Introduction
The lung alveolar epithelium covers 99% of the
internal surface area of the lung and is composed of two
morphological and functional distinct types of cells: alveolar type
I (ATI) and type II (ATII). ATI cells have a broad flat shape,
covering 95% of the alveolar surface and accounting for 40% of the
alveolar epithelium and 8% of the peripheral lung cells. ATII
cells, however, have a small cuboidal shape, covering only 5% of
the alveolar surface and accounting for 60% of the alveolar
epithelium and 15% of peripheral lung cells (1–3).
ATII cells are also distinguished by the presence of lamellar
bodies (LBs), intracellular organelles that store and secrete
surfactant protein-C (SP-C). Functionally, ATII cells regulate
alveolar fluid levels and contribute to host defense and the immune
response (4,5). Notably, accumulating evidence
indicates that ATII cells are the progenitors of ATI cells in the
alveoli; ATII cells are believed to play a pivotal role in
maintaining tissue homeostasis via epithelium restoration (1,3,6).
Once ATI cells become damaged by lung injury, ATII
cells proliferate and transdifferentiate into ATI cells, thereby
facilitating the repair of lung epithelium (1,3,6).
Transfusion of exogenous ATII cells derived from human embryonic
stem cells was reported to efficiently repair acute lung injury in
a mouse model (7). Using a rat
lung ischemia-reperfusion injury (LIRI) model, we also discovered
that ATII cells displayed a self-repair capacity. However, the
process was protracted and insufficient (8). Therefore, research efforts are
focused on developing methods to endogenously enhance the repair
capacity of ATII cells or exogenously increase ATII cells (1,3).
Statins, 3-hydroxy-3-methylglutaryl coenzyme A
(HMG-CoA) reductase inhibitors, were initially developed as
antimicrobials but quickly gained popularity for their efficacious
lipid-lowering effects. Their widespread clinical use as first-line
drugs for hyperlipidemia revealed potential pleiotropic
pharmacological effects, and extensive research studies have
demonstrated that the statins elicit anti-inflammatory and
angiogenic effects and improve vascular endothelial cell functions
(9,10). The HMG-CoA reductase pathway (also
known as the mevalonate pathway) is essential for the synthesis of
a number of isoprenoids, such as the prenylation-inducing farnesyl
pyrophosphate and geranylgeranyl pyrophosphate, and mevalonate
pathway-derived isoprenylation (and associated membrane
localization) is a prerequisite for ligand-induced activation of
several proteins. Since statins are able to inhibit HMG-CoA
conversion to mevalonate, the pleiotropic effects of statins are
usually mevalonate pathway-dependent (11). In addition, statin-induced
inhibition of the mevalonate pathway may lead to the activation of
the phosphatidylinositol-3 kinase (PI3K)/Akt pathway, which tightly
controls cell fate processes, including proliferation and apoptosis
and has been proposed as a mechanism of the protective effects of
statin (12).
In terms of lung injury, Naidu et al
(13) were the first to
demonstrate that simvastatin was able to ameliorate LIRI and
determined that the mechanism involved a modulation of the
endothelial nitric oxide synthase (eNOS). Müller et al
(14) later reported that
high-dose simvastatin was able to attenuate ventilator-induced
injury in mice by reducing pulmonary inflammation and
hyperpermeability. However, clinical trials have not yet completely
confirmed the protective effects of statins in lung injury. One
trial discovered that statins had no influence on the progression
of lung injury (15), but two
others reported that statins improved organ dysfunction in acute
lung injury (16) and decreased
the lipopolysaccharide-induced pulmonary inflammation in healthy
volunteers (17). Therefore,
research to comprehensively elucidate under what conditions and by
what mechanisms statins attenuate acute lung injury is required.
Recently, simvastatin was shown to improve lung function by
improving alveolar epithelial cell proliferation in a chronic
obstructive pulmonary disease mouse model (18). We, therefore, hypothesized that
simvastatin may also enhance the proliferation of ATII cells, which
repairs the lung epithelium and thereby attenuates LIRI.
In this study, we demonstrated that simvastatin
restores the function of impaired ATII cells both in vitro
and in vivo, by using ATII cell lines (human A549 and mouse
MLE-12) and a rat LIRI model, respectively. The data revealed that
the restoration of ATII cell function involves the activation of
PI3K/Akt signaling in a mevalonate pathway-dependent manner.
Materials and methods
Cell culture and the
hypoxia-reoxygenation (H/R) injury model
The human alveolar epithelium-derived type II cell
line A549 (CCL-185) and mouse lung epithelial type II cell line
MLE-12 (CRL-2110) were obtained from the American Type Culture
Collection (ATCC, Manassas, VA, USA). The cells were respectively
cultured in RPMI-1640 (Hyclone) and DMEM/F-12 media (Sigma-Aldrich,
St. Louis, MO, USA) supplemented with 10% fetal bovine serum (FBS;
Invitrogen Life Technologies), penicillin (100 U/ml) and
streptomycin (10 μg/ml) in a humidified atmosphere of 5%
CO2 at 37°C. Logarithmically growing cells were seeded
at a density of 5×104 cells/ml for the following
experiments. A hypoxic condition was created by incubating cells in
a hypoxia chamber (Billups-Rothenberg, Del Mar, CA, USA) with an
atmosphere of 95% N2/5% CO2 at 37°C for 2 h
for each cell group.
Cell proliferation assay
Cell proliferation was determined using the Cell
Counting Kit-8 (CCK-8; Dojindo, Kumamoto, Japan) assay according to
the manufacturer’s protocol. Cells were suspended at a final
concentration of 5×103 cells/well and cultured in
96-well flat bottom tissue culture plates. Cells were randomly
divided into 7 groups, including the control and 6 simvastatin
(Sigma-Aldrich) treatment groups (5, 10, 20, 30, 50 and 100
μmol/l). Cells were pretreated with simvastatin for 30 min
prior to incubation in hypoxic conditions. The proliferation assay
was performed at 0, 2, 4, 6, 12 and 24 h after reoxygenation. The
proliferation ability of cells was evaluated by absorbance
measurements at 450 nm using an automated microplate reader
(CliniBio 128C; Tecan, Grödig, Austria). Final data from each cell
group were normalized by subtracting the absorbance values of the
blank group (cells without any treatment) and the experiments were
performed in triplicates/group for each time point.
Flow cytometry
The ratio of apoptotic cells to SP-C-positive ATII
cells was determined by flow cytometry (FACSCalibur; BD
Biosciences, Franklin Lakes, NJ, USA) according to the
manufacturer’s instructions. Cells were suspended at a final
concentration of 3×105 cells/well and cultured in 6-well
flat bottom tissue culture plates. Cells were randomly divided into
6 groups including the control (H/R injury only) and 4 simvastatin
treatment groups (5, 10, 20 and 50 μmol/l). For apoptosis
assay, cells were harvested at 4 h after reoxygenation and analyzed
by Annexin V (AV), propidium iodide (PI) and an apoptosis detection
kit (BD Biosciences). For detecting SP-C-positive ATII cells, cells
were harvested at 24 h after reoxygenation and immunoreacted with
primary antibody rabbit polyclonal anti-SP-C and secondary antibody
Cy-3-labeled goat anti-rabbit IgG (Santa Cruz Biotechnology, Inc.,
Santa Cruz, CA, USA).
