Introduction
Diabetes mellitus (DM) is a metabolic disease
characterized by hyperglycemia due to impaired insulin secretion
and/or insulin dysfunction (1). In
recent years, the incidence of DM has markedly increased and
endangers human health. DM is affected by multiple factors,
including genetic factors, autoimmune system defects and viral
infections. Pancreatic islet β-cell dysfunction decreases insulin
secretion and elevates blood glucose levels, eventually resulting
in the occurrence of DM (2,3). DM is thought to be closely associated
with islet inflammation caused by immune dysregulation. It has been
suggested that inflammatory factors involved in the inflammatory
response exert a crucial role in the occurrence and development of
DM by inducing islet β-cell apoptosis and insulin secretion defects
(4). The islet cells do not release
inflammatory factors; however, macrophages that infiltrate islet
cells release inflammatory factors, including interleukin-1β
(IL-1β) and tumor necrosis factor-α (TNF-α), which may damage islet
β cells. Activated T cells also produce inflammatory factors and
induce apoptosis, leading to the death of islet β-cells (5). Studies have indicated that insulin
induces AKT phosphorylation through the phosphoinositide 3-kinase
(PI3K)/AKT signaling pathway, which promotes glucose uptake and
synthesis of glycogen (6,7). The synthesis of glycogen further
affects the absorption and metabolism of blood glucose and
increases insulin sensitivity.
MicroRNAs (miRNAs/miRs) are a class of highly
evolutionarily conserved, single-stranded, non-coding RNAs. miRNAs
degrade mRNA or inhibit translation of target genes via binding to
target mRNAs (8). A complex
regulatory network is formed by an individual miRNA with multiple
target genes, to participate in cell differentiation,
proliferation, apoptosis and metabolism (9,10).
Hence, miRNAs are important in the differentiation, development,
regulation of the quantity and maintenance of the function of
islets (11,12). A previous study demonstrated that
neuron navigator 1 (NAV1) was elevated under high-glucose
conditions in isolated human pancreatic islets, indicating that
NAV1 was involved in the pathogenesis of diabetes mellitus
(13). It was also reported that
miR-18 is involved in the regulation of the occurrence, invasion
and metastasis of multiple tumor types (14). However, few studies have explored the
function of miR-18 in islet β-cells. The present study aimed to
explore the role of miR-18 in diabetes mellitus and the underlying
mechanisms, providing novel ideas for the treatment of
diabetes.
Materials and methods
Cell culture and transfection
The mouse islet β-cell line MIN6 was obtained from
the American Type Culture Collection (Manassas, VA, USA). Cells
were cultured in Dulbecco's modified Eagle's medium (DMEM; HyClone;
GE Healthcare, Little Chalfont, UK) containing 15% fetal bovine
serum (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA), 50
µM β-mercaptoethanol, 100 U/ml penicillin, 10 µg/ml streptomycin,
10 mM HEPES, 1 mM sodium pyruvate and 25 mM glucose. Cell passage
was performed every 4 days. MIN6 cells were induced with a mixture
of cytokines for 24 h at 37°C, including IL-1β (10 ng/ml; cat. no.
GMP-101-1B; PrimeGene, Shanghai, China), TNF-α (10 ng/ml; cat. no.
