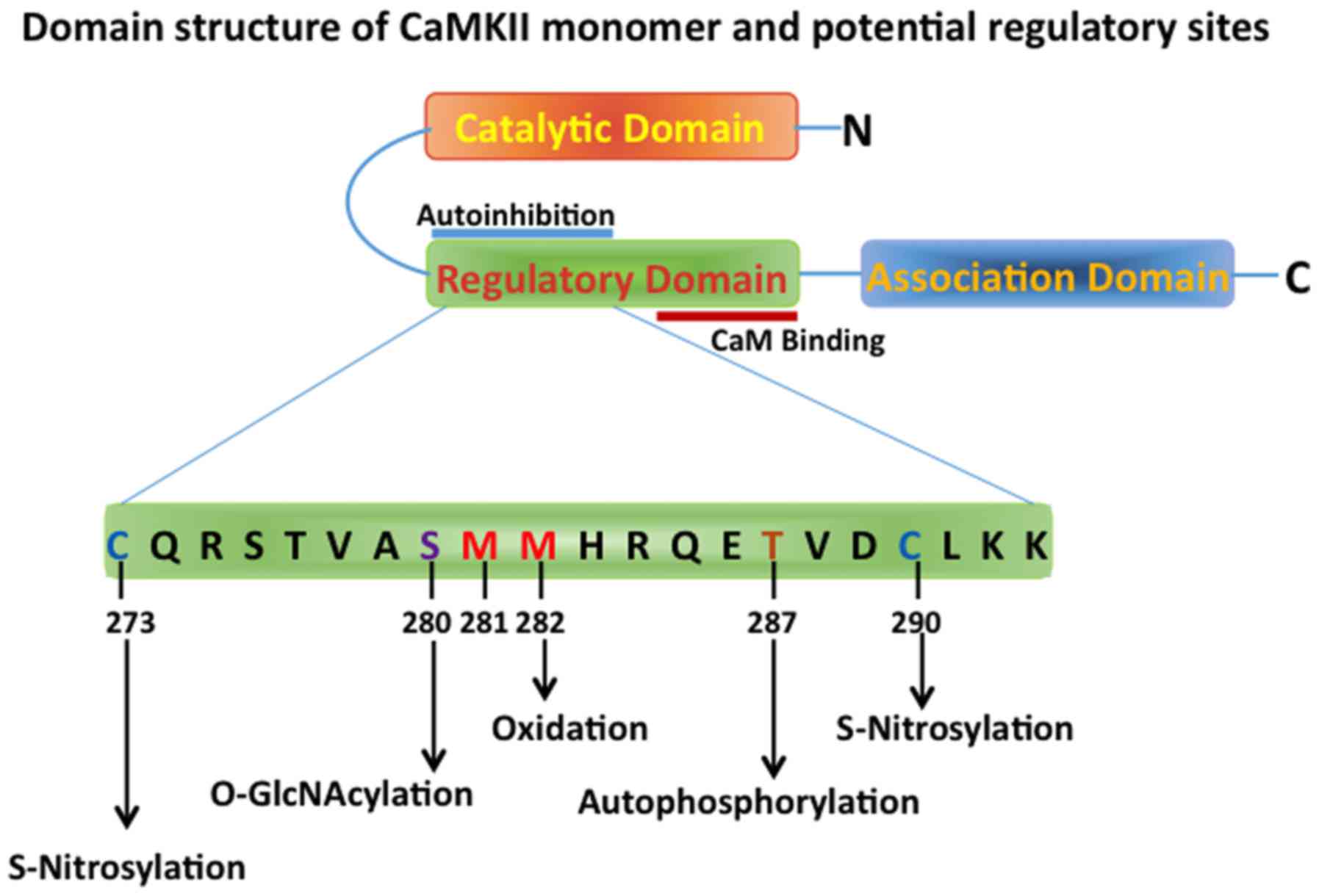
(18). There are three main domains
in the CaMKII monomer-N-terminal catalytic domain, regulatory
domain and the C-terminal association domain (Fig. 1) (19). The regulatory domain, which interacts
with the catalytic site, maintains the catalytic activity low under
unstimulated basal conditions, and contains binding sites for
Ca2+ and calmodulin. The C-terminal association domain
participates in the multimerization process, thus forming the
mature dodecameric holoenzyme, with two hexameric stacked rings
(20). Complex of calmodulin and
Ca2+ binds with the regulatory domain and this displaces
this domain from the catalytic domain thereby restoring the
activity of the enzyme (activation) and also exposes certain other
regulatory binding sites, which can influence the CaMKII activity.
