Introduction
Axenfeld-Rieger syndrome (ARS), occurring in
~1/200,000 individuals, is a rare autosomal-dominant genetic
disease characterized by anterior segment disorders and systemic
abnormalities. The offspring of ARS patients have a 50% chance of
inheriting the disease. Common anterior segment abnormalities
include abnormalities in the anterior chamber angle structure, as
well as the cornea and iris. Approximately 50% of ARS patients
eventually progress to glaucoma, which may induce blindness within
a small number of years. In addition to the ocular phenotype,
systemic developmental abnormalities, including craniofacial
hypoplasia (midface hypoplasia, hypertelorism and telecanthus),
dental abnormalities (microdontia, oligodontia, adontia and
hypodontia), pituitary abnormalities, hearing loss, kidney
abnormalities, cardiovascular outflow tract malformation, redundant
periumbilical skin, and variable neurological and skeletal
anomalies, may also be associated with ARS (1,2).
ARS heterozygotes have less severe and more variable
phenotypes than null homozygotes. Affected individuals display
overlapping phenotypes, including Axenfeld-Rieger anomaly (ARA) and
ARS (3). As a result of the abnormal
migration of neural crest cells, ARA encompasses a spectrum of
ocular disorders, including iridocorneal adhesions, prominent and
anteriorly displaced Schwalbe's line, and atrophic iris. ARS
patients present with ocular and systemic malformations.
Two major genes, forkhead box C1 (FOXC1) and
pituitary homeobox 2 (PITX2), have important roles in embryonic
development and have been demonstrated to cause ARS.
Disease-causing copy number changes and point mutations in these
two genes have been identified in 40–70% of ARS patients (4). Mutations in the FOXC1 gene, a member of
the FOX family of transcription factors, in ARS patients are more
likely to be associated with glaucoma (5). PITX2 is a member of the homeobox gene
family and regulates the proliferation and differentiation of
various ocular anterior segment tissues and certain non-ocular
tissues, including branchial arches and myocardium (6). The mutations in PITX2 are more likely
to induce systemic abnormalities than mutations in FOXC1 (4,7).
In the present study, a novel genetic mutation
(c.1494delG) was identified in a Chinese family with ARS and the
clinical features of the patients were described.
Materials and methods
Patient recruitment and
evaluation
A Chinese Han family from southern China presented
with an autosomal-dominant inheritance pattern for ARS (Fig. 1A and 1B) that affected 2 of the 7
living family members. The Chinese family was recruited in November
2017. Data on the medical and ophthalmic medical history were
collected and ophthalmological examinations were performed. All
procedures were performed according to the guidelines of the
Declaration of Helsinki for research involving human subjects, and
were approved by the ethics committee of the Chinese PLA General
Hospital (Beijing, China). Written informed consent, including for
DNA extraction, gene analysis and ophthalmological examinations,
was obtained from all family members. In addition, 150 normal
controls recruited for the present study had provided written
informed consent for DNA extraction and gene analysis.
DNA library preparation
Genomic DNA from patients was quantified by agarose
gel electrophoresis and Nanodrop™ 2000 (Thermo Fisher Scientific,
Inc.). Libraries were prepared using an Illumina standard protocol
(Illumina, Inc.). In brief, 3 µg genomic DNA was fragmented by
nebulization (E220; Covaris, Inc.). The fragmented DNA was then
repaired and an ‘A’ was ligated to the 3′ end. Next, Illumina
adapters (Illumina, Inc.) were ligated to the fragments, and the
sample was size-selected, aiming for a 350–400-base-pair product.
The size-selected product was amplified using PCR (each sample was
tagged with a unique index during this procedure), and the final
product was validated using the Agilent Bioanalyzer 2100 (Agilent
Technologies, Inc.).