GMP-103-01; PrimeGene) and IFN-γ (10 ng/ml; cat. no. 224-09;
PrimeGene). Prior to transfection, cells were seeded in 24-well
plates for 24 h until the cell confluency reached 80–90%. miRNA
negative control or miR-18 mimics and inhibitor (GenePharma,
Shanghai, China) were diluted with serum-free and antibiotic-free
medium and then mixed with Lipofectamine® 2000
(Invitrogen; Thermo Fisher Scientific, Inc.). The following
sequences were used: miRNA negative control forward,
5′-UUCUCCGAACGUGUCCGUTT-3′ and reverse,
5′-ACGUGACACGUUCGGAGAATT-3′; miR-18 mimics forward,
5′-UAAGGUGCAUCUAGUGCAGAUAG-3′ and reverse,
5′-AUCUGCACUAGAUGCACCUUAUU-3′; miR-18 inhibitor forward,
5′-CUAUCUGCACUAGAUGCACCUUA-3′, and reverse,
5′-AUGUACGGUAUAUAGACCUGCGA-3′. After the mixture was maintained at
room temperature for 20 min, it was added to each well, and the
cells were cultured in a humidified atmosphere containing 5%
CO2 at 37°C for 4–6 h, then the medium was replaced. The
subsequent experiments were performed after 24 h of
transfection.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted with 400 µl TRIzol
(Invitrogen; Thermo Fisher Scientific, Inc.) and 80 µl chloroform
was added, followed by centrifugation at 4°C and 4,500 × g/min for
15 min. The supernatant was collected and incubated with 200 µl
isopropanol, followed by another round of centrifugation at 4°C and
4,500 × g/min for 15 min. The RNA extract was further purified
using 75% ethanol. The RT procedure was performed using the Takara
PrimeScript RT Master Mix kit (Takara Bio Inc., Tokyo, Japan)
according to the manufacturer's protocol. RT-qPCR was performed
using SYBR® Green Master Mix (Takara Bio Inc.) according
to the manufacturer's protocol. The primer sequences used in this
experiment were as follows: Proinsulin forward,
5′-GCAGCCTTTGTGAACCAACAC-3′ and reverse,
5′-CCCCGCACACTAGGTAGAGA-3′; GAPDH forward,
5′-ACCCACTCCTCCACCTTTGA-3′ and reverse,
5′-CTGTTGCTGTAGCCAAATTCGT-3′; NAV1 forward,
5′-GCCTCAGACAATCTCAGTTCAG-3′ and reverse,
5′-ACCACTGTCGTACTCCAGTTTT-3′; B-cell lymphoma 2 (Bcl-2)-associated
X protein (Bax) forward, 5′-CATATAACCCCGTCAACGCAG-3′ and reverse,
5′-GCAGCCGCCACAAACATAC-3′; Bcl-2 forward,
5′-GTCTTCGCTGCGGAGATCAT-3′ and reverse,
5′-CATTCCGATATACGCTGGGAC-3′; miR-18 forward,
5′-CAGTAAAGGTAAGGAGAGCTCAATCTG-3′ and reverse,
5′-CATACAACCACTAAGCTAAAGAATAATCTGA-3′. The qPCR was performed
according to the miScript SYBR Green PCR kit instructions (Qiagen
GmbH, Hilden, Germany). The relative RNA expression levels were
analyzed using the 2−ΔΔCq method (15).
Glucose-stimulated insulin secretion
(GSIS) assay
MIN6 cells were washed with Krebs-Ringer bicarbonate
HEPES (KRBH) buffer once and incubated with 0.2 ml glucose-free
KRBH buffer for 1 h. After discarding the supernatant, MIN6 cells
were maintained in low-glucose KRBH buffer for 1 h, followed by
incubation with high-glucose KRBH buffer for another hour. Prior to
the GSIS assay, 200 µl 10% ethanol hydrochloride solution was added
in each well and released insulin levels were detected by
ELISA.
ELISA
Corresponding reagents were prepared and placed at
room temperature. Sample or standard solution (100 µl) was added in
the sample wells or standard wells, respectively. Corresponding
antibodies, including proinsulin antibody (1:500; cat. no. ab8304;
Abcam, Cambridge, MA, USA) and insulin antibody (1:500; cat. no.
ab100578; Abcam) were then added for incubation at room temperature
for a total of 2 h. Subsequently, horseradish peroxidase-labeled
antibodies were added (1:1,000; cat. no. ab150074; Abcam). After
incubation for 1 h at room temperature, 100 µl substrate was added,
followed by colour reaction in the dark for 10 min. The absorbance
at the wavelength of 450 nm was detected by a microplate reader
(Bio-Rad Laboratories, Hercules, CA, USA).
Terminal deoxynucleotidyl transferase
(TdT)-mediated deoxyuridine triphosphate nick end labeling (TUNEL)
assay
MIN6 cells were fixed in 4% formaldehyde and then
washed with PBS containing 2% hydrogen peroxide at room
temperature. Two drops of TdT enzyme buffer (Beyotime Institute of
Biotechnology, Haimen, China) were then added to the cells and
allowed to react at room temperature for 1 h prior to termination.