Animals and experimental design
Male Sprague-Dawley rats were obtained from the
Laboratory Animal Center of Zhejiang Province (Hangzhou, China).
All animals received human care in compliance with The Principles
of Laboratory Animal Care, formulated by the National Society of
Medical Research and Guide for the Care and Use of Laboratory
Animals, published by the US National Institutes of Health
(Publication no. 85–23, revised 1996). The experimental protocol
was approved by the Animal Care and Scientific Committee of Nanjing
Medical University. The animals were randomly divided into 4 groups
(n=6 for each group at each time point): sham (no hilar clamping);
I/R (left hilar clamping only); LSIM (orally treated with a low
dose of 0.5 mg/kg/day simvastatin for 3 days prior to surgery and
until the end of the experiments); HSIM (high dose of 5 mg/kg/day
simvastatin, same schedule as LSIM). The rat LIRI model was
established as previously described (8). Briefly, animals of the I/R group
underwent warm ischemia for 60 min by left pulmonary hilar
occlusion. Animals of the sham group underwent identical procedures
without left pulmonary hilar occlusion. All animals of the 4 groups
were sacrificed at baseline and 4 h and 3 days after
reperfusion.
Lung injury evaluation
Lung injury was evaluated by hematoxylin and eosin
(H&E) staining and myeloperoxidase (MPO) activity assay. The
specimens of the upper portion of the left lung were fixed in
normal 4% buffered formalin for 48 h, followed by dehydration in an
ascending series of alcohol and embedded in paraffin wax. Sections
(∼4 μm) were stained with H&E. Neutrophil infiltration
and other inflammatory changes were examined in detail under light
microscopy. MPO activity assay was used to compare the relative
neutrophil sequestration in lung tissue of the experimental animals
as previously described (8). Lung
tissue MPO activity was expressed as the change in absorbance/g of
protein/min.
Harvest of bronchoalveolar lavage (BAL)
fluid
At the end of each experiment, BAL fluid was
obtained by cannulating the trachea with a blunt 20-gauge needle
and lavaging the lungs 3 times with 1 ml of ice-cold PBS. The BAL
was immediately centrifuged at 1000 rpm for 15 min to remove all
cells and cellular debris. The supernatant was stored at −80°C
until the protein concentrations were measured.
Double immunofluorescence staining
The left lung of each rat was collected and embedded
in paraffin. Specimens were cut into 4-μm serial sections,
incubated overnight at 50°C and double stained for SP-C and
proliferating cell nuclear antigen immunofluorescence. FITC-labeled
goat anti-mouse IgG and Cy-3-labeled goat anti-rabbit IgG were
employed as secondary antibodies for SP-C (rabbit polyclonal
anti-SP-C; Santa Cruz Biotechnology, Inc.) and proliferating cell
nuclear antigen (mouse monoclonal anti-PCNA; Abcam), respectively.
Sections were examined using a Leica microscope equipped with a
reflected light fluorescence device. Positively stained cells were
counted by an investigator in a blinded manner. The number of
positive cells was determined in 6 random areas in a high
magnification field (×400)/section.
Ultrastructural analysis
At the end of each experiment, the lower portion of
the left lung (1 mm3) was collected and fixed in 2.5%
glutaraldehyde for >12 h before processing for transmission
electron microscopy (TEM). After primary fixation, pieces of each
section were washed in 0.1 M cacodylate buffer, postfixed in 1%
osmium tetroxide (OsO4) in buffer for 1 h, washed in
buffer, dehydrated through an ascending acetone series and
flat-embedded in Epon-Araldite. Thin sections were cut from at
least 3 blocks of embedded lungs/animal using a Reichert UM 2
Ultramicrotome. The sections were mounted onto copper grids and
stained using 5% uranyl acetate and Reynold’s lead citrate. They
were examined using a Joel 1010 microscope at ×10,000 magnification
and operated at 80 kV; images were captured for ultrastructural
analysis.
Stereological analysis
Stereological analysis does not involve assumptions
and therefore fulfills the criteria for design-based or unbiased
stereology. Following systematic uniformly random sampling on
ultrathin sections, 10 ATII cells/section at ×10,000 magnification
were randomly identified under TEM. Digital micrographs of these
cells subsequently underwent stereological analysis utilizing an
image analysis system (AnalySIS; Soft Imaging System, Germany) as
previously described (8). The
morphometric parameters of LBs included volume density (Vv-LB),
surface area density (Sv-LB), surface area and volume ratio
(Rsv-LB) and number density (Nv-LB). Each parameter was calculated
based on previously described formulas (8).
Western blot analysis
Cells, tissue specimens and BAL fluid were harvested
and incubated for 10 min on ice with lysis buffer. The lysates were
then centrifuged at 15,000 rpm for 15 min at 4°C and the
supernatants were collected. Protein concentration in the lysates
was measured using a Beckman DU 800 Protein Assay kit (Thermo
Fisher Scientific, Waltham, MA, USA). Equal amounts of protein
samples were separated by sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) and then transferred onto a
polyvinylidene difluoride membrane (Millipore). Nonspecific binding
sites were blocked in 5% non-fat milk and 0.1% Tween-20 in TBS for
overnight shaking at 4°C. The membranes were then incubated in a
primary antibody buffer overnight, with gentle shaking at 4°C
followed by 3 washes in 0.1% TBST, after which they were incubated
with HRP-labeled secondary antibodies at room temperature for 2 h
and again washed 3 times. The blots were then detected using
SuperSignal West Pico Chemiluminescent Substrate ECL (Pierce
Biotechnology, Inc. Thermo Fisher Scientific). The primary
antibodies against SP-C and tubulin were purchased from Santa Cruz
Biotechnology, Inc. PI3K/Akt signal pathway-related protein (Akt,
phosphorylated(p)-Akt, P70, p-P70, mTOR, caspase-3) antibodies were
purchased from Cell Signaling Technology, Inc. (Beverly, MA, USA).
Protein density on scanned western blots was analyzed using Image J
1.44 software, and we performed semi-quantitative analysis for the
results of the western blot analysis.
Statistical analysis
All data are represented as the means ± SD. Data
were analyzed using a commercially available statistical software
package (SPSS for Windows, version 13.0; Chicago, IL, USA). The
one-way analysis of variance (ANOVA) was used to analyze all the
results statistically. Post hoc comparisons were performed
using the Tukey’s test or Dunnett’s T3 test. A P-value of <0.05
was considered to indicate a statistically significant
difference.
Results
Only low dose simvastatin improves the
proliferation and inhibits the apoptosis of ATII cell lines after
H/R injury
In order to evaluate whether simvastatin protects
H/R-injured ATII cells, the CCK-8 Kit assay was used to determine
cell proliferation at 0, 2, 4, 6, 12 and 24 h after reoxygenation,
and cell apoptosis was determined by AV/PI double-staining flow
cytometry. Protein levels of SP-C at 4 and 24 h after reoxygenation
were determined by western blot analysis. The ratio of
SP-C-positive ATII cells at 24 h after reoxygenation was analyzed
by flow cytometry. Only doses of simvastatin that were <20
μM increased the proliferation of ATII cells and inhibited
apoptosis. Both MLE-12 and A549 cells reacted similarly to the low
dose simvastatin treatment (Fig.