Targeted gene enrichment and
sequencing
The amplified DNA was captured with an ocular
disease-associated gene panel with biotinylated oligo-probes using
MyGenostics GenCap Enrichment technologies (MyGenostics, Inc.). The
probes were designed to tile along 662 ocular disease-associated
genes. The capture experiment was performed according to
manufacturer's protocol. In brief, 1 µg DNA library was mixed with
Buffer BL and GenCap gene panel probe (MyGenostics Inc.), and
heated at 95°C for 7 min and 65°C for 2 min in a PCR machine 2720
(Applied Biosystems; Thermo Fisher Scientific, Inc.). Next, 23 µl
of the 65°C pre-warmed Buffer HY (MyGenostics Inc.) was added to
the mix, and the mixture was kept at 65°C with the PCR heat lid on
for 22 h for hybridization. MyOne beads (50 µl; Thermo Fisher
Scientific, Inc.) were washed in 500 µl 1X binding buffer 3 times
and resuspended in 80 µl 1X binding buffer. Subsequently, 64 µl 2X
binding buffer was added to the hybrid mix and transferred to the
tube with 80 µl MyOne beads. The mix was rotated for 1 h on a
rotator. The beads were then washed with WB1 buffer at room
temperature for 15 min once and WB3 buffer at 65°C for 15 min three
times. The bound DNA was then eluted with Buffer Elute. The eluted
DNA was finally amplified in 15 cycles using the following program:
Initial denaturation at 98°C for 30 sec, 15 cycles of 98°C for 25
sec, 65°C for 30 sec and 72°C for 30 sec, followed by a hold at
72°C for 5 min. The PCR product was purified using SPRI beads
(Beckman Coulter, Inc.) according to the manufacturer's protocol.
The enrichment libraries were sequenced on an Illumina HiSeq 500
sequencer (Illumina, Inc.) for paired-read 100 bp.
The variations were detected by a hereditary eye
disease enrichment panel and validated by Sanger sequencing. PCR
primer sets were designed using Primer 6.0 (forward,
5′-TACTCTCTGCCTCCGGTCAC-3′ and reverse, 5′-TGCTTTGGGGTTCGATTTAG-3′;
chr6:1611971-1612380, 410 bp). The products were sequenced using a
Bigdye terminator v3.1 cycle sequencing kit (Thermo Fisher
Scientific, Inc.) and analyzed on an ABI 3730XL Genetic Analyzer
(Thermo Fisher Scientific, Inc.).
Bioinformatics analysis
Following HiSeq 500 sequencing, high-quality reads
were retrieved from raw reads by filtering out the low-quality
reads and adaptor sequences using the Solexa QA package and the
Cutadapt program (https://cutadapt.readthedocs.io/en/stable/),
respectively (8). The BWA program
(http://bio-bwa.sourceforge.net/) was
then used to align the clean-read sequences to the human reference
genome hg19 (9). After the PCR
duplicates were removed using Picard software (http://broadinstitute.github.io/picard/), the single
nucleotide polymorphisms (SNPs) and small insertions/deletions
(InDels) were identified using the GATK HaplotypeCaller program
(https://software.broadinstitute.org/gatk/). The SNPs
and InDels identified were annotated using the ANNOVAR program
(http://annovar.openbioinformatics.org/en/latest/)
(10). MagicViewer (http://bioinformatics.zj.cn/magicviewer/index.php) was
used to view the short-read alignment and validate the candidate
SNPs and InDels. Non-synonymous variants were evaluated using 4
algorithms, Ployphen (http://genetics.bwh.harvard.edu/pph2/), SIFT
(http://sift.jcvi.org/), PANTHER (www.pantherdb.org) and Pmut (http://mmb.pcb.ub.es/PMut/), as previously described,
to determine pathogenicity. The Human Gene Mutation Database was
used to support the novelty of the results (http://www.biobase-international.com/product/hgmd).