CaMKII can phosphorylate several proteins involved in
Ca2+ homeostasis, the well-studied protein targets being
LTCC, RyR2, voltage-gated Na+ channel and K+
channels (21,22) and also ATP-sensitive K+
channels (23) and chloride channels
(24), which have been shown to be
important for cardiac arrhythmias. Regulation and activity level of
CaMKII depends upon its holoenzymic state and post-translational
modifications including phosphorylation, glycosylation and
oxidation. CaMKII is known to autophosphorylate itself at
Thr286/287 residue of the calmodulin-Ca2+ bound
catalytic domain, mediated by another adjacent catalytic domain.
This autophosphorylation renders the catalytic domain to maintain
its activity even in the absence of calmodulin and Ca2+
(25,26).
Organization of CaMKII in
cardiomyocytes
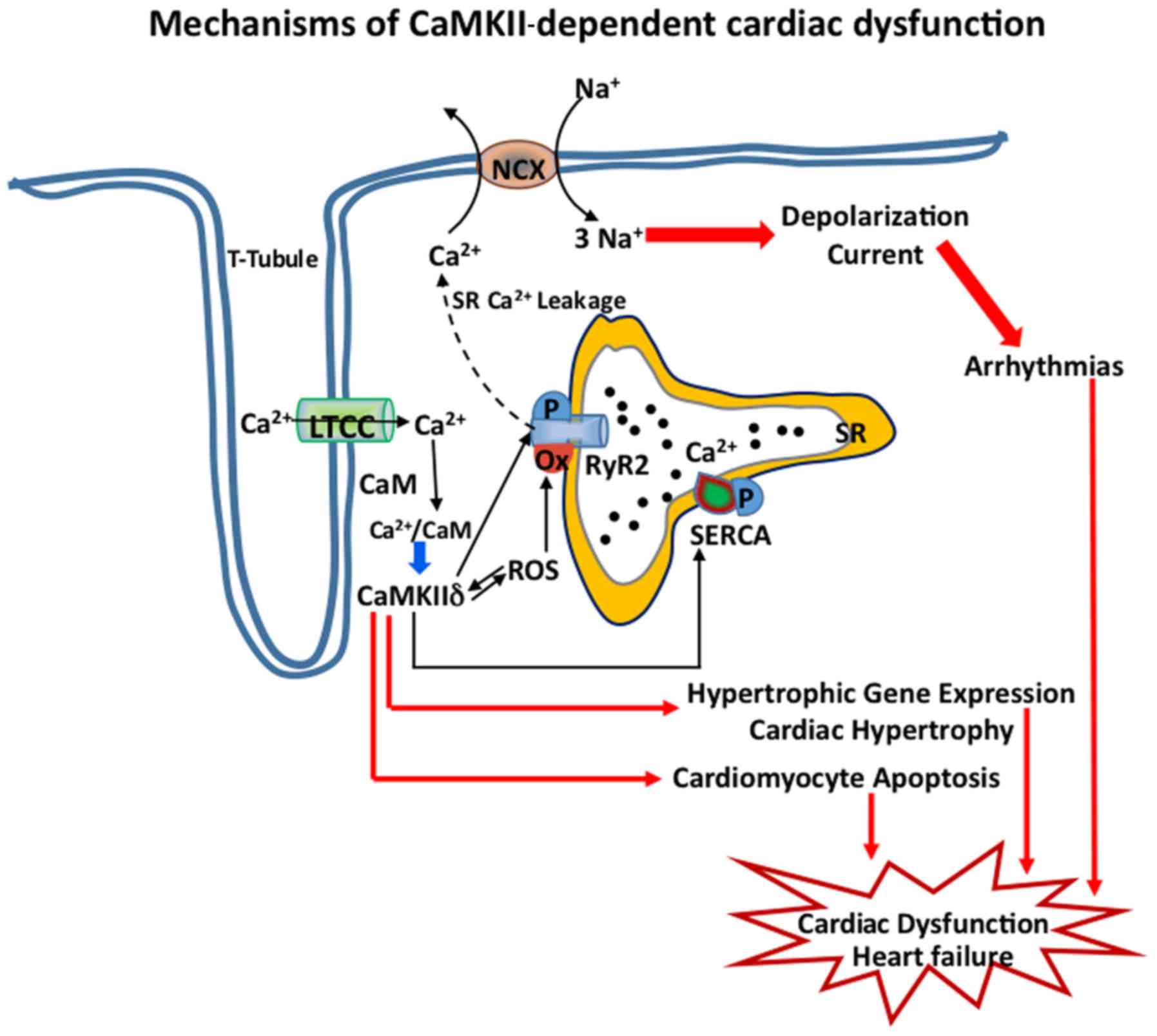
Subcellular localization is critical for the
maintenance of membrane excitability and CaMKII is found to be
distributed in high density near the t-tubules of cardiomyocyte,
close to LTCC (Cav1.2) and to RyR2 channels of SR, which regulate
the Ca-induced Ca release intracellularly (Fig. 2). Thus, phosphorylation of S2814 of
RyR2 by CaMKII leads to dysregulated intracellular Ca2+
homeostasis, which in turn cause perturbation of maladaptive stress
response and proarrhythmic events, thus further aggravating the HF
(Figs. 1 and 2). Thus, mouse models which express RyR2
with S2814A mutation and thus are not phosphorylated by CaMKII, are
protected from pressure overload in vivo (27). CaMKII is also found in mitochondria,
nucleus and near the intercalated disc (17). CaMKII subcellular localization
appears to be dependent on the nature of the target and its
location and the presence of interacting domains on the target.
Thus α- and β-subunits of LTCC, which are phosphorylated by CaMKII,
bind with CaMKII, because of the homology between the
phosphorylation sites and the auto-inhibitory region of the CaMKII
(21,28). A similar homology domain, as seen in
the LTCC β-subunit, is also found in the actin-associated protein,
βIV-spectrin, to which CaMKII is known to bind. This interaction is
a prerequisite for the CaMKII-mediated phosphorylation of the
voltage-gated Na+ channels at the intercalated disc in
cardiomyocytes (29).
Evidence for CaMKII as a therapeutic target
in heart disease
CaMKII acts as a molecular nexus that connects
neurohumoral stimulation to HF and cardiac remodeling (20). There has been a significant
development in our understanding of the role of CaMKII in