1A). A response was noted by MLE-12 cells to 20 μM
simvastatin and by A549 cells starting at 5 μM simvastatin
with regards to proliferation at time 0; this may be due to the
nature of different cell types. After reoxygenation in the control
group, the proliferative capability was increased at 2 h, slightly
decreased at 4 and 6 h and increased at 12 and 24 h. As compared
with the control group, the low dose (≤20 μM)
simvastatin-treated groups exhibited dose-dependent increases in
the proliferative capacity of ATII cells. It was notable that the
ATII cells treated with 20 μM simvastatin demonstrated
statistically significant increases at all time points examined.
High dose simvastatin (50 and 100 μM) treatments did not
significantly increase the proliferative capacity of ATII cells at
any of the time points examined. In fact, proliferation of MLE-12
and A549 cells was significantly lower compared to that in the
control groups at 12 and 24 h after reoxygenation, and the
proliferation of A549 cells was markedly lower in response to the
100 μM simvastatin treatment.
AV/PI double staining was used to identify cell
apoptosis, which is presented as a histogram indication of
FITC-AV-positive cells and includes both early and late apoptotic
cells. Since our previous study demonstrated that the apoptosis of
ATII cells usually increases at 4 h after LIRI (8), we selected the 4 h time point after
reoxygenation for the observations of cell apoptosis after H/R. For
both MLE-12 and A549 cells the apoptotic cell ratio was markedly
lower in the low dose simvastatin-treated groups (5, 10 and 20
μM) compared to the control groups (Fig. 1B). In contrast, the apoptotic cell
ratio in the high dose simvastatin-treated groups (50 and 100
μM) was significantly increased as compared with the low
dose groups. The 100 μM simvastatin-treated groups in both
MLE-12 and A549 cells exhibited markedly increased numbers of
apoptotic cells, up to 50% more than that in the control
groups.
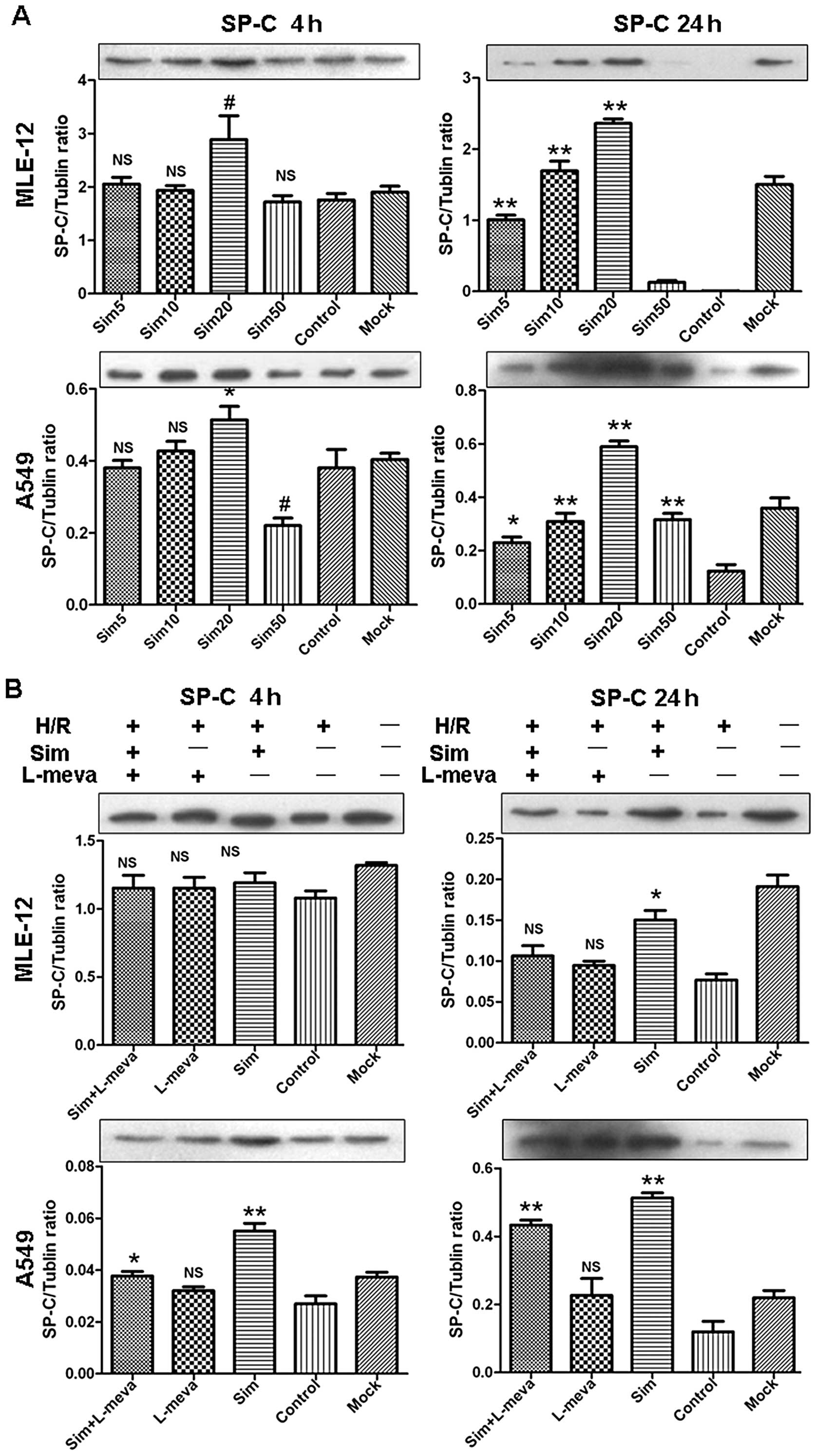
Finally, the simvastatin-induced changes in the
percentage of SP-C-positive ATII cells and SP-C protein levels were
detected (Figs. 2 and 3A, respectively). The MLE-12 and A549
cell lines again demonstrated similar trends in response to
simvastatin treatments. At 4 h after reoxygenation, there was no
significant difference observed in the percentage of SP-C-positive
cells between the simvastatin-treated and the control groups (data
not shown). This finding was consistent with the SP-C protein
detected by western blot analysis (Fig. 3A). Furthermore, at 24 h after
reoxygenation, the percentage of SP-C-positive cells was
significantly increased in the low dose simvastatin-treated groups
(5, 10 and 20 μM), compared with the control groups. In
contrast, the percentage was markedly decreased in the 50 μM
simvastatin-treated groups, and was even lower than the control
group for the A549 cell line. These results were confirmed by
western blot analysis.