Results
Phenotyping
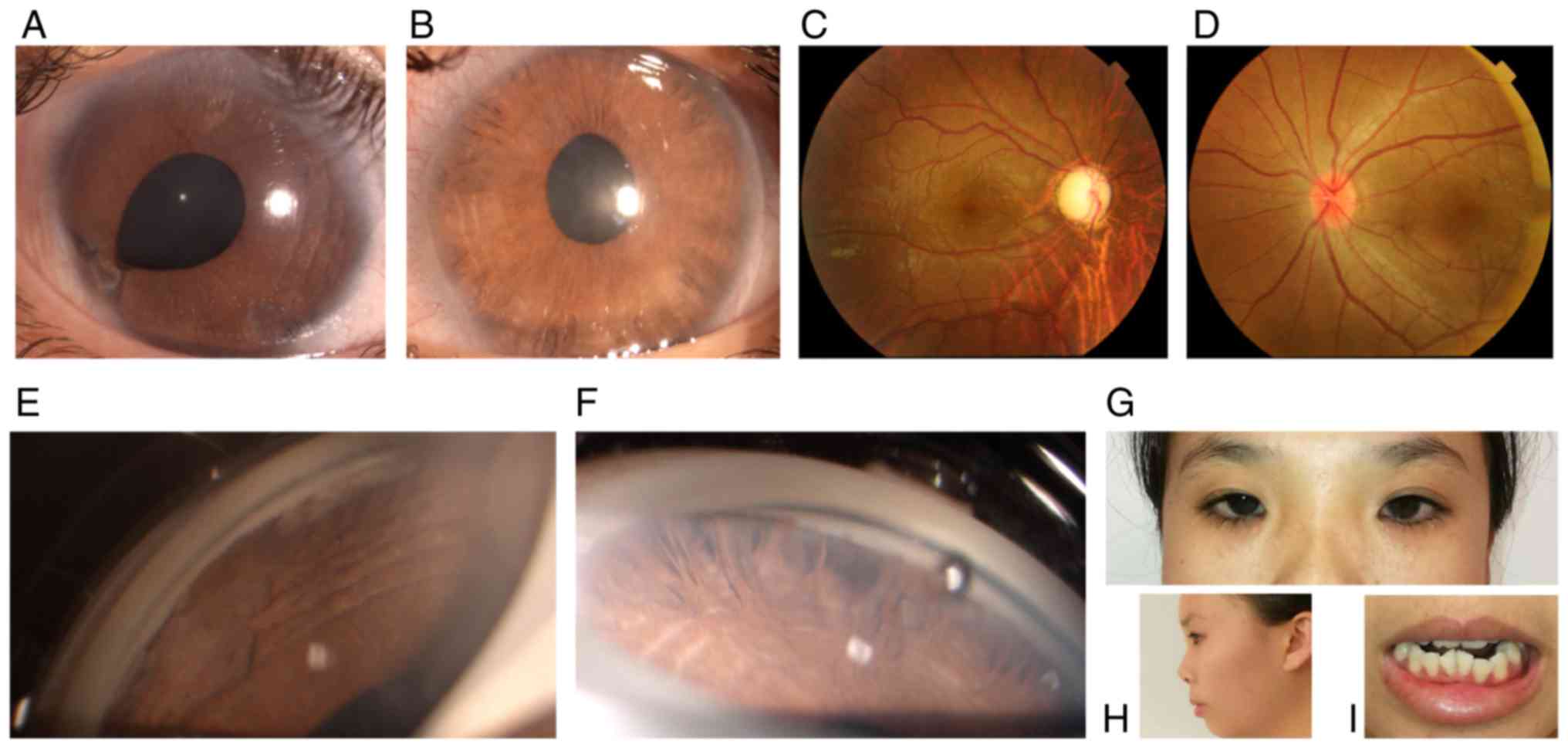
Case III:2 (the proband)
The 20-year-old female attended the outpatient
clinic of our hospital in November 2017. The proband complained of
blurred vision for the past 2 weeks in the right eye. Upon initial
examination, the patient's best corrected visual acuity (BCVA) was
HM/10 cm OD and 20/100 OS. The intraocular pressure (IOP) measured
by Goldmann tonometry reached 50 mmHg OD and 33 mmHg OS. Slit-lamp
microscopic examination indicated characteristic corneal posterior
embryotoxon in the bilateral eyes. Stromal hypoplasia with
corectopia were present in each eye. Iridocorneal adhesion was
noted in the right eye. Gonioscopy of the bilateral eyes indicated
open angles, peripheral broad-based synechiae, anterior insertion
of the iris into the trabecular meshwork and prominent iris
processes. By fundoscopy, the cup-to-disk ratio was determined to
be 0.9 in the right eye and 0.2 in the left. The corneal diameter
of each eye was 12 mm. The corneal endothelial cells in each eye
were normal. The central corneal thickness was 589 µm in the right
eye and 643 µm in the left. The axial globe length was 25.4 mm in
the right eye and 20.9 mm in the left. The eyes of the patient were
treated by topical anti-glaucoma medications containing brimonidine
tartrate and travoprost. The proband's IOP in the bilateral eyes
was well controlled and the BCVA in the right eye improved to
20/200. The visual field defect was severe in the right eye but the
visual field in the left eye was normal. Further physical
examination revealed midface hypoplasia, hypertelorism, telecanthus
and dental malocclusion. The umbilicus was normal. The proband was
diagnosed with ARS based on the characteristic ocular and systemic
signs (Fig. 2A-H).
Case II:2 (the proband's father)
The proband's father was 45 years old. This
patient's BCVA was 20/20 in each eye. IOP measured with Goldmann
tonometry was 23 mmHg in the right eye and 22 mmHg in the left eye.
The iris was atrophic in the bilateral eyes. Gonioscopy indicated
open angles in each eye, with prominent iris processes and anterior
