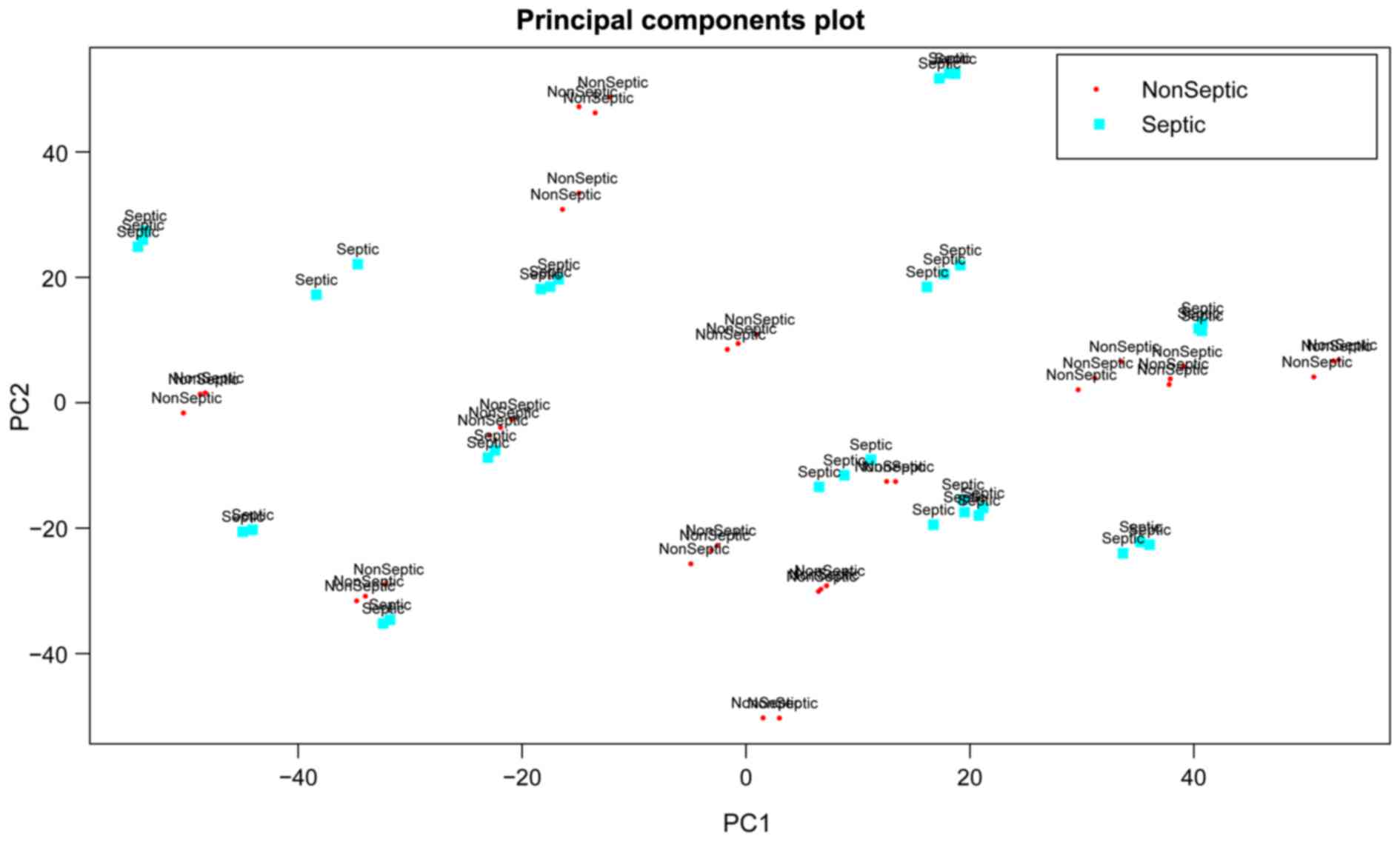
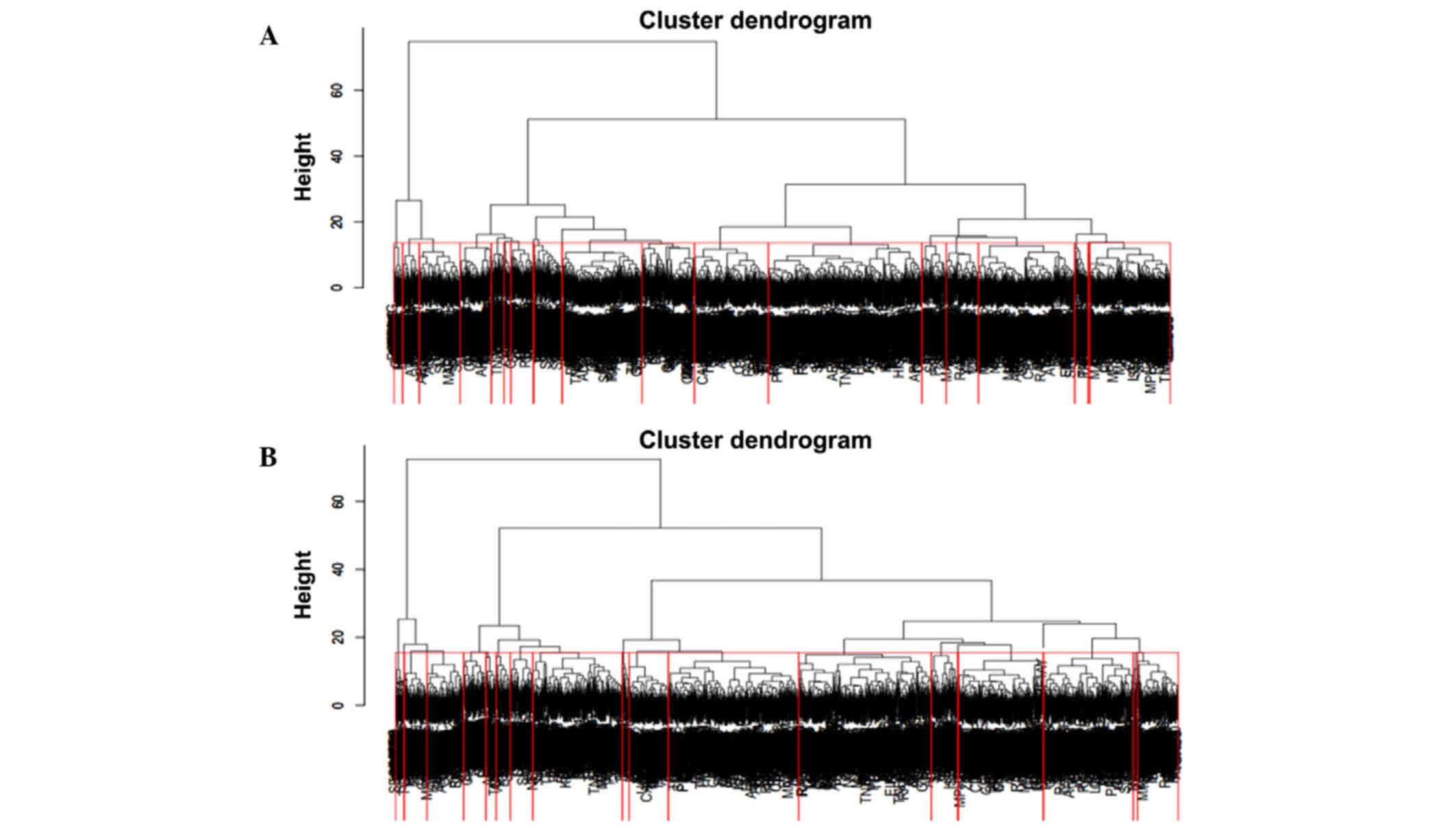
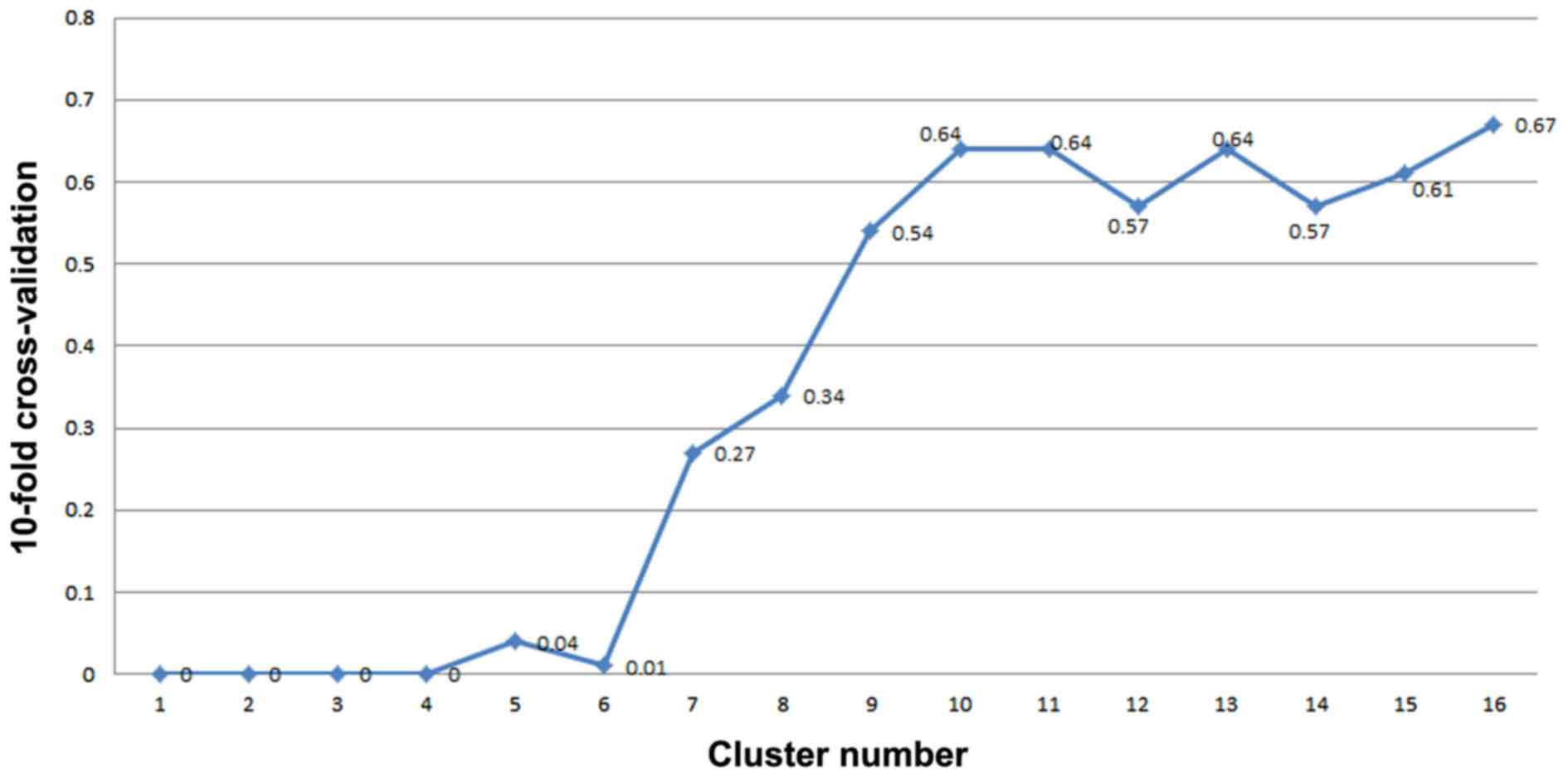
|
1
|
Pierrakos C and Vincent JL: Sepsis
biomarkers: A review. Crit Care. 14:R152010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
American College of Chest
Physicians/Society of Critical Care Medicine Consensus Conference:
Definitions for sepsis and organ failure and guidelines for the use
of innovative therapies in sepsis. Crit Care Med. 20:864–874. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Soong J and Soni N: Sepsis: Recognition
and treatment. Clin Med. 12:276–280. 2012. View Article : Google Scholar
|
|
4
|
Levy MM, Dellinger RP, Townsend SR,
Linde-Zwirble WT, Marshall JC, Bion J, Schorr C, Artigas A, Ramsay
G and Beale R: Surviving Sepsis Campaign: The surviving sepsis
campaign: Results of an international guideline-based performance
improvement program targeting severe sepsis. Crit Care Med.
38:367–374. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mayr FB, Yende S and Angus DC:
Epidemiology of severe sepsis. Virulence. 5:4–11. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Venet F and Chung CS: Increased
circulating regulatory T cells (CD4(+)CD25 (+)CD127 (−)) contribute
to lymphocyte anergy in septic shock patients. Intensive Care Med.