cardiovascular diseases and several reports over the past two
decades have suggested such roles, making CaMKII a potential
therapeutic target. Thus, it has been noted that cytosolic CaMKIIδC
isoform as well as the nuclear CaMKIIδB isoform were found to be
elevated in the two ventricles of patients with ischemic
cardiomyopathy (30). There is also
a significant elevation of autonomous activity of CaMKII and its
expression, in patients with advanced and end stage HF (31). As the upregulation of CaMKII is
associated with heart disease and failure by promoting apoptosis,
inflammation that leads to cardiac dysfunction (32,33), the
possibility that inhibition of this enzyme activity can have
therapeutic effects has been considered. Experimental transgenic
animal models, overexpressing CaMKII have been found to suffer from
HF (34), whereas CaMKII knockout
mice were protected from HF induced by transaortic constriction
(35). Additionally, mice expressing
a mutant CaMKII (S2814D), which is constitutively active, suffered
exacerbated mortality (36).
CaMKIIδγ knockout mice with total deletion of heart specific
isoforms CaMKII, are protected from pressure overload and
β-adrenergic stimulation-induced cardiac dysfunction and
interstitial fibrosis (32,37). Similarly, the elevated activity of
CaMKII is also associated with atrial fibrillation and sinus node
disease (38) and several other HF
contributory diseases such as inherited arrhythmias (39,40).
Oxidation of 281/282 methionine residue in CaMKII is
susceptible to oxidative stress and this oxidation leads to the
activation of CaMKII and it has been shown that this oxidation is
particularly important in cardiomyocytes as it may relate to
conditions of ischemia/reperfusion injury (41). Met281/282 oxidation prevents the
re-association of the inhibitory regulatory domain with the
catalytic domain of CaMKII (Fig. 1)
(42). Angiotensin II and
aldosterone are shown to mediate their activation effects on CaMKII
via oxidation (43), as
cardiomyocytes expressing oxidation-resistant mutant CaMKII were
protected from angiotensin II-induced apoptosis (41). Also, diabetic mice expressing an
oxidation-resistant CaMKII mutant (MM281/282VV) were found to be
protected from MI (44). In fact, it
has been noted that increased oxidation status of CaMKII seen after
MI in diabetic patients appears to be associated with higher
mortality, than in non-diabetic individuals, which again emphasizes
the detrimental effects of CaMKII activation, particularly when the
heart is stressed.