insertion of the iris into the trabecular meshwork. The patient's
visual field test was normal. Further physical examination revealed
midface hypoplasia, hypertelorism and telecanthus. The umbilicus
was normal. The diagnosis of ARS was made based on the
characteristic clinical findings (Fig.
3A-H).
Case I:1 (the proband's grandfather,
who had passed away)
The proband's grandfather had passed away at the age
85. The family members of this case reported that he had gone blind
due to glaucoma at the age 72. The grandfather was likely to be a
gene variant, which was supported by his medical history and gene
analysis of other family members.
Genotyping
The 662 inheritable genetic ocular genes were
captured and sequenced by next-generation sequencing using genomic
DNA from the proband (III:2). DNA sequencing revealed that two
affected members in the family harbored a novel deletion mutation
(Fig. 1B). The nucleotide deletion
(c.1494delG) within exon 1 in the FOXC1 gene resulted in a
frameshift (p.G499Afs*20) mutation, with the destruction of
forkhead domain (FHD) structure, which is supposed to result in ARS
through haploinsufficiency.
None of the 5 unaffected individuals (Fig. 1B: I:2, II:1, II:3, II:4 and III:1)
had this mutation in the FOXC1 gene. Since case no. I:1 had passed
away, gene analysis was not available for this patient. ARS is
transmitted by an autosomal-dominant mode of inheritance, so case
no. I:1 was likely to be a variant.
The frameshift mutation (p.G499Afs*20) in the
C-terminal transactivation domain (aa 466–553) induced a shorter
mutant FOXC1 protein (517aa). FOXC1 protein sequence alignment
(amino acids 499–517) across species indicates evolutionary
conservation at the C-terminal fragment (Fig. 4). The prediction of the protein
structure revealed a series of α-helices in the C-terminal
transactivation domain of the wild-type protein, which was missing
in the mutant (Fig. 5). Loss of the
series of α-helices may have impaired FOXC1 function through
altering the transactivation activity, thereby inducing ARS in this
family.