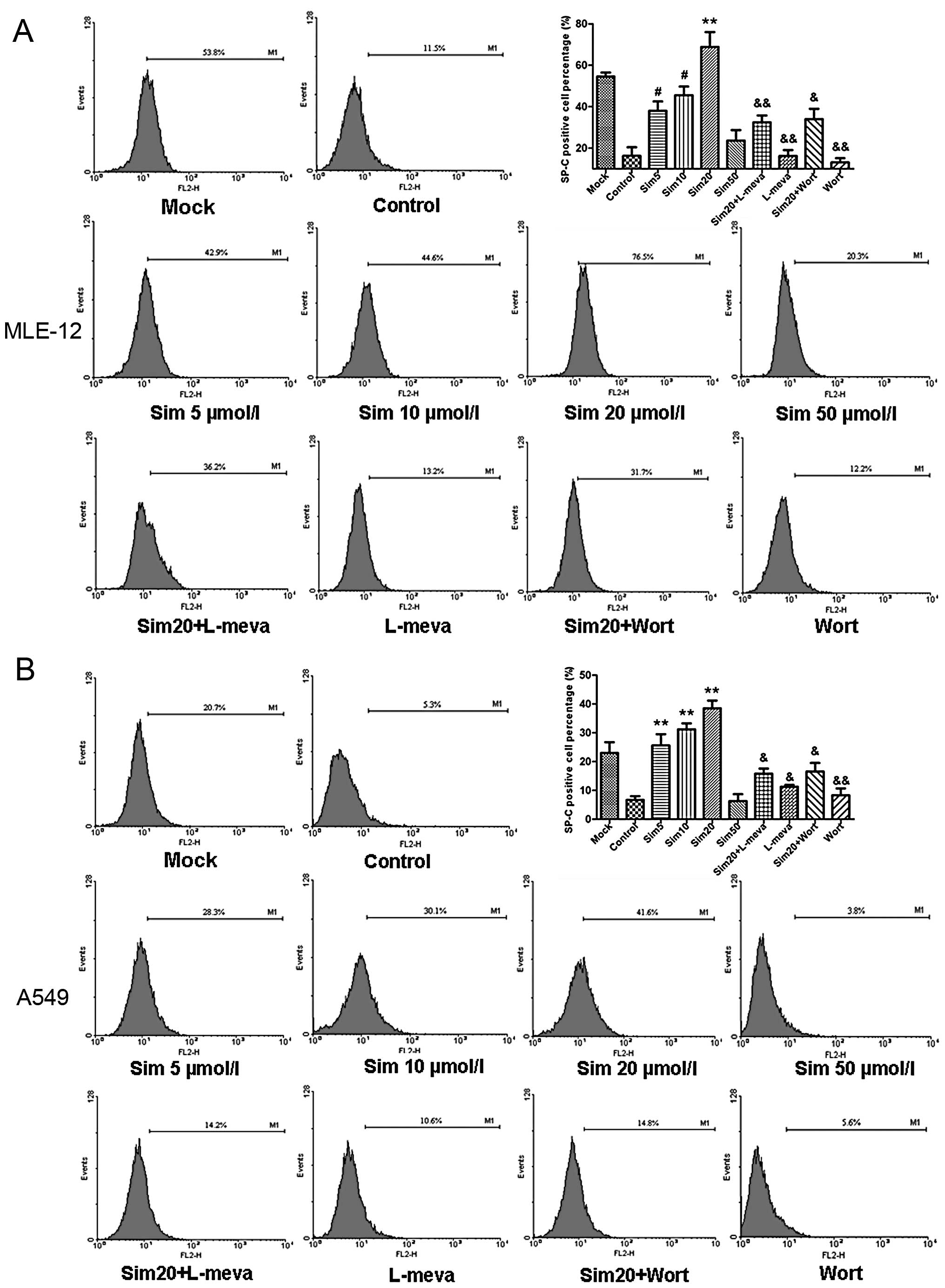 | Figure 2Effects and mechanisms of simvastatin
(sim) on the SP-C-positive cell ratio in hypoxia-reoxygenation
(H/R)-injured ATII cell lines, MLE-12 (A) and A549 (B).
SP-C-positive cell ratio was determined by flow cytometry at 24 h
after reoxygenation. SP-C-positive cell percentage was
significantly increased in low dose simvastatin-treated groups (5,
10 and 20 μM), as compared with the control groups, whereas
it was markedly decreased in the 50 μM simvastatin-treated
groups. Administration of L-mevalonate (L-meva, 20 μM)
blocked the increasing SP-C-positive cell number by simvastatin by
rescuing simvastatin-induced mevalonate depletion. Similarly,
wortmannin (Wort), a competitive PI3K inhibitor, reversed the
simvastatin-induced increase of the percentage of SP-C positive
cells. Data are expressed as means ± SD. *P<0.05,
#P<0.01 and **P<0.001 vs. control
group; &P<0.05, &&P<0.01
vs. sim20 group. |
Restoration of ATII cell function in
vitro by low dose simvastatin is mediated by the mevalonate and the
PI3K/Akt signaling pathways
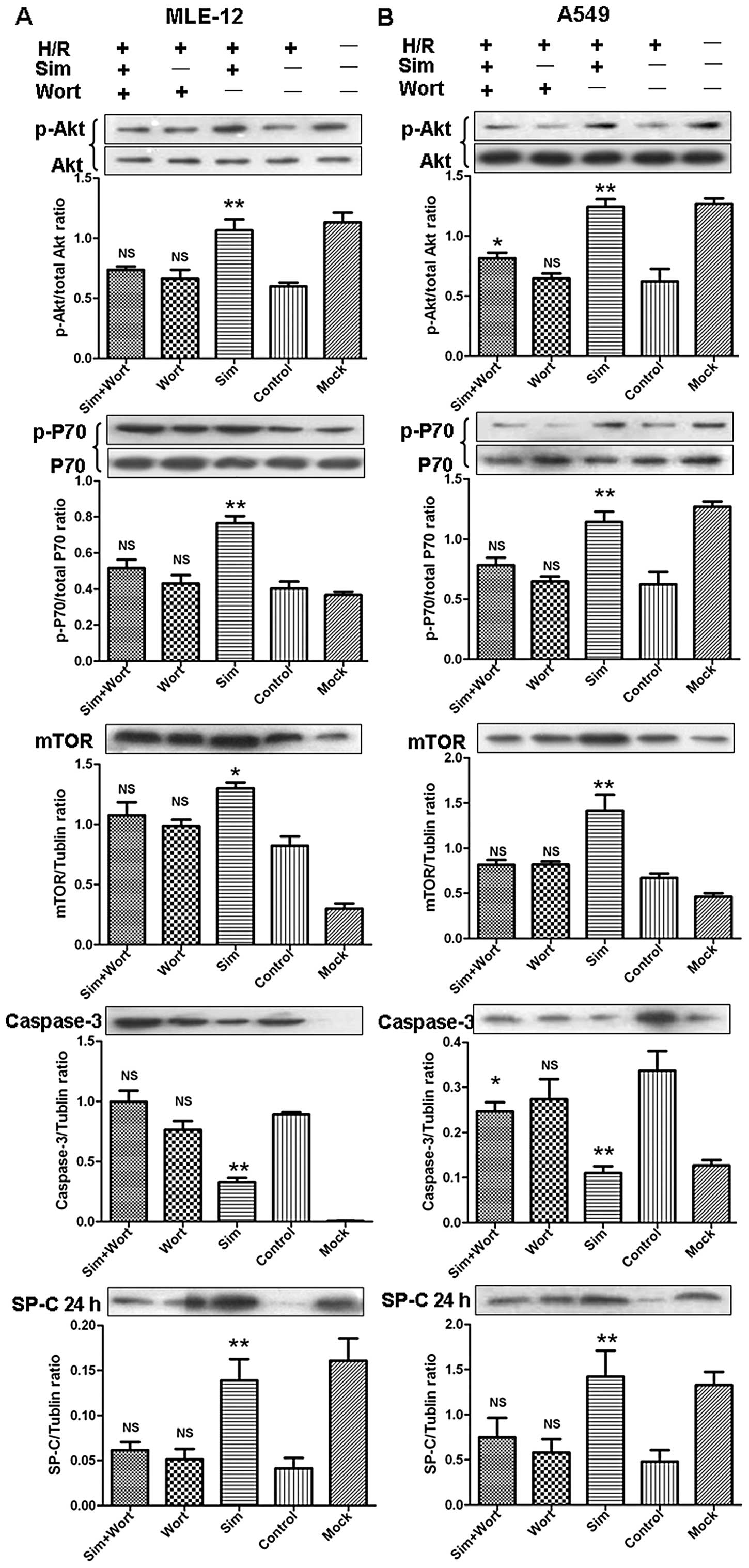
In order to investigate whether the mevalonate
pathway and the downstream PI3K/Akt pathway are involved in the
restoration of ATII cell function induced by statins, L-mevalonate
(L-meva) and wortmannin (Wort) were used in the following
experiments. L-meva restores mevalonate depletion induced by
simvastatin and Wort is a competitive inhibitor of the PI3K
protein. Administration of L-meva (20 μM) significantly
blocked the SP-C-increasing effect of simvastatin by rescuing
simvastatin-induced mevalonate depletion in the MLE-12- and
A549-treated cells, as compared to the simvastatin only treated
groups at 24 h after reoxygenation (Figs. 2 and 3B). Similarly, treatment with Wort (100
nM) in combination with simvastatin reversed the
simvastatin-induced increase in the percentage of SPC-positive
cells and the SP-C protein levels at 24 h after reoxygenation in
both cell lines (Figs. 2 and
4). Moreover, cells treated with
20 μM simvastatin showed increased levels of p-Akt and
downstream p-P70 and m-TOR and suppressed caspase-3 levels at 4 h
after reoxygenation (Fig. 4).