Of note, CaMKII oxidation and activity is found to
be much less following MI in mice with deletion of the MyD88
gene, an important mediator of inflammatory signaling. These
MyD88-knockout mice also show lower post MI inflammatory cell
infiltration, cardiomyocyte death and fibrosis. Oxidized CaMKII can
in turn enhance the transcription of proinflammatory genes by
enhancing NF-κB activity (45).
Other post-translational modifications of CaMKII that cause its
activation and are involved in the pathology of HF include
nitrosylation and O-GlcNAcylation, which are important under
hyperglycemic conditions seen in diabetes (46).
Therapeutic measures against CaMKII
Inasmuch as the activation of CaMKII is involved
with heart disease, several studies have focused on developing
CaMKII inhibitors that have the potential to have therapeutic
effects in HF and heart diseases. Most of the currently available
inhibitors are for research purposes and lack specificity and/or
potency. For example, KN-93, which is a commonly used CaMKII
inhibitor, also directly affects many other ion channel including
LTCC (47). Administration of KN-93
to mice with structural heart disease, for 3 weeks led to chronic
inhibition of CaMKIIδ and resulted in a dose-dependent improvement
in left ventricular function (48).
Similarly, peptide molecules (AIP and AC3-I) that mimic the
autoinhibitory-regulating domain of CaMKII, also have several
limitations regarding their specificity and delivery. Among the
several inhibitors tested, the most promising is the endogenous
inhibitor, known as CaMKIIN and its derivatives, which bind to the
active kinase at the B/C sites, which also prevent protein-protein
interactions of CaMKII with other targeting proteins (47). It has been recently shown that
targeting CaMKII/ERK interaction in heart muscle using selective
CaMKII peptide inhibitor AntCaNtide was able to prevent hypertrophy
in spontaneously hypertensive rats (49). The first generation CaMKII inhibitors
based on targeting the ATP binding to catalytic site and the recent
availability of crystal structures of CaMKII holoenzyme both in its
autoinhibited as well as active states may be useful in the
development of more specific and potent inhibitors for this enzyme.
Furthermore, blockade of activating pathways such as O-GlcNAc
modification were also found to be effective in preventing
arrhythmogenesis in diabetic animals by inhibiting the hexosamine
biosynthetic pathway using the inhibitor DON (50). Thus, there are formidable
difficulties in achieving the required specificity for developing
CaMKII inhibitors that can be developed for therapeutic
applications (47).
In addition to pharmacological inhibitors, exercise,
which has proven beneficial cardiovascular effects, seems to have
the ability to antagonize the negative effects of CaMKIIδ in
failing heart. Thus, aerobic training caused a reduction in CaMKIIδ
activity and improved heart function in diabetic mice compared to
non-exercising diabetic mice (51).
Notably, it has been demonstrated that swimming exercise may
obliterate the O-GlcNAcylation-mediated activation of CaMKII in
type I diabetic mice with the resultant improvement in heart
condition (52). Thus, of note is
along with pharmacological approaches, lifestyle changes can be
beneficial in protecting from the CaMKII-mediated aggravation of
injured or stressed heart. As such, the development of specific
drugs that target heart isoforms of CaMKII seems a far-reaching
goal and further work is needed in understanding structure-activity
relationships of these isoenzymes to accomplish this task.
Conclusions
HF involves altered gene expression, disturbed
signaling pathways and altered Ca2+ homeostasis. Chronic
activation of CaMKII isoforms in heart further aggravates the
injury to myocardium and the expression and activity of CaMKII is
elevated in myocardium in different heart diseases and stress
conditions. CaMKII regulates many cellular pathways such as E-C
coupling and relaxation events in heart, cardiomyocyte apoptosis,
transcriptional activation of genes related to cardiac hypertrophy,
inflammation, and arrhythmias. CaMKII and reactive oxygen species,
which mutually activate each other, increase CICR from SR, which
leads to cardiomyocyte membrane depolarization and arrhythmias. All
these CaMKII-mediated changes in heart ultimately culminate in
dysfunctional myocardium and HF. Despite significant leaps in
understanding the structural details of CaMKII, which is a very
complicated and multimeric modular protein, and genetic studies
implicating CaMKII in the pathogenesis of HF, currently there is no
specific and potent inhibitor of this enzyme, that can be developed
for therapeutic purposes and further study is needed in this
direction.