Discussion
ARS is characterized by anomalies in the anterior
segment of the eye and systemic signs, and its diagnosis is based
on its typical clinical features and genetic defects. There are
three types of ARS and each has a different genetic cause. ARS type
1 is caused by mutations in the PITX2 gene. Type 3 is caused by
mutations in the FOXC1 gene. The gene that causes Axenfeld-Rieger
syndrome type 2 is not known, but it is located on chromosome 13
(11). The two patients in the
present study had type 3 ARS.
At times, ARS requires to be distinguished from
other anterior segment dysgenesis (ASD) disorders and iridocorneal
endothelial syndrome (ICE). ASD disorders encompass a variety of
clinical presentations, including ARS, aniridia, Peters anomaly, as
well as syndromal associations. ICE is an acquired disorder, is
usually unilateral and predominantly affects females in early to
middle adulthood. On specular microscopy, corneal endothelial cells
of ICE patients were reduced in number and revealed variable
degrees of pleomorphism in size and shape, with loss of hexagonal
margins. The specular microscopy results of the two patients of the
present study were normal.
Most ARS patients presenting with ocular features
and with full penetrance are associated with mutations in the FOXC1
and PITX2 genes (11). PR/SET domain
5 and cytochrome P450 family 1 subfamily B member 1 mutations were
also reported to be associated with ARS (2,12). In
addition, the association between ARS and PAX6 deletion that was
previously reported has recently been demonstrated to be incorrect
(13). Two other loci on chromosomes
16q23-24 and 13q14 have been considered to be associated with ARS
(11). The most common FOXC1 defects
leading to ARS are point mutations (14).
Biochemical analyses of FOXC1 Arg127His demonstrated
that although this mutation significantly perturbed the protein
function, a patient with this mutation had less severe clinical
manifestations than a patient with the biochemically milder
mutation FOXC1 Pro79Thr (15,16).
Functional analyses of the p.Pro79Thr and p.Pro79Leu mutations
indicated that they equivalently inhibited the function of FOXC1,
but the impairment of the ARS patient with the FOXC1 Pro79Thr
mutation appeared to be more serious than that of the patient with
the Pro79Leu mutation (17,18). Therefore, there is no apparent
correlation between the functional impairment caused by the
missense mutation of FOXC1 and the severity of the phenotype. The
phenotype of ARS varies considerably among cases, and even between
the two eyes of the same patient (14,19,20).
This is consistent with the presentation of the Chinese ARS
patients of the present study. The proband presented with glaucoma
and severe visual field defect in the right eye but a normal visual
field in the left eye. The ocular examinations revealed notable
corneal posterior embryotoxon, corectopia and iridocorneal adhesion
in the right eye. Gonioscopy of the two eyes indicated peripheral
broad-based synechiae and iris strands bridging the iridocorneal
angle to the trabecular meshwork. In addition, the proband
presented with a flat face, a flat nasal bridge, hypertelorism and
telecanthus. The proband was diagnosed with ARS due to genetic
defect. The phenotype of the proband's father was considerably
milder than that of the proband, and he had normal visual function
of the bilateral eyes. Ocular examination of the proband's father
indicated slight IOP elevation, atrophy of the iris and anterior
insertion of the iris into the trabecular meshwork in each eye.
Further physical examination revealed midface hypoplasia,
hypertelorism, telecanthus and flat broad nasal bridge. Based on
this presentation and the results of the genetic test, the
proband's father was diagnosed with ARS.
Genetic defects in PITX2 differ widely and include
coding-region frameshift, splice-site, nonsense and missense
mutations. FOXC1 and PITX2 interact with each other functionally
and physically. FOXC1 is negatively regulated by PITX2. Functional
protein dosage alteration, duplication of FOXC1 and deletion of
PITX2 are thought to cause similar phenotypes of ARS (21,22). ARS
patients with duplication mutations of FOXC1 primarily present with
iris hypoplasia and glaucoma, while those with missense mutations
have various manifestations with extraocular phenotypes (18).