Co-administration of Wort and simvastatin resulted in markedly
suppressed p-Akt levels leading to observed decreases in m-TOR and
P70 levels, as well as to the downstream increases in caspase-3
levels.
Simvastatin dose-dependently attenuates
the LIRI, promotes proliferation of ATII cells and restores their
function in vivo
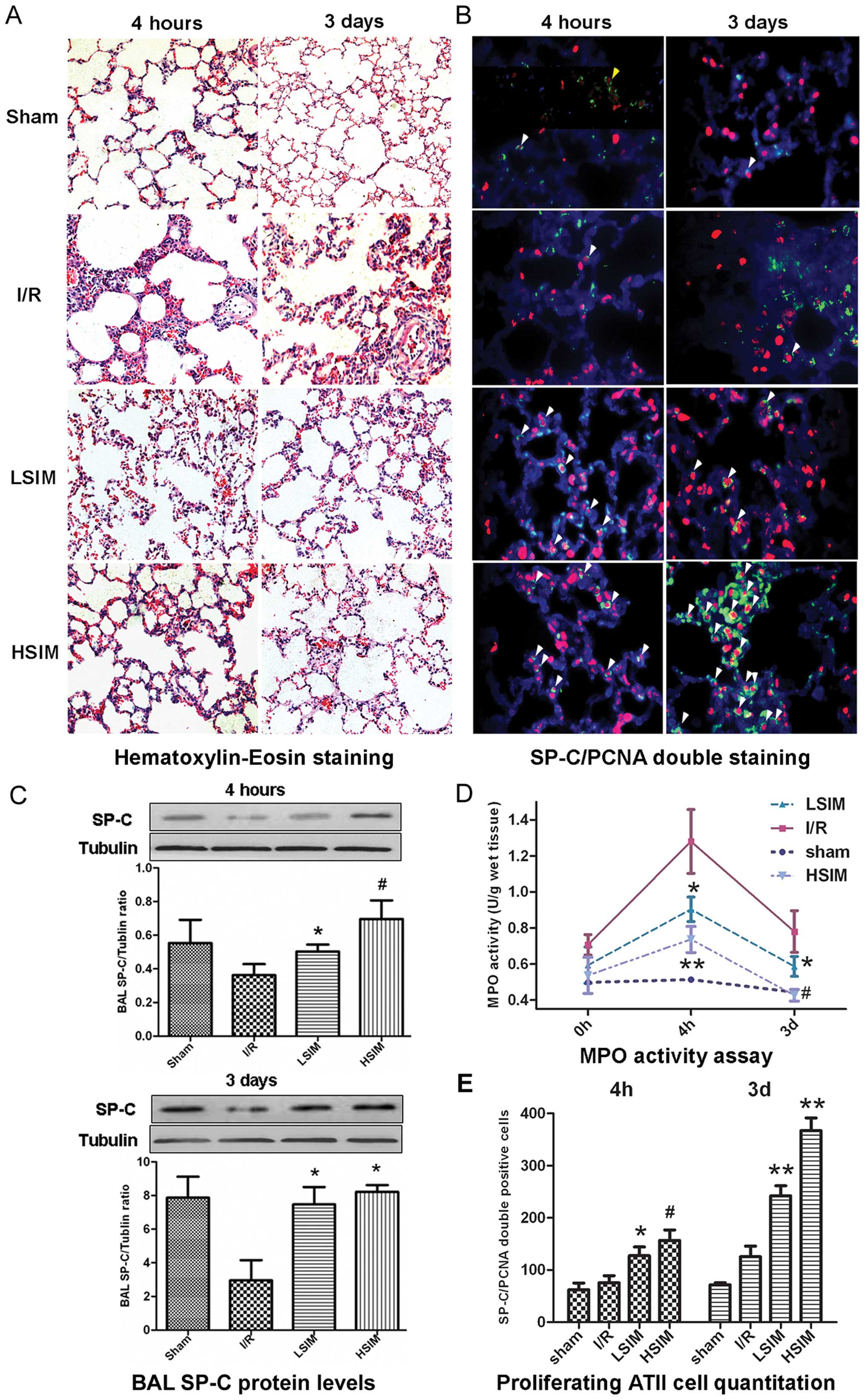
To evaluate the protective effect of simvastatin on
LIRI, H&E staining and MPO activity assay were performed on rat
lung tissues. As shown in Fig.
5A, serious LIRI was achieved in the I/R group, as evidenced by
infiltration of neutrophils and small areas of intra-alveolar edema
at 4 h after reperfusion, which slightly recovered 3 days after
reperfusion. In the simvastatin-treated groups, less neutrophil
infiltration was observed and the areas of intra-alveolar edema
were less than that in the I/R group. In the HSIM group, the
pulmonary structure was found to be mostly recovered to normal 3
days after reperfusion. The MPO activity assay confirmed the
observations of H&E staining (Fig. 5D). Compared with the baseline of
the sham group, the MPO activities of the I/R group were elevated
at 4 h and slightly declined at 3 days after reperfusion. However,
the simvastatin-treated groups displayed significant and
dose-dependent decreases of MPO activities at both 4 h and 3 days
after reperfusion, as compared with the I/R group.
Furthermore, SP-C protein levels in BAL fluid were
determined and SP-C/PCNA double immunofluorescence staining of lung
tissues was performed to evaluate the proliferation and function of
ATII cells. In both LSIM and HSIM groups, SP-C protein levels in
BAL fluid were significantly increased at 4 h and 3 days after
reperfusion as compared with the I/R group. The FITC-SP-C and
Cy-3-PCNA double-positive cells were counted in a high
magnification field (×400). As shown by the representative images
in Fig. 5B and the quantitative
analysis in Fig. 5E, SP-C/PCNA
double-positive cell numbers in the LSIM and HSIM groups were found
to be significantly increased at 4 h and further increased (by
>2-fold) at 3 days after reperfusion, as compared with the I/R
group.