35:678–686. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tidswell M, Tillis W, Larosa SP, Lynn M,
Wittek AE, Kao R, Wheeler J, Gogate J and Opal SM: Phase 2 trial of
eritoran tetrasodium (E5564), a Toll-like receptor 4 antagonist, in
patients with severe sepsis. Crit Care Med. 38:72–83. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Machado JR, Soave DF, Da SM, de Menezes
LB, Etchebehere RM, Monteiro ML, dos Reis MA, Corrêa RR and Celes
MR: Neonatal sepsis and inflammatory mediators. Mediators Inflamm.
2014:269681. 2014.PubMed/NCBI
|
|
9
|
Nancy B, Diana S, Kerstin H, Oliver K,
Katrin L, Michael B, Martin BF, Diana I and Michael K: C-Terminal
Alpha-1 Antitrypsin Peptide: A New Sepsis Biomarker with
Immunomodulatory Function. Mediators Inflamm. 2016:1–13. 2016.
|
|
10
|
Chi YF, Chai JK, Yu YM, Luo HM, Zhang QX
and Feng R: Association between PAI-1 polymorphisms and plasma
PAI-1 level with sepsis in severely burned patients. Genet Mol Res.
14:10081–10086. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang JF, Yu MG, Bian JJ, Deng XM, Wan XJ
and Zhu KM: Serum miR-146a and miR-223 as potential new biomarkers
for sepsis. Biochem Biophys Res Commun. 394:184–188. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Póvoa P, Coelho L, Almeida E, Fernandes A,
Mealha R, Moreira P and Sabino H: C-reactive protein as a marker of
infection in critically ill patients. Clin Microbiol Infect.
11:101–108. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Schmit X and Vincent JL: The time course
of blood C-reactive protein concentrations in relation to the
response to initial antimicrobial therapy in patients with sepsis.
Infection. 36:213–219. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Luzzani A, Polati E, Dorizzi R,
Rungatscher A, Pavan R and Merlini A: Comparison of procalcitonin
and C-reactive protein as markers of sepsis. Crit Care Med.
31:1737–1741. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gabay C and Kushner I: Acute-phase
proteins and other systemic responses to inflammation. N Engl J
Med. 340:448–454. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hanriot D, Bello G, Ropars ADC, Poitevin
G, Grosjean S, Latger-Cannard V, Devaux Y, Zannad F, Regnault V and
Lacolley P: C-reactive protein induces pro- and anti-inflammatory
effects, including activation of the liver X receptor alpha, on
human monocytes. Thromb Haemost. 99:558–569. 2008.PubMed/NCBI
|
|
17
|
Culley FJ, Bodmansmith KB, Ferguson MA,
Nikolaev AV, Shantilal N and Raynes JG: C-reactive protein binds to
phosphorylated carbohydrates. Glycobiology. 10:59–65. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Assicot M, Gendrel D, Carsin H, Raymond J,
Guilbaud J and Bohuon C: High serum procalcitonin concentrations in
patients with sepsis and infection. Lancet. 341:515–518. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
O'Grady NP, Barie PS, Bartlett JG, Bleck
T, Carroll K, Kalil AC, Linden P, Maki DG, Nierman D, Pasculle W
and Masur H: American College of Critical Care Medicine; Infectious
Diseases Society of America: Guidelines for evaluation of new fever
in critically ill adult patients: 2008 update from the American
college of critical care medicine and the infectious diseases
society of America. Crit Care Med. 36:1330–1349. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Snider RH Jr, Nylen ES and Becker KL:
Procalcitonin and its component peptides in systemic inflammation:
Immunochemical characterization. J Investig Med. 45:552–560.