FOXC1, a 553-amino-acid protein, is characterized by
a conserved 110-amino-acid motif known as the FHD. The nuclear
localization signals at either end of the FHD support the transfer
of FOXC1 to the nucleus and binding to DNA, thereby regulating the
downstream target genes. FOXC1 has a phosphorylated inhibitory
domain and two transactivation domains located outside the FHD.
FOXC1 is highly expressed in the heart, kidney and skeletal
muscles, and has a role in tumor development, tissue-specific gene
expression and embryogenesis. To the best of our knowledge, 54
different genetic mutations of FOXC1 have been detected in ARS
patients, including missense mutations (n=31), InDels/duplications
(n=17) and nonsense mutations (n=6) (9). Most missense mutations affect the amino
acids within the FHD, which impair the FOXC1 protein function by
altering the protein structure, nuclear localization,
transactivation activity, DNA-binding ability and protein stability
(16,19). The deletions and duplications of
FOXC1 induce ARS by affecting the FOXC1 protein level required for
the normal development of ocular tissues. Mutations at the
C-terminal transactivation domain are less characterized. It has
been reported that FOXC1 is a short-lived transcription factor (the
half life t1/2<30 min) that is degraded through the
ubiquitin 26 S proteasome pathway. Amino acid residues 367–553,
which include the C-terminal transactivation domain, are essential
for this ubiquitin incorporation and proteolysis, indicating that
FOXC1 protein levels and activity are regulated by
post-translational modifications (23). The frameshift mutation
(p.G499Afs*20), located at the C-terminal transactivation domain,
probably influences FOXC1 protein ubiquitination. It may therefore
be suggested that it is the underlying cause of ARS in the Chinese
family of the present study.
ARS was diagnosed in this family based on ocular
findings and craniofacial features. An increased IOP was detected
in the two ARS patients. Glaucoma was diagnosed in the female
patient (case III.2). The grandfather of this proband (case I.1)
had gone blind due to glaucoma; he was likely to be a gene variant
and passed on the mutation to his son (case II.2). Characteristic
ocular features were observed in the proband, including iris
insertion into the trabecular meshwork, corectopia, iridocorneal
adhesions, posterior embrytoxon and atrophic iris. Although the
severity of glaucoma is linked to the level of iris insertion into
the trabecular meshwork, physical occlusion of the angle structure
is not a prerequisite for glaucoma in ARS patients (24). The female patient of the present
study responded well to topical anti-glaucoma combination therapy.
Trabeculotomy-trabeculectomy surgery has been proved to be a safe
and effective procedure for ARA patients with glaucoma (25). It has been reported that a relatively
low proportion of patients with glaucoma and either FOXC1 or PITX2
mutations (18%) respond to medication or surgery. In comparison
with other mutation types, FOXC1 duplication mutants have been
associated with a relatively poor prognosis regarding glaucoma
development (16).
In the present study, a novel mutation was
identified in a Chinese family with ARS and the clinical
presentation and eye examination results of the affected patients,
including craniofacial and ocular abnormalities, were described. A
heterozygous deletion of the FOXC1 gene (c.1494delG) was detected.
This 1-bp deletion caused a frameshift mutation, p.G499Afs*20, in
the C-terminal transactivation domain, disrupted the secondary
structure of the FOXC1 protein and influenced protein
ubiquitination. The present results further supported the critical
control of FOXC1 in ocular development and broadened the spectrum
of FOXC1 mutations in Chinese patients with ARS.
Acknowledgements
Not applicable.
Funding
This study was supported by grants from the Special
Research and Trial Production Project in Sanya (grant no.
2017KS03), the Science and Technology Achievement Transformation
Project in Sanya (grant no. 2017CZ13), the Key Research Plan of
Hainan Province (grant no. ZDYF2018139), the Innovation project for
Medical and Health Science and Technology in Sanya (grant no.