The cells were incubated in TdT buffer for 1 h at 37°C. After
washing with PBS for 3 times, cells were incubated with the
peroxidase-labeled anti-digoxigenin antibody (1:200; cat. no.
ab150155; Abcam) in a wet box at room temperature for 30 min. TUNEL
staining was observed and recorded under an optical microscope
(IX70; Olympus, Tokyo, Japan).
Dual-luciferase reporter gene
assay
Through a bioinformatics prediction (genemania.org), NAV1 was screened out as a target gene
of miR-18. MIN6 cells were inoculated into a 48-well plate and cell
density was allowed to reach 40–60% on the next day. The cells were
transfected using Lipofectamine 2000. Transfection experiments were
performed using NC mimics or microRNA-18 mimics, NC inhibitor or
microRNA-18 inhibitor, NAV1-WT or NAV1-MUT plasmid (0.2 µg; cat.
no. k801-200; AmyJet Scientific, Inc., Wuhan, China), and an
internal reference plasmid PRL-SV40 (0.004 µg; cat. no. k803-500;
AmyJet Scientific, Inc.). At 24 h after cell transfection, cells
were lysed using a dual luciferase reporter gene assay system
solution (Promega Corporation, Madison, WI, USA) to detect
fluorescence intensity. Comparison with Renilla luciferase
activity was used for normalization.
Western blot analysis
Total protein was extracted from treated cells with
radioimmunoprecipitation assay buffer, BCA method (Beyotime
Institute of Biotechnology) was used for quantification of total
protein. A total of 10 µl protein was loaded and separated by
SDS-PAGE (12% gel) electrophoresis and transferred to a
polyvinylidene difluoride membrane (Roche, Basel, Switzerland).
Membranes were washed with Tris-buffered saline containing Tween-20
(TBST), and blocked with 5% skimmed milk with TBST at 25°C for 1 h.
After incubation with primary antibodies, including NAV1 (cat. no.
ab65166), PI3K (cat. no. ab151549), phosphorylated (p)-PI3K (cat.
no. ab182651), AKT (cat. no. ab8805), p-AKT (cat. no. ab38449),
Bcl-2 (cat. no. ab59348), BAX (cat. no. ab32503) and GAPDH (cat.
no. ab8245; all from Abcam) at 4°C overnight and secondary
antibodies (1:1,000; cat. no. ab6940; Abcam). Enhanced
chemiluminescence reagent (Thermo Fisher Scientific, Inc.) was used
to detect the signal on the membrane. Quantity One (version 4.0;
Bio-Rad Laboratories) was used for quantification of western
blotting signals.
Statistical analysis
The SPSS 21.0 statistical software package (IBM
Corp., Armonk, NY, USA) was used for data analysis. Values are
expressed as the mean ± standard deviation. Comparison between
multiple groups was performed using one-way analysis of variance
followed by a least significant difference post-hoc test. P<0.05
was considered to indicate statistical significance.
Results
Inflammatory factors promote miR-18
expression in islet β-cells
Multiple inflammatory factors are involved in the
occurrence and progression of DM through a complex regulatory
network. In the present study, MIN6 cells were induced with a
mixture of cytokines (IL-1β, TNF-α and IFN-γ). After 24 h of
incubation, miR-18 expression was markedly elevated in MIN6 cells
(Fig. 1A). To further examine the
effect of inflammatory factors on miR-18 levels, MIN6 cells were
induced with 10 ng/ml IL-1β, TNF-α, IFN-γ or a combination of these
cytokines. The results indicated that the levels of miR-18 were
upregulated by induction with IL-1β or a combination of these
cytokines, suggesting that IL-1β markedly induced miR-18
expression, while TNF-α and IFN-γ did not (Fig. 1B). Subsequently, miR-18 mimics and
inhibitor were constructed and their transfection efficacies in
MIN6 cells were verified by RT-qPCR (Fig. 1C).
miR-18 inhibits insulin
production
The results of the RT-qPCR analysis indicated that
transfection of miR-18 mimics caused a downregulation of proinsulin
levels in MIN6 cells (Fig. 2A).