1997.PubMed/NCBI
|
|
21
|
Tang BM, Eslick GD, Craig JC and McLean
AS: Accuracy of procalcitonin for sepsis diagnosis in critically
ill patients: Systematic review and meta-analysis. Lancet Infect
Dis. 7:210–217. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wacker C, Prkno A, Brunkhorst FM and
Schlattmann P: Procalcitonin as a diagnostic marker for sepsis: A
systematic review and meta-analysis. Lancet Infect Dis. 13:426–435.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Irizarry RA, Hobbs B, Collin F,
Beazer-Barclay YD, Antonellis KJ, Scherf U and Speed TP:
Exploration, normalization and summaries of high density
oligonucleotide array probe level data. Biostatistics. 4:249–264.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bicciato S, Luchini A and Di Bello C: PCA
disjoint models for multiclass cancer analysis using gene
expression data. Bioinformatics. 19:571–578. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Peri S, Navarro JD, Kristiansen TZ,
Amanchy R, Surendranath V, Muthusamy B, Gandhi TK, Chandrika KN,
Deshpande N, Suresh S, et al: Human protein reference database as a
discovery resource for proteomics. Nucleic Acids Res. 32:(Database
issue). D497–D501. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cameron DA, Middleton FA, Chenn A and
Olson EC: Hierarchical clustering of gene expression patterns in
the Eomes+lineage of excitatory neurons during early neocortical
development. BMC Neurosci. 13:902012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mansmann U and Meister R: Testing
differential gene expression in functional groups. Goeman's global
test versus an ANCOVA approach. Methods Inf Med. 44:449–453.
2005.PubMed/NCBI
|
|
29
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al: Gene ontology: Tool for the unification of biology. The gene
ontology consortium. Nat Genet. 25:25–29. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kanehisa M and Goto S: KEGG: Kyoto
encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
da Huang W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liu J, Li SC and Luo X: Iterative
reweighted noninteger norm regularizing SVM for gene expression
data classification. Comput Math Methods Med. 2013:7684042013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jawad I, Lukšić I and Rafnsson SB:
Assessing available information on the burden of sepsis: Global
estimates of incidence, prevalence and mortality. J Glob Health.
2:0104042012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Geörg M, Maudsdotter L, Tavares R and
Jonsson AB: Meningococcal resistance to antimicrobial peptides is
mediated by bacterial adhesion and host cell RhoA and Cdc42
signalling. Cell Microbiol. 15:1938–1954. 2013.PubMed/NCBI
|
|
35
|
Rauch PJ, Chudnovskiy A, Robbins CS, Weber
GF, Etzrodt M, Hilgendorf I, Tiglao E, Figueiredo JL, Iwamoto Y,
Theurl I, et al: Innate response activator B cells protect against
microbial sepsis. Science. 335:597–601. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Durand M and Thomas SL: Incidence of
infections in patients with giant cell arteritis: A cohort study.
Arthritis Care Res (Hoboken). 64:581–588. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Metukuri MR, Reddy CM, Reddy PR and
Reddanna P: Bacterial LPS-mediated acute inflammation-induced
spermatogenic failure in rats: Role of stress response proteins and
mitochondrial dysfunction. Inflammation. 33:235–243. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sista F, Schietroma M, Santis GD, Mattei
A, Cecilia EM, Piccione F, Leardi S, Carlei F and Amicucci G:
Systemic inflammation and immune response after laparotomy vs
laparoscopy in patients with acute cholecystitis, complicated by
peritonitis. World J Gastrointest Surg. 5:73–82. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Benarafa C: The SerpinB1 knockout mouse a
model for studying neutrophil protease regulation in homeostasis
and inflammation. Methods Enzymol. 499:135–148. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ormsby T, Schlecker E, Ferdin J, Tessarz
AS, Angelisová P, Köprülü AD, Borte M, Warnatz K, Schulze I,
Ellmeier W, Horejsí V and Cerwenka A: Btk is a positive regulator
in the TREM-1/DAP12 signaling pathway. Blood. 118:936–945. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ganter U, Bauer J, Schulz-Huotari C,
Gebicke-Haerter PJ, Beeser H and Gerok W: Repression of alpha
2-macroglobulin and stimulation of alpha 1-proteinase inhibitor
synthesis in human mononuclear phagocytes by endotoxin. Eur J
Biochem. 169:13–20. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Buttenschoen K, Buttenschoen DC, Berger D,
Vasilescu C, Schafheutle S, Goeltenboth B, Seidelmann M and Beger
HG: Endotoxemia and acute-phase proteins in major abdominal
surgery. Am J Surg. 181:36–43. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Su L, Zhou R, Liu C, Wen B, Xiao K, Kong
W, Tan F, Huang Y, Cao L and Xie L: Urinary proteomics analysis for
sepsis biomarkers with iTRAQ labeling and two-dimensional liquid
chromatography-tandem mass spectrometry. J Trauma Acute Care Surg.