References
|
1
|
Nabel EG and Braunwald E: A tale of
coronary artery disease and myocardial infarction. N Engl J Med.
366:54–63. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Daniels L, Bell JR, Delbridge LM, McDonald
FJ, Lamberts RR and Erickson JR: The role of CaMKII in diabetic
heart dysfunction. Heart Fail Rev. 20:589–600. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mathers CD and Loncar D: Projections of
global mortality and burden of disease from 2002 to 2030. PLoS Med.
3:e4422006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Joseph SM, Cedars AM, Ewald GA, Geltman EM
and Mann DL: Acute decompensated heart failure: Contemporary
medical management. Tex Heart Inst J. 36:510–520. 2009.PubMed/NCBI
|
|
5
|
McMurray JJ, Adamopoulos S, Anker SD,
Auricchio A, Böhm M, Dickstein K, Falk V, Filippatos G, Fonseca C,
Gomez-Sanchez MA, et al: ESC Committee for Practice Guidelines: Esc
guidelines for the diagnosis and treatment of acute and chronic
heart failure 2012: The task force for the diagnosis and treatment
of acute and chronic heart failure 2012 of the European Society of
Cardiology. Developed in collaboration with the heart failure
association (hfa) of the esc. Eur Heart J. 33:1787–1847. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hill JA and Olson EN: Cardiac plasticity.
N Engl J Med. 358:1370–1380. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bers DM: Cardiac sarcoplasmic reticulum
calcium leak: Basis and roles in cardiac dysfunction. Annu Rev
Physiol. 76:107–127. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cho GW, Altamirano F and Hill JA: Chronic
heart failure: Ca(2+), catabolism, and catastrophic cell death.
Biochim Biophys Acta. 1862:763–777. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
van Oort RJ, Brown JH and Westenbrink BD:
CaMKII confirms its promise in ischaemic heart disease. Eur J Heart
Fail. 16:1268–1269. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Grandi E, Edwards AG, Herren AW and Bers
DM: CaMKII comes of age in cardiac health and disease. Front
Pharmacol. 5:1542014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bers DM:
Ca2+-calmodulin-dependent protein kinase II regulation
of cardiac excitation-transcription coupling. Heart Rhythm.
8:1101–1104. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Couchonnal LF and Anderson ME: The role of
calmodulin kinase II in myocardial physiology and disease.
Physiology (Bethesda). 23:151–159. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Luczak ED and Anderson ME: CaMKII
oxidative activation and the pathogenesis of cardiac disease. J Mol
Cell Cardiol. 73:112–116. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bell JR, Vila-Petroff M and Delbridge LM:
CaMKII-dependent responses to ischemia and reperfusion challenges
in the heart. Front Pharmacol. 5:962014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Grimm M, Ling H and Brown JH: Crossing
signals: Relationships between β-adrenergic stimulation and CaMKII
activation. Heart Rhythm. 8:1296–1298. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhao Z, Fefelova N, Shanmugam M, Bishara
P, Babu GJ and Xie LH: Angiotensin II induces afterdepolarizations
via reactive oxygen species and calmodulin kinase II signaling. J
Mol Cell Cardiol. 50:128–136. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Swaminathan PD, Purohit A, Hund TJ and
Anderson ME: Calmodulin-dependent protein kinase II: Linking heart
failure and arrhythmias. Circ Res. 110:1661–1677. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mishra S, Gray CB, Miyamoto S, Bers DM and
Brown JH: Location matters: Clarifying the concept of nuclear and
cytosolic CaMKII subtypes. Circ Res. 109:1354–1362. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hoelz A, Nairn AC and Kuriyan J: Crystal
structure of a tetradecameric assembly of the association domain of
Ca2+/calmodulin-dependent kinase II. Mol Cell.
11:1241–1251. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Anderson ME, Brown JH and Bers DM: CaMKII
in myocardial hypertrophy and heart failure. J Mol Cell Cardiol.