Furthermore, miR-18 mimics reduced released insulin levels, as
detected by ELISA (Fig. 2B). To
confirm the regulatory effect of miR-18 on islet β-cells, MIN6
cells were induced with 10 ng/ml IL-1β after transfection with
miR-18 inhibitor. The results demonstrated that miR-18 knockdown
partially abrogated the IL-1β-induced reduction of proinsulin
(Fig. 2C). The GSIS assay indicated
an inhibitory effect of miR-18 on insulin secretion (Fig. 2D). However, the suppressed insulin
secretion capacity in IL-1β-induced MIN6 cells was partially
abrogated by miR-18 knockdown (Fig.
2E).
miR-18 promotes apoptosis of islet
β-cells
The TUNEL assay indicated that miR-18 overexpression
promotes apoptosis of islet β-cells, whereas miR-18 knockdown
produced the opposite results (Fig.
3A). miR-18 mimics caused a marked upregulation of the
pro-apoptotic gene Bax, while downregulating the anti-apoptotic
gene Bcl-2 in MIN6 cells (Fig. 3B and
C).
miR-18 inhibits NAV1 expression in
islet β-cells
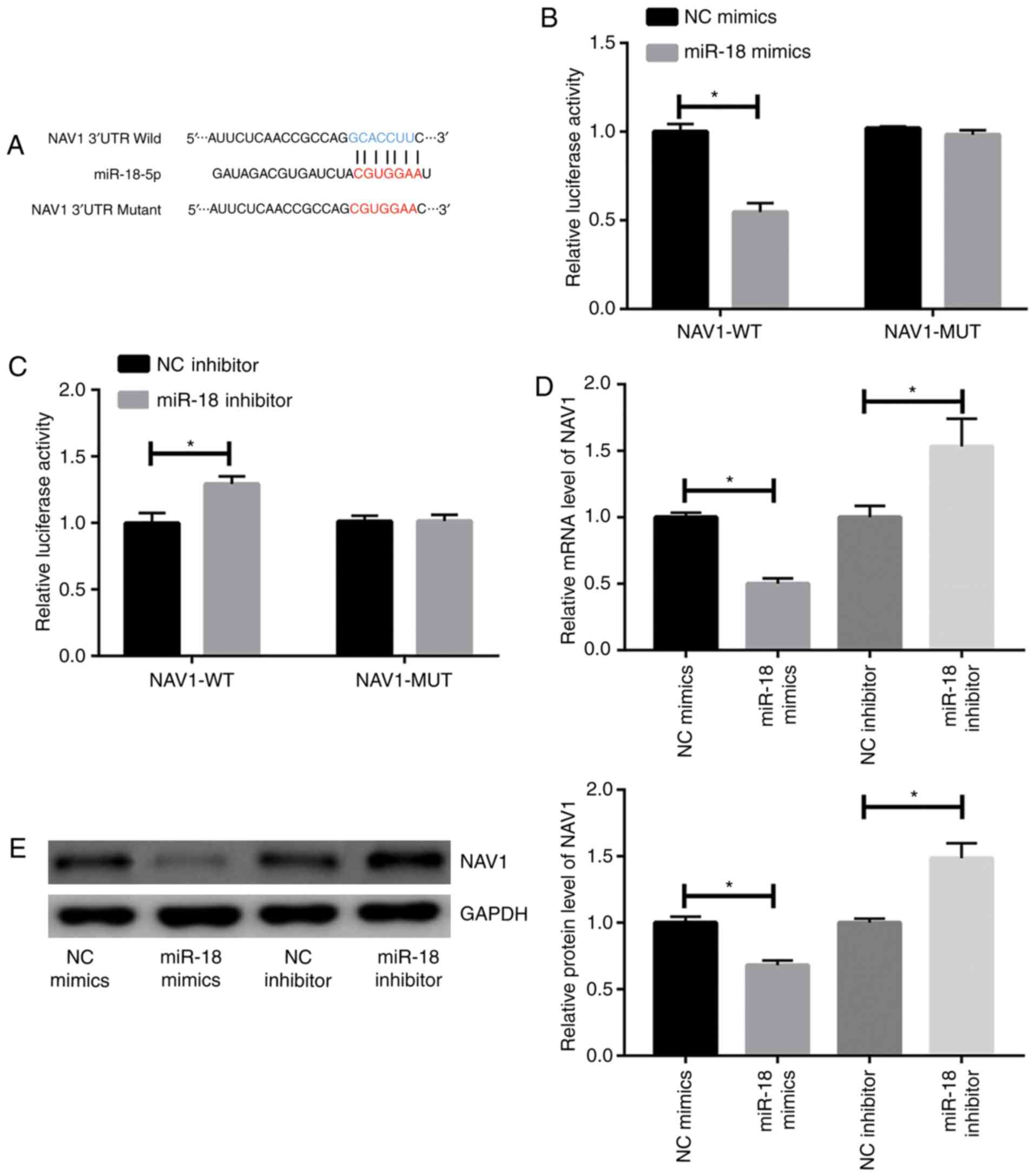
Through a bioinformatics prediction, NAV1 was
screened out as a target gene of miR-18. Wild-type NAV1 and
mutant-type NAV1 sequences were then constructed to verify the
binding interaction of miR-18 and NAV1 (Fig. 4A). Decreased luciferase activity was
identified in MIN6 cells co-transfected with miR-18 mimics and
wild-type NAV1 reporter plasmid. However, no significant change in
luciferase activity was identified after co-transfection of miR-18
mimic and mutant-type NAV1 reporter plasmid (Fig. 4B). Furthermore, cells co-transfected
with miR-18 inhibitor and wild-type NAV1 reporter plasmid exhibited
a higher luciferase activity compared with those co-transfected
with miR-18 inhibitor and mutant-type NAV1 reporter plasmid
(Fig. 4C). To further verify the
interaction between miR-18 and NAV1, the expression levels of NAV1
were detected after transfection of miR-18 mimics or inhibitor. The
results indicated that mRNA and protein levels of NAV1 were