Finally, ultrastructural examination and
stereological analysis were performed to evaluate the function of
ATII cells in the rat models. Representative images of the
ultrastructural findings and stereological analysis results for
each treatment group are shown in Fig. 6. The nuclei and LBs of ATII cells
were found to be intact in the sham group at 4 h and 3 days after
reperfusion (Fig. 6A). Apoptotic
ATII cells were also present at 4 h after reperfusion. Numerous
apoptotic bodies (ApoB), dilations of nuclear, scattered chromatin
and empty LBs (eLBs) were observed. Gradual self-repair of ATII
cells was observed until 3 days after reperfusion, and the number
and volume of LBs remained low and small. However, in the LSIM and
HSIM groups, the ApoB and eLB structures were markedly absent at 4
h after reperfusion and numerous LBs were noted at 3 days after
reperfusion. The results of quantitative comparison by
stereological analysis are shown in Fig. 6B. At 4 h after reperfusion, as
compared with the I/R group, both the Vv-LB and Nv-LB were
significantly higher in LSIM and HSIM groups. The Rsv-LB levels
were markedly lower in LSIM and HSIM groups. In particular, at 3
days after reperfusion, the Nv-LB levels in LSIM and HSIM groups
were drastically higher compared to those in the I/R group (∼2- to
3-fold).
 | Figure 6Transmission electron microscopy of
the ultrastructure of ATII cells in rat lung tissues. TEM images
were obtained under magnification ×10,000. (A) Representative
ultrastructural images of ATII cells in each group at 4 h and 3
days after I/R injury. (B) Stereological analysis of lamellar
bodies in ATII cells from each group. The morphometric parameters
of lamellar bodies included volume density (Vv-LB), surface area
density (Sv-LB), surface area and volume ratio (Rsv-LB) and number
density (Nv-LB). Both the Vv-LB and Nv-LB were found to be
significantly increased in the simvastatin-treated groups. eLb,
empty lamellar bodies; LB, lamellar bodies; Nu, nucleus; ApoB,
apoptotic bodies; I/R, ischemia-reperfusion group; LSIM, low dose
simvastatin (0.5 mg/kg/day) group; HSIM, high dose simvastatin (5
mg/kg/day) group. *P<0.05 and **P<0.001
vs. I/R group. NS, no significance. |
PI3K/Akt signaling pathway is involved in
simvastatin-contributing protective effects on ATII cells in
vivo
In order to investigate whether the PI3K/Akt pathway
is involved in the in vivo protective effects of statins,
the relative levels of PI3K/Akt signaling pathway-related proteins
were determined by western blot analysis. At 4 h after reperfusion,
p-Akt levels in the LSIM and HSIM groups were significantly higher
compared to the I/R group (Fig.
7). Subsequently, the downstream p-P70 level was also markedly
increased. At 3 days after reperfusion, p-Akt levels further
increased in the LSIM and HSIM groups, as did the downstream p-P70
levels. There appeared to be no difference in the caspase-3 level
between each group; however, the SP-C levels in the LSIM and HSIM
groups increased markedly.
Discussion
Although the protective effects of statins on LIRI
have been investigated previously (13), to the best of our knowledge, our
study is the first to provide evidence that lung ATII cells are the
potential target of statin therapy in LIRI. First, our data
demonstrated a biphasic effect of simvastatin on ATII cells in
vitro. Only a low dose of simvastatin (≤20 μM) improved
proliferation and inhibited apoptosis of ATII cells, whereas a high
dose of simvastatin (≥50 μM) promoted apoptosis. Second, the
in vitro protective effects of simvastatin were observed to
be reversed by L-meva and Wort, a competitive inhibitor of the PI3K
pathway. Third, the in vivo rat LIRI model also demonstrated
that the number of proliferative ATII cells was significantly
increased and the number and volume density of their functional
cell organelle LBs was dose-dependently enhanced by simvastatin.
Thus, our results provide a new understanding of the mechanisms
underlying statin-induced attenuation of LIRI and the facilitation
of tissue repair, whereby the function of ATI progenitor cells,
ATII cells, were restored by simvastatin via activation of the PI3K
pathway in a mevalonate pathway-dependent manner. These findings
provide further insights into the utility and mechanism of
simvastatin treatment of LIRI, and may benefit future research into
improved clinical applications of simvastatin.
During the past decades, several reports have been
published on the pleiotropic effects of statins, involving various
disease processes of different organs. Statins are well-recognized
for their properties of lowering lipid content, protecting against
IR injury (19,20), quelling inflammation (21–24), improving tissue remodeling
(25,26), modulating angiogenesis (27), promoting cell proliferation
(18), and inhibiting cancer cell
proliferation and inducing apoptosis (28–32). However, nearly as many reports
exist that present experimental data that contradict the protective
effects of statins (15). In the
present study, we also obtained contradictory results in that
simvastatin produced biphasic effects on ATII cell proliferation
and apoptosis according to the dose. Furthermore, we observed that
a low dose of simvastatin (20 μM) was able to activate
PI3K/Akt signaling, which contradicts results from other studies
that indicated that statins inhibit signaling of this pathway
(25,28–32). In order to better elucidate these
paradoxical phenomena, we reviewed the results of several
representative studies on the pleiotropic effects of statins.
Firstly, the dosage of statins may be an important
factor that impacts their biological effects. Previous
investigations have reported findings of dose-dependent biphasic
effects of statins on tissue angiogenesis. Weis et al
(27) demonstrated that low
concentrations of statins (0.005–0.01 μM) enhanced
endothelial cell (EC) proliferation, migration and differentiation,
which were significantly inhibited by high concentrations (0.05–1
μM). Similarly, pitavastatin was demonstrated to enhance the
migration, proliferation and viability of human microvessel ECs at
a low concentration (0.01 μM) but inhibits these processes
at a high concentration (1 μM) (33). Furthermore, a recent study of
ischemic retinopathies demonstrated a biphasic effect of
simvastatin (34), in which low
concentrations (0.01–0.1 μM) significantly increased
proliferation, migration, sprouting and tubulogenesis of retinal
microvascular ECs but high concentrations (1–10 μM) caused
cell death and apoptosis. Our present data indicated that low dose
simvastatin (5–20 μM) promoted ATII cell proliferation and
inhibited apoptosis, but the effects were completely opposite with
high doses (50–100 μM), similar to the above-mentioned
published studies. Notably, the concentration of statins (5–100
μM) used in our in vitro experiments was higher than
that previously used in the other published studies (0.01–10
μM). We think that the different cell type used in our
experiments had a unique requirement for higher doses of
simvastatin to elicit the observed effects. The MLE-12 cell line
used in our study is a SV-40 virus immortalized mouse ATII cell
line and the A549 ATII cell line originated from a lung
adenocarcinoma tissue. There are obvious disparities between the
pharmacokinetics of these two cell lines and that of primary
cultured human ECs. This may be one explanation of why the statin
concentrations used in this study were higher compared to those in
the previous studies.