51:468–473. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bers DM and Morotti S: Ca(2+) current
facilitation is CaMKII-dependent and has arrhythmogenic
consequences. Front Pharmacol. 5:1442014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mustroph J, Maier LS and Wagner S: CaMKII
regulation of cardiac K channels. Front Pharmacol. 5:202014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sierra A, Zhu Z, Sapay N, Sharotri V,
Kline CF, Luczak ED, Subbotina E, Sivaprasadarao A, Snyder PM,
Mohler PJ, et al: Regulation of cardiac ATP-sensitive potassium
channel surface expression by calcium/calmodulin-dependent protein
kinase II. J Biol Chem. 288:1568–1581. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sellers ZM, De Arcangelis V, Xiang Y and
Best PM: Cardiomyocytes with disrupted CFTR function require CaMKII
and Ca(2+)-activated Cl(−) channel activity to maintain contraction
rate. J Physiol. 588:2417–2429. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Braun AP and Schulman H: The
multifunctional calcium/calmodulin-dependent protein kinase: From
form to function. Annu Rev Physiol. 57:417–445. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dupont G, Houart G and De Koninck P:
Sensitivity of CaM kinase II to the frequency of Ca2+
oscillations: A simple model. Cell Calcium. 34:485–497. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Respress JL, van Oort RJ, Li N, Rolim N,
Dixit SS, deAlmeida A, Voigt N, Lawrence WS, Skapura DG, Skårdal K,
et al: Role of RyR2 phosphorylation at S2814 during heart failure
progression. Circ Res. 110:1474–1483. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Anderson ME: Sticky fingers: CaMKII finds
a home on another ion channel. Circ Res. 104:712–714. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Makara MA, Curran J, Little SC, Musa H,
Polina I, Smith SA, Wright PJ, Unudurthi SD, Snyder J, Bennett V,
et al: Ankyrin-G coordinates intercalated disc signaling platform
to regulate cardiac excitability in vivo. Circ Res. 115:929–938.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sossalla S, Fluschnik N, Schotola H, Ort
KR, Neef S, Schulte T, Wittköpper K, Renner A, Schmitto JD, Gummert
J, et al: Inhibition of elevated
Ca2+/calmodulin-dependent protein kinase II improves
contractility in human failing myocardium. Circ Res. 107:1150–1161.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Maier LS: CaMKIIdelta overexpression in
hypertrophy and heart failure: Cellular consequences for
excitation-contraction coupling. Braz J Med Biol Res. 38:1293–1302.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Weinreuter M, Kreusser MM, Beckendorf J,
Schreiter FC, Leuschner F, Lehmann LH, Hofmann KP, Rostosky JS,
Diemert N, Xu C, et al: CaM kinase II mediates maladaptive
post-infarct remodeling and pro-inflammatory chemoattractant
signaling but not acute myocardial ischemia/reperfusion injury.
EMBO Mol Med. 6:1231–1245. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mattiazzi A, Bassani RA, Escobar AL,
Palomeque J, Valverde CA, Petroff M Vila and Bers DM: Chasing
cardiac physiology and pathology down the CaMKII cascade. Am J
Physiol Heart Circ Physiol. 308:H1177–H1191. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang T, Maier LS, Dalton ND, Miyamoto S,
Ross J Jr, Bers DM and Brown JH: The deltaC isoform of CaMKII is
activated in cardiac hypertrophy and induces dilated cardiomyopathy
and heart failure. Circ Res. 92:912–919. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Backs J, Backs T, Neef S, Kreusser MM,
Lehmann LH, Patrick DM, Grueter CE, Qi X, Richardson JA, Hill JA,
et al: The delta isoform of CaM kinase II is required for
pathological cardiac hypertrophy and remodeling after pressure
overload. Proc Natl Acad Sci USA. 106:2342–2347. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
van Oort RJ, McCauley MD, Dixit SS,
Pereira L, Yang Y, Respress JL, Wang Q, De Almeida AC, Skapura DG,
Anderson ME, et al: Ryanodine receptor phosphorylation by
calcium/calmodulin-dependent protein kinase II promotes
life-threatening ventricular arrhythmias in mice with heart