negatively regulated by miR-18 (Fig. 4D
and E).
miR-18 inhibits the PI3K/AKT pathway
in islet β-cells
It was speculated that the PI3K/AKT pathway is
involved in the regulation of islet β cells. The results indicated
that miR-18 mimics caused a marked downregulation of the levels of
p-AKT and p-PI3K, as well as the p-AKT/AKT and the p-PI3K/PI3K
ratio, in MIN6 cells (Fig. 5A).
miR-18 knockdown produced the opposite results (Fig. 5B).
Discussion
Epidemiological studies have indicated that the
increased incidence of DM worldwide is closely associated with the
increased number of obese individuals. Obesity is involved in the
pathogenesis of DM through inflammation (16–18). It
is currently thought that islet-β cell dysfunction has a key role
in the occurrence and development of DM. Dysfunctional β-cells and
apoptosis are the leading causes of DM. Relative studies have
proved the significant role of cytokines in regulating islet cell
function, of which TNF-α and IL-1β are crucial in the pathogenesis
of DM. TNF-α is produced by macrophages and adipose cells. It
promotes lipid decomposition and release of free fatty acids, thus
leading to insulin resistance. TNF-α is also an important cytokine
involved in cell apoptosis. It is the initiator of the classic
death receptor and caspase-8 apoptotic pathway. Abundant
peroxynitrite and free radicals are further released to promote
cell apoptosis (19). IL-1β inhibits
insulin secretion and damage islet cell function through nuclear
factor-κB, c-Jun N-terminal kinase and suppressor of cytokine
signaling-3 pathways, eventually leading to the occurrence of DM
(20). A variety of inflammatory
factors, including TNF-α, IL-1β and INF-γ, are involved in the
development of DM (21). In the
present study, IL-1β treatment caused a marked upregulation of
miR-18 expression in MIN6 cells.
Various studies have focused on the effects of a
high-fat diet in gene transcription and protein translation.