Different diseases or injuries have different
physiopatho-logical processes. Thus, statins may also elicit
opposite effects under various disease conditions in the context of
different organs. It has been proposed that statins do not simply
inhibit or activate signaling pathways, but modulate them in order
to alter cell fate. Among the numerous signaling pathways described
as targets of statins, the PI3K/Akt pathway plays a crucial role in
cell survival, apoptosis and proliferation (35). In conditions of tissue
ischemia-reperfusion (I/R) injury (25,26) or inflammatory disorders, such as
airway hyperreactivity (21),
renal glomerular podocyte injury (22) or traumatic brain injury (23), statins usually exert their
protective effects by promoting cell proliferation and inhibiting
apoptosis through the inhibition of upstream RhoA-kinase and the
activation of downstream PI3K/Akt signaling. However, in conditions
of cancer, statins appear to significantly decrease cancer cell
proliferation, migration and metastasis via the inhibition of
PI3K/Akt signaling (28–32). In fact, I/R or inflammation may
induce apoptosis and inhibit proliferation via the downregulation
of cell proliferation through any of the PI3K/Akt, ERK, or MAPK
pathways and the upregulation of apoptosis through the Bcl-2, Bax
and caspase families. However, cell survival-related pathways are
usually upregulated in several types of solid cancers. Our data
indicated that low concentrations of statins uniquely reverse H/R
injured ATII cell function in vitro via a mevalonate
pathway-dependent activation of PI3K/Akt signaling. Administration
of PI3K inhibitor, wortmannin, reversed the protective effects of
simvastatin by decreasing the levels of p-Akt and those of the
downstream factors mTOR and p-P70 and by subsequently activating
caspase-3. This modulated signaling cascade coincides with previous
studies regarding the protective effect of statins on I/R.
Consequently, we speculate that statins may maintain the balance of
normal or abnormal cell fate by modulating the PI3K/Akt signaling
pathway.
Finally, despite our notable findings, this study is
subject to several experimental limitations. First, although MLE-12
and A549 cells have been used as in vitro models in previous
ATII studies (36–38), these two cell lines cannot
completely simulate the in vivo physiology of ATII cells. In
future studies, primary cultured ATII cells may provide more direct
evidence for the protective effect of statins. Second, in the in
vivo experiments, the entire lung tissue specimen was used in
the western blot analysis of PI3K/Akt signaling pathway-related
proteins, which may have masked some subtle effects on the PI3K/Akt
signaling pathway of ATII cells or detected changes that were
induced by molecular interactions with non-ATII cells. In addition,
statins were administered just prior to the onset of injury. It
remains unknown whether administration of statins post-LIRI also
has the same protective effect on ATII cells. More experiments are
required to further identify this issue. Finally, we did not
examine whether the repairing capacity of ATII cells was enhanced
by simvastatin in the present study. Recently, the transforming
growth factor β1 (TGF-β1)/Smad pathway was demonstrated to
contribute to the transdifferentiation process from ATII to ATI
cells (39). It is possible that
statins may also promote ATII cell transdifferentiation into ATI
cells via activation of the TGF-β1/Smad pathway, which may be
dependent on or independent of the mevalonate pathway. These
questions will be addressed in our future studies.
In summary, to the best of our knowledge, we for the
first time demonstrated that LIRI-induced injury of ATII cells may
be reversed by relatively low dose simvastatin in vivo and
in vitro. The protective effects of simvastatin were at
least partially due to promotion of ATII cell proliferation and
SP-C expression and inhibition of apoptosis through the activation
of the PI3K/Akt signaling pathway in a mevalonate pathway-dependent
manner. These findings provide new insights into the molecular
mechanisms underlying the action of statins in LIRI. Administration
of statins may provide a promising therapeutic strategy to rapidly
regenerate epithelium in damaged lungs by endogenously increasing
ATII cells.
Acknowledgements
This study was supported by grants
from the Human Resource Summit Grant of Jiangsu Province (10-D-107
to Dr R.Y. and 10-D-105 to Dr F.J.) and the Natural Science
Foundation of Jiangsu Province (BK2010589 to Dr L.X. and BK2011578
to Dr R.Y.).
References
|
1.
|
N FujinoH KuboT SuzukiIsolation of
alveolar epithelial type II progenitor cells from adult human
lungsLab Invest91363378201110.1038/labinvest.2010.18721079581
|
|
2.
|
EL HerzogAR BrodyTV ColbyR MasonMC
WilliamsKnowns and unknowns of the alveolusProc Am Thorac
Soc5778782200810.1513/pats.200803-028HR18757317
|
|
3.
|
D WangDL HavilandAR BurnsE ZsigmondRA
WetselA pure population of lung alveolar epithelial type II cells
derived from human embryonic stem cellsProc Natl Acad Sci
USA10444494454200710.1073/pnas.070005210417360544
|
|
4.
|
AV AndreevaMA KutuzovTA
Voyno-YasenetskayaRegulation of surfactant secretion in alveolar
type II cellsAm J Physiol Lung Cell Mol
Physiol293L259L271200710.1152/ajplung.00112.200717496061
|
|
5.
|
RJ MasonBiology of alveolar type II
cellsRespirology11SupplS12S15200610.1111/j.1440-1843.2006.00800.x
|
|
6.
|
IP NeuringerSH RandellStem cells and
repair of lung injuriesRespir Res56200410.1186/1465-9921-5-6
|
|
7.
|
D WangJE MoralesDG CalameJL AlcornRA
WetselTransplantation of human embryonic stem cell-derived alveolar
epithelial type II cells abrogates acute lung injury in miceMol
Ther18625634201010.1038/mt.2009.31720087316
|
|
8.
|
D FengS ZhangZ HuF FanF JiangR YinL
XuDynamic investigation of alveolar type II cell function in a
long-term survival model of rat lung ischemia-reperfusion
injuryScand J Clin Lab
Invest70364373201010.3109/00365513.2010.49541520560845
|
|
9.
|
JG ShanesKN MinadeoA MoretM GronerSA
TabaieStatin therapy in heart failure: prognostic effects and
potential mechanismsAm Heart
J154617623200710.1016/j.ahj.2007.05.02017892981
|
|
10.
|
GC MakrisG GeroulakosMC MakrisDP
MikhailidisME FalagasThe pleiotropic effects of statins and omega-3
fatty acids against sepsis: a new perspectiveExpert Opin Investig
Drugs19809814201010.1517/13543784.2010.49083020470189
|
|
11.
|
NR VeillardV BraunersreutherC ArnaudF
BurgerG PelliS SteffensF MachSimvastatin modulates chemokine and
chemokine receptor expression by geranylgeranyl isoprenoid pathway
in human endothelial cells and
macrophagesAtherosclerosis1885158200610.1016/j.atherosclerosis.2005.10.015
|
|
12.
|
A UndasM Celinska-LowenhoffM KaczorJ
MusialNew nonlipid effects of statins and their clinical relevance
in cardiovascular diseaseThromb Haemost9110651077200415175791
|
|
13.
|
BV NaiduSM WoolleyAS FarivarR ThomasC
FragaMS MulliganSimvastatin ameliorates injury in an experimental
model of lung ischemia-reperfusionJ Thorac Cardiovasc
Surg126482489200310.1016/S0022-5223(03)00699-812928648
|
|
14.