2014YW35) and the Natural Science Foundation of Beijing
Municipality (grant no. 7162180).
Availability of data and materials
The data supporting the conclusions of the present
study are contained within the manuscript. All raw data
generated/used during the present study are available from the
corresponding author on reasonable request.
Authors' contributions
XW and NHX performed the analysis of data and
drafted the manuscript. HZL and JDW participated in the analysis
and interpretation of data. TW, WL, LLC and YW performed the
acquisition of data. BHH made substantial contributions to the
conception and design of the study and revision of the manuscript.
All authors approved the final manuscript as submitted and agree to
be accountable for all aspects of the work.
Ethics approval and consent to
participate
The study was approved by the Ethics Committee of
the Chinese PLA General Hospital (Beijing, China) and the
experiments followed the tenets of the Declaration of Helsinki.
Patient consent for publication
Written informed consent was obtained from all
patients to publish their cases and the photos in this study.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ARS
|
Axenfeld-Rieger syndrome
|
|
FOXC1
|
forkhead box C1
|
|
PITX2
|
pituitary homeobox 2
|
|
FHD
|
forkhead domain
|
|
ARA
|
Axenfeld-Rieger anomaly
|
|
IOP
|
intraocular pressure
|
|
ICE
|
iridocorneal endothelial syndrome
|
|
BCVA
|
best corrected visual acuity
|
|
ASD
|
anterior segment dysgenesis
|
|
SNPs
|
single nucleotide polymorphisms
|
|
InDels
|
small insertions/deletions
|
References
|
1
|
Hjalt TA and Semina EV: Current molecular
understanding of Axenfeld-Rieger syndrome. Expert Rev Mol Med.
7:1–17. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Millá E, Mañé B, Duch S, Hernan I, Borràs
E, Planas E, Dias Mde S, Carballo M and Gamundi MJ; Spanish
Multicenter Glaucoma Group-Estudio Multicéntrico Español de
Investigación Genética del Glaucoma, EMEIGG, : Survey of familial
glaucoma shows a high incidence of cytochrome P450, family 1,
subfamily B, polypeptide 1 (CYP1B1) mutations in non-consanguineous
congenital forms in a Spanish population. Mol Vis. 19:1707–1722.
2013.PubMed/NCBI
|
|
3
|
Espinoza HM, Cox CJ, Semina EV and Amendt
BA: Amolecular basis for differential developmental anomalies in
Axenfeld-Rieger syndrome. Hum Mol Genet. 11:743–753. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Reis LM, Tyler RC, Volkmann Kloss BA,
Schilter KF, Levin AV, Lowry RB, Zwijnenburg PJ, Stroh E, Broeckel
U, Murray JC and Semina EV: PITX2 and FOXC1 spectrum of mutations
in ocular syndromes. Eur J Hum Genet. 20:1224–1233. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Challa P: Glaucoma genetics. Int
Ophthalmol Clin. 48:73–94. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gage PJ and Camper SA: Pituitary homeobox
2, a novel member of the bicoid-related family of homeobox genes,
is a potential regulator of anterior structure formation. Hum Mol
Genet. 6:457–464. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tümer Z and Bach-Holm D: Axenfeld-Rieger
syndrome and spectrum of PITX2 and FOXC1 mutations. Eur J Hum
Genet. 17:1527–1539. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cox MP, Peterson DA and Biggs PJ:
SolexaQA: At-a-glance quality assessment of Illumina
second-generation sequencing date. BMC Bioinformatics. 11:4852010.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li H and Durbin R: Fast and accurate short
read alignment with Burrows-Wheeler transform. Bioinformatics.