74:940–945. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Butchar JP, Mehta P, Justiniano SE,
Guenterberg KD, Kondadasula SV, Mo X, Chemudupati M, Kanneganti TD,
Amer A and Muthusamy N: Reciprocal regulation of activating and
inhibitory Fc{gamma} receptors by TLR7/8 activation: implications
for tumor immunotherapy. Clin Cancer Res. 16:2065–2075. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hu WC: Sepsis is a syndrome with
hyperactivity of TH17-like innate immunity and hypoactivity of
adaptive immunity. ArXiv Preprint ArXiv:. 1311:47472013.
|
|
46
|
da Pinheiro Silva F, Aloulou M, Skurnik D,
Benhamou M, Andremont A, Velasco IT, Chiamolera M, Verbeek JS,
Launay P and Monteiro RC: CD16 promotes Escherichia coli sepsis
through an FcR gamma inhibitory pathway that prevents phagocytosis
and facilitates inflammation. Nat Med. 13:1368–1374. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Iacobone E, Bailly-Salin J, Polito A,
Friedman D, Stevens RD and Sharshar T: Sepsis-associated
encephalopathy and its differential diagnosis. Crit Care Med.
37:(Suppl 10). S331–S336. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Hsu AA, Fenton K, Weinstein S, Carpenter
J, Dalton H and Bell MJ: Neurological injury markers in children
with septic shock. Pediatr Crit Care Med. 9:245–251. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Vodounon CA, Chabi CB, Skibo YV, Ezin V,
Aikou N, Kotchoni SO, Akpona SA, Babamoussa L and Abramova ZI:
Influence of the programmed cell death of lymphocytes on the
immunity of patients with atopic bronchial asthma. Allergy Asthma
Clin Immunol. 10:1–11. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Perl M, Chung CS, Swan R and Ayala A: Role
of programmed cell death in the immunopathogenesis of sepsis. Drug
Discov Today Dis Mech. 4:223–230. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kurko J, Vähä-Mäkilä M, Tringham M, Tanner
L, Paavanen-Huhtala S, Saarinen M, Näntö-Salonen K, Simell O,
Niinikoski H and Mykkänen J: Dysfunction in macrophage toll-like
receptor signaling caused by an inborn error of cationic amino acid
transport. Mol Immunol. 67:416–425. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Bermpohl D, Halle A, Freyer D, Dagand E,
Braun JS, Bechmann I, Schröder NW and Weber JR: Bacterial
programmed cell death of cerebral endothelial cells involves dual
death pathways. J Clin Invest. 115:1607–1615. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Bergsbaken T, Fink SL and Cookson BT:
Pyroptosis: Host cell death and inflammation. Nat Rev Microbiol.
7:99–109. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Into T, Kiura K, Yasuda M, Kataoka H,
Inoue N, Hasebe A, Takeda K, Akira S and Shibata K: Stimulation of
human Toll-like receptor (TLR) 2 and TLR6 with membrane
lipoproteins of Mycoplasma fermentans induces apoptotic cell death
after NF-kappa B activation. Cell Microbiol. 6:187–199. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Armstrong L, Medford AR, Hunter KJ,
Uppington KM and Millar AB: Differential expression of Toll-like
receptor (TLR)-2 and TLR-4 on monocytes in human sepsis. Clin Exp
Immunol. 136:312–319. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Menasche G, Feldmann J, Houdusse A,
Desaymard C, Fischer A, Goud B and De SBG: Biochemical and
functional characterization of Rab27a mutations occurring in
Griscelli syndrome patients. Blood. 101:2736–2742. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Ménasché G, Pastural E, Feldmann J,
Certain S, Ersoy F, Dupuis S, Wulffraat N, Bianchi D, Fischer A, Le
Deist F and de Saint Basile G: Mutations in RAB27A cause Griscelli
syndrome associated with haemophagocytic syndrome. Nat Genet.
25:173–176. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
58
|
Johnson JL, Hong H, Monfregola J and Catz
SD: Increased survival and reduced neutrophil infiltration of the
liver in Rab27a- but not Munc13-4-deficient mice in
lipopolysaccharide-induced systemic inflammation. Infect Immun.
79:3607–3618. 2011. View Article : Google Scholar : PubMed/NCBI
|