Although post-transcriptional and translational regulations are
also crucial, their regulatory effects on β-cell damage induced by
a high-fat diet have been rarely reported. miRNAs exert their
crucial roles by regulating their target genes at the
post-transcriptional and translational levels. It has been
predicted that >60% of the human genome is regulated by miRNAs
(22). Previous studies have
indicated that certain miRNAs regulate the differentiation and
development of mouse islet β-cells (23). Knockdown of miR-146a or miR-34a in
mouse islet β-cells was demonstrated to remarkedly decrease
palmitate-induced apoptosis, while it did not affect the insulin
release function (24). The present
study indicated that inflammatory factors cause an upregulation of
miR-18 expression in islet β-cells and that miR-18 markedly
inhibits insulin production.
Studies have suggested that the number of islet
β-cells is progressively reduced during the disease course of DM,
which may be explained by the excessive apoptosis occurring
(25). The amount of islet β-cells
is markedly decreased in patients with type 2 DM. Abundant
apoptosis is observed in DM patients, whereas proliferation of
islet β-cells is under normal control, indicating the significant
role of apoptosis in the occurrence and progression of DM. Studies
on human islet amyloid polypeptide transgenic mice have indicated
that the increase in β-cell apoptosis exceeds the increase in cell
replication, leading to β-cell loss (26). Furthermore, high levels of cell
apoptosis are encountered at the early stage of DM. Other studies
indicated that the apoptotic rate of β-cells in patients with type
1 or type 2 DM is 3–10 times higher than that of healthy controls
(4,12), while the proliferation rate of
β-cells is maintained at a normal level. In the present study,
miR-18 markedly promoted MIN6 cell apoptosis.
miR-18 is widely expressed in human and mouse
tissues, which serves as a tumor-suppressor gene via targeting
K-Ras (27). The present study
demonstrated that NAV1 is a target gene of miR-18. The PI3K/AKT
pathway is one of the classical signaling pathways involved in
suppression of apoptosis and promotion of proliferation (28). In the present study, miR-18 was
indicated to regulate DM development via inhibiting the PI3K/AKT
pathway.
In conclusion, miR-18 expression is upregulated by
IL-1β induction in islet β-cells. miR-18 promotes apoptosis of
islet β-cells, at least in part, by inhibiting NAV1 expression and
insulin production via suppression of the PI3K/AKT pathway.
However, miR-18 has multiple target genes and furthermore, the
association between NAV-1 and the apoptosis of islet β-cell
requires further investigation. In addition, the present study only
provided evidence from in vitro experiments, and a further
in vivo study may be required.
Acknowledgements
Not applicable.
Funding
No funding received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
HF and MS designed the study; LC, ZW and LS
performed the experiments; ZW and LS collected the data; HF and MS
analyzed the data; and HF and LC prepared the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Report of the expert committee on the
diagnosis and classification of diabetes mellitus, . Diabetes Care.
20:1183–1197. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gomis R, Artola S, Conthe P, Vidal J,
Casamor R and Font B; investigadores del Grupo de Estudio OBEDIA, :
Prevalence of type 2 diabetes mellitus in overweight or obese
outpatients in Spain. OBEDIA Study. Med Clin (Barc). 142:485–492.
2014.(In Spanish). View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Runkel M, Muller S, Brydniak R and Runkel
N: Downgrading of type 2 diabetes mellitus (T2DM) after obesity
surgery: Duration and severity matter. Obes Surg. 25:494–499. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cnop M, Welsh N, Jonas JC, Jörns A, Lenzen
S and Eizirik DL: Mechanisms of pancreatic beta-cell death in type
1 and type 2 diabetes: Many differences, few similarities.
Diabetes. 54 (Suppl 2):S97–S107. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bergman RN, Finegood DT and Kahn SE: The
evolution of beta-cell dysfunction and insulin resistance in type 2
diabetes. Eur J Clin Invest. 32 (Suppl 3):S35–S45. 2002. View Article : Google Scholar
|
|
6
|
Li X, Li X, Wang G, Xu Y, Wang Y, Hao R
and Ma X: Xiao Ke Qing improves glycometabolism and ameliorates
insulin resistance by regulating the PI3K/Akt pathway in KKAy mice.