25:1754–1760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang K, Li M and Hakonarson H: ANNOVAR:
Functional annotation of genetic variants from high-throughput
sequencing data. Nucleic Acids Res. 38:e1642010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Seifi M and Walter MA: Axenfeld-Rieger
syndrome. Clin Genet. 93:1123–1130. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Micheal S, Siddiqui SN, Zafar SN,
Venselaar H, Qamar R, Khan MI and den Hollander AI: Whole exome
sequencing identifies a heterozygous missense variant in the PRDM5
gene in a family with Axenfeld-Rieger syndrome. Neurogenetics.
17:17–23. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Riise R, D'haene B, De Baere E, Grønskov K
and Brøndum-Nielsen K: Rieger syndrome is not associated with PAX6
deletion: A correction to Acta. Ophthalmol Scand 2001:79: 201–203.
Acta Ophthalmol. 87:9232009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kim GN, Ki CS, Seo SW, Yoo JM, Han YS,
Chung IY, Park JM and Kim SJ: A novel forkhead box C1 gene mutation
in a Korean family with Axenfeld-Rieger syndrome. Mol Vis.
19:935–943. 2013.PubMed/NCBI
|
|
15
|
Kawase C, Kawase K, Taniguchi T, Sugiyama
K, Yamamoto T, Kitazawa Y, Alward KL, Stone EM, Nishimura DY and
Sheffield VC: Screening for mutations of Axenfeld-Rieger syndrome
caused by FOXC1 gene in Japanese patients. J Glaucoma. 10:477–482.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Saleem RA, Banerjee-Basu S, Berry FB,
Baxevanis AD and Walter MA: Structural and functional analyses of
disease-causing missense mutations in the forkhead domain of FOXC1.
Hum Mol Genet. 12:2993–3005. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Suzuki T, Takahashi K, Kuwahara S, Wada Y,
Abe T and Tamai M: A novel (Pro79Thr) mutation in the FKHL7 gene in
a Japanese family with Axenfeld-Rieger syndrome. Am J Ophthalmol.
132:572–575. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Strungaru MH, Dinu I and Walter MA:
Genotype-phenotype correlations in Axenfeld-Rieger malformation and
glaucoma patients with FOXC1 and PITX2 mutations. Invest Ophthalmol
Vis Sci. 48:228–237. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Seifi M, Footz T, Taylor SA and Walter MA:
Comparison of bioinformatics prediction, molecular modeling, and
functional analyses of FOXC1 mutations in patients with
Axenfeld-Rieger syndrome. Hum Mutat. 38:169–179. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Honkanen RA, Nishimura DY, Swiderski RE,
Bennett SR, Hong S, Kwon YH, Stone EM, Sheffield VC and Alward WL:
A family with Axenfeld-Rieger syndrome and peters anomaly caused by
a point mutation (Phe112Ser) in the FOXC1 gene. Am J Ophthalmol.
135:368–75. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Berry FB, Lines MA, Oas JM, Footz T,
Underhill DA, Gage PJ and Walter MA: Functional interactions
between FOXC1 and PITX2 underlie the sensitivity to FOXC1 gene dose
in Axenfeld-Rieger syndrome and anterior segment dysgenesis. Hum
Mol Genet. 15:905–919. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Seifi M, Footz T, Taylor SA, Elhady GM,
Abdalla EM and Walter MA: Novel PITX2 gene mutations in patients
with Axenfeld-Rieger syndrome. Acta Ophthalmol. 94:e571–e579. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Berry FB, Mirzayans F and Walter MA:
Regulation of FOXC1 stability and transcriptional activity by an
epidermal growth factor-activated mitogen-activated protein kinase
signaling cascade. J Biol Chem. 281:10098–10104. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Schachtschabel DO, Binninger EA and Rohen
JW: In vitro cultures of trabecular meshwork cells of the human eye
as a model system for the study of cellular aging. Arch Gerontol
Geriatr. 9:251–262. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mandal AK and Pehere N: Early-onset
glaucoma in Axenfeld-Rieger anomaly: Long-term surgical results and
visual outcome. Eye (Lond). 30:936–942. 2016. View Article : Google Scholar : PubMed/NCBI
|