failure. Circulation. 122:2669–2679. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kreusser MM, Lehmann LH, Keranov S, Hoting
MO, Oehl U, Kohlhaas M, Reil JC, Neumann K, Schneider MD, Hill JA,
et al: Cardiac CaM kinase II genes δ and γ contribute to adverse
remodeling but redundantly inhibit calcineurin-induced myocardial
hypertrophy. Circulation. 130:1262–1273. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wu Y and Anderson ME: CaMKII in sinoatrial
node physiology and dysfunction. Front Pharmacol. 5:482014.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
DeGrande S, Nixon D, Koval O, Curran JW,
Wright P, Wang Q, Kashef F, Chiang D, Li N, Wehrens XH, et al:
CaMKII inhibition rescues proarrhythmic phenotypes in the model of
human ankyrin-B syndrome. Heart Rhythm. 9:2034–2041. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Liu N, Ruan Y, Denegri M, Bachetti T, Li
Y, Colombi B, Napolitano C, Coetzee WA and Priori SG: Calmodulin
kinase II inhibition prevents arrhythmias in RyR2(R4496C+/−) mice
with catecholaminergic polymorphic ventricular tachycardia. J Mol
Cell Cardiol. 50:214–222. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Erickson JR, Joiner ML, Guan X, Kutschke
W, Yang J, Oddis CV, Bartlett RK, Lowe JS, O'Donnell SE,
Aykin-Burns N, et al: A dynamic pathway for calcium-independent
activation of CaMKII by methionine oxidation. Cell. 133:462–474.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chao LH, Pellicena P, Deindl S, Barclay
LA, Schulman H and Kuriyan J: Intersubunit capture of regulatory
segments is a component of cooperative CaMKII activation. Nat
Struct Mol Biol. 17:264–272. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Rueda JO Velez, Palomeque J and Mattiazzi
A: Early apoptosis in different models of cardiac hypertrophy
induced by high renin-angiotensin system activity involves CaMKII.
J Appl Physiol 1985. 112:2110–2120. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Luo M, Guan X, Luczak ED, Lang D, Kutschke
W, Gao Z, Yang J, Glynn P, Sossalla S, Swaminathan PD, et al:
Diabetes increases mortality after myocardial infarction by
oxidizing CaMKII. J Clin Invest. 123:1262–1274. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Singh MV, Swaminathan PD, Luczak ED,
Kutschke W, Weiss RM and Anderson ME: MyD88 mediated inflammatory
signaling leads to CaMKII oxidation, cardiac hypertrophy and death
after myocardial infarction. J Mol Cell Cardiol. 52:1135–1144.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Mollova MY, Katus HA and Backs J:
Regulation of CaMKII signaling in cardiovascular disease. Front
Pharmacol. 6:1782015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Pellicena P and Schulman H: CaMKII
inhibitors: From research tools to therapeutic agents. Front
Pharmacol. 5:212014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhang R, Khoo MS, Wu Y, Yang Y, Grueter
CE, Ni G, Price EE Jr, Thiel W, Guatimosim S, Song LS, et al:
Calmodulin kinase II inhibition protects against structural heart
disease. Nat Med. 11:409–417. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
49
|
Cipolletta E, Rusciano MR, Maione AS,
Santulli G, Sorriento D, Del Giudice C, Ciccarelli M, Franco A,
Crola C, Campiglia P, et al: Targeting the CaMKII/ERK interaction
in the heart prevents cardiac hypertrophy. PLoS One.
10:e01304772015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Erickson JR, Pereira L, Wang L, Han G,
Ferguson A, Dao K, Copeland RJ, Despa F, Hart GW, Ripplinger CM, et
al: Diabetic hyperglycaemia activates CaMKII and arrhythmias by
O-linked glycosylation. Nature. 502:372–376. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Stølen TO, Høydal MA, Kemi OJ, Catalucci
D, Ceci M, Aasum E, Larsen T, Rolim N, Condorelli G, Smith GL, et
al: Interval training normalizes cardiomyocyte function, diastolic
Ca2+ control, and SR Ca2+ release
synchronicity in a mouse model of diabetic cardiomyopathy. Circ
Res. 105:527–536. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Bennett CE, Johnsen VL, Shearer J and
Belke DD: Exercise training mitigates aberrant cardiac protein
O-GlcNAcylation in streptozotocin-induced diabetic mice. Life Sci.
92:657–663. 2013. View Article : Google Scholar : PubMed/NCBI
|