Front Med. 12:388–396. 2018. View Article : Google Scholar
|
|
7
|
Gao L, Li SL and Li YK: Liraglutide
promotes the osteogenic differentiation in MC3T3-E1 cells via
regulating the expression of Smad2/3 through PI3K/Akt and
wnt/β-catenin pathways. DNA Cell Biol. Nov 7–2018.(Epub ahead of
print). View Article : Google Scholar
|
|
8
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xie MB, Sui XQ, Pei D, Yao Q and Huang Q:
Study on the expression and mechanism of plasma microRNA-21 in
patients with ischemic cardiomyopathy. Eur Rev Med Pharmacol Sci.
21:4649–4653. 2017.PubMed/NCBI
|
|
10
|
Sayed D and Abdellatif M: MicroRNAs in
development and disease. Physiol Rev. 91:827–887. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Guay C and Regazzi R: Role of islet
microRNAs in diabetes: Which model for which question?
Diabetologia. 58:456–463. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Plaisance V, Waeber G, Regazzi R and
Abderrahmani A: Role of microRNAs in islet beta-cell compensation
and failure during diabetes. J Diabetes Res. 2014:6186522014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Schrimpe-Rutledge AC, Fontes G, Gritsenko
MA, Norbeck AD, Anderson DJ, Waters KM, Adkins JN, Smith RD,
Poitout V and Metz TO: Discovery of novel glucose-regulated
proteins in isolated human pancreatic islets using LC-MS/MS-based
proteomics. J Proteome Res. 11:3520–3532. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sanchez-Mejias A, Kwon J, Chew XH, Siemens
A, Sohn HS, Jing G, Zhang B, Yang H and Tay Y: A novel
SOCS5/miR-18/miR-25 axis promotes tumorigenesis in liver cancer.
Int J Cancer. 144:311–321. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Seidell JC: Obesity, insulin resistance
and diabetes-a worldwide epidemic. Br J Nutr. 83 (Suppl 1):S5–S8.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kahn BB and Flier JS: Obesity and insulin
resistance. J Clin Invest. 106:473–481. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gupta D, Krueger CB and Lastra G:
Over-nutrition, obesity and insulin resistance in the development
of beta-cell dysfunction. Curr Diabetes Rev. 8:76–83. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Borst SE: The role of TNF-alpha in insulin
resistance. Endocrine. 23:177–182. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Febbraio MA: Role of interleukins in
obesity: Implications for metabolic disease. Trends Endocrinol
Metab. 25:312–319. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Donath MY, Størling J, Maedler K and
Mandrup-Poulsen T: Inflammatory mediators and islet beta-cell
failure: A link between type 1 and type 2 diabetes. J Mol Med
(Berl). 81:455–470. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Friedman RC, Farh KK, Burge CB and Bartel
DP: Most mammalian mRNAs are conserved targets of microRNAs. Genome
Res. 19:92–105. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lynn FC, Skewes-Cox P, Kosaka Y, McManus
MT, Harfe BD and German MS: MicroRNA expression is required for
pancreatic islet cell genesis in the mouse. Diabetes. 56:2938–2945.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lovis P, Roggli E, Laybutt DR, Gattesco S,
Yang JY, Widmann C, Abderrahmani A and Regazzi R: Alterations in
microRNA expression contribute to fatty acid-induced pancreatic
beta-cell dysfunction. Diabetes. 57:2728–2736. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Clark A, Wells CA, Buley ID, Cruickshank
JK, Vanhegan RI, Matthews DR, Cooper GJ, Holman RR and Turner RC:
Islet amyloid, increased A-cells, reduced B-cells and exocrine
fibrosis: Quantitative changes in the pancreas in type 2 diabetes.
Diabetes Res. 9:151–159. 1988.PubMed/NCBI
|
|
26
|
Butler AE, Janson J, Bonner-Weir S, Ritzel
R, Rizza RA and Butler PC: Beta-cell deficit and increased
beta-cell apoptosis in humans with type 2 diabetes. Diabetes.
52:102–110. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tsang WP and Kwok TT: The miR-18a*
microRNA functions as a potential tumor suppressor by targeting on
K-Ras. Carcinogenesis. 30:953–959. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Huang XL, Cui GH and Zhou KY: Correlation
of PI3K-Akt signal pathway to apoptosis of tumor cells. Ai Zheng.
27:331–336. 2008.(In Chinese). PubMed/NCBI
|