|
HC MullerK HellwigS RosseauSimvastatin
attenuates ventilator-induced lung injury in miceCrit
Care14R143201010.1186/cc920920673352
|
|
15.
|
DJ KorR IscimenM YilmazMJ BrownDR BrownO
GajicStatin administration did not influence the progression of
lung injury or associated organ failures in a cohort of patients
with acute lung injuryIntensive Care
Med3510391046200910.1007/s00134-009-1421-8
|
|
16.
|
TR CraigMJ DuffyM ShyamsundarC McDowellCM
O’KaneJS ElbornDF McAuleyA randomized clinical trial of
hydroxymethylglutaryl-coenzyme a reductase inhibition for acute
lung injury (The HARP Study)Am J Respir Crit Care
Med183620626201110.1164/rccm.201003-0423OC20870757
|
|
17.
|
M ShyamsundarT CraigC O’KaneD
McAuleyComment on ‘Statin administration did not influence the
progression of lung injury or associated organ failures in a cohort
of patients with acute lung injury’Intensive Care
Med35149414952009
|
|
18.
|
S TakahashiH NakamuraM SekiReversal of
elastase-induced pulmonary emphysema and promotion of alveolar
epithelial cell proliferation by simvastatin in miceAm J Physiol
Lung Cell Mol
Physiol294L882L890200810.1152/ajplung.00238.200718310229
|
|
19.
|
S WolfrumA DendorferM SchuttSimvastatin
acutely reduces myocardial reperfusion injury in vivo by activating
the phosphatidylinositide 3-kinase/Akt pathwayJ Cardiovasc
Pharmacol44348355200410.1097/01.fjc.0000137162.14735.3015475833
|
|
20.
|
RM BellDM YellonAtorvastatin, administered
at the onset of reperfusion, and independent of lipid lowering,
protects the myocardium by up-regulating a pro-survival pathwayJ Am
Coll Cardiol41508515200310.1016/S0735-1097(02)02816-412575984
|
|
21.
|
AA ZekiL FranziJ LastNJ KenyonSimvastatin
inhibits airway hyperreactivity: implications for the mevalonate
pathway and beyondAm J Respir Crit Care
Med180731740200910.1164/rccm.200901-0018OC19608720
|
|
22.
|
B BussolatiMC DeregibusV FonsatoStatins
prevent oxidized LDL-induced injury of glomerular podocytes by
activating the phosphatidylinositol 3-kinase/AKT-signaling pathwayJ
Am Soc Nephrol1619361947200510.1681/ASN.200408062915843472
|
|
23.
|
H WuD LuH JiangSimvastatin-mediated
upregulation of VEGF and BDNF, activation of the PI3K/Akt pathway,
and increase of neurogenesis are associated with therapeutic
improvement after traumatic brain injuryJ
Neurotrauma25130139200810.1089/neu.2007.036918260796
|
|
24.
|
M EtoT KozaiF CosentinoH JochTF
LüscherStatin prevents tissue factor expression in human
endothelial cells: role of Rho/Rho-kinase and Akt
pathwaysCirculation10517561759200210.1161/01.CIR.0000015465.73933.3B11956113
|
|
25.
|
N TakedaM KondoS ItoY ItoK ShimokataH
KumeRole of RhoA inactivation in reduced cell proliferation of
human airway smooth muscle by simvastatinAm J Respir Cell Mol
Biol35722729200610.1165/rcmb.2006-0034OC16858009
|
|
26.
|
L Taraseviciene-StewartR ScerbaviciusKH
ChoeSimvastatin causes endothelial cell apoptosis and attenuates
severe pulmonary hypertensionAm J Physiol Lung Cell Mol
Physiol291L668L676200610.1152/ajplung.00491.200516698853
|
|
27.
|
M WeisC HeeschenAJ GlassfordJP
CookeStatins have biphasic effects on
angiogenesisCirculation105739745200210.1161/hc0602.10339311839631
|
|
28.
|
JI HanaiN DoroAT SasakiS KobayashiLC
CantleyP SethVP SukhatmeInhibition of lung cancer growth: ATP
citrate lyase knockdown and statin treatment leads to dual blockade
of mitogen-actiated protein kinase (MAPK) and
phosphatidylinositol-3- kinase (PI3K)/AKT pathwaysJ Cell
Physiol22717091720201210.1002/jcp.2289521688263
|
|
29.
|
UK KhanzadaOE PardoC MeierJ DownwardMJ
SecklA ArcaroPotent inhibition of small-cell lung cancer cell
growth by simvastatin reveals selective functions of Ras isoforms
in growth factor
signallingOncogene25877887200610.1038/sj.onc.120911716170339
|
|
30.
|
A HoriguchiM SumitomoJ AsakumaT AsanoT
AsanoM Hayakawa3-Hydroxy-3-methylglutaryl-coenzyme A reductase
inhibitor, fluvastatin, as a novel agent for prophylaxis of renal
cancer metastasisClin Cancer
Res1086488655200410.1158/1078-0432.CCR-04-1568
|
|
31.
|
T SanliC LiuA RashidLovastatin sensitizes
lung cancer cells to ionizing radiation: modulation of molecular
pathways of radioresistance and tumor suppressionJ Thorac
Oncol6439450201110.1097/JTO.0b013e3182049d8b21258249
|
|
32.
|
SA GlynnD O’SullivanAJ EustaceM ClynesN
O’DonovanThe 3-hydroxy-3-methylglutaryl-coenzyme A reductase
inhibitors, simvastatin, lovastatin and mevastatin inhibit
proliferation and invasion of melanoma cellsBMC
Cancer89200810.1186/1471-2407-8-9
|
|
33.
|
M KatsumotoT ShinguR KuwashimaA NakataS
NomuraK ChayamaBiphasic effect of HMG-CoA reductase inhibitor,
pitavastatin, on vascular endothelial cells and angiogenesisCirc
J6915471555200510.1253/circj.69.154716308507
|
|
34.
|
RJ MedinaCL O’NeillAB DevineTA GardinerAW
StittThe pleiotropic effects of simvastatin on retinal
microvascular endothelium has important implications for ischaemic
retinopathiesPLoS One3e2584200810.1371/journal.pone.0002584
|
|
35.
|
LC CantleyThe phosphoinositide 3-kinase
pathwayScience29616551657200210.1126/science.296.5573.165512040186
|
|
36.
|
KA FosterCG OsterMM MayerML AveryKL
AudusCharacterization of the A549 cell line as a type II pulmonary
epithelial cell model for drug metabolismExp Cell
Res243359366199810.1006/excr.1998.41729743595
|
|
37.
|
Y HoshinoT MioS NagaiH MikiI ItoT
IzumiCytotoxic effects of cigarette smoke extract on an alveolar
type II cell-derived cell lineAm J Physiol Lung Cell Mol
Physiol281L509L516200111435227
|
|
38.
|
S KannanH HuangD SeegerAlveolar epithelial
type II cells activate alveolar macrophages and mitigate P.
Aeruginosa infectionPLoS
One4e4891200910.1371/journal.pone.000489119305493
|
|
39.
|
M BhaskaranN KolliputiY WangD GouNR
ChintagariL LiuTrans-differentiation of alveolar epithelial type II
cells to type I cells involves autocrine signaling by transforming
growth factor beta 1 through the Smad pathwayJ Biol
Chem28239683976200710.1074/jbc.M609060200
|