Introduction
Leukemia is one of the most comment malignant
diseases worldwide. Epidemiological studies have shown that in the
United States, among males aged >40 years, leukemia is the most
common fatal cancer; among females, leukemia is the leading cause
of cancer-related mortality before the age of 20. In children
(newborns to 14 years of age) almost one third of cancer cases are
diagnosed are leukemia (particularly acute lymphocytic leukemia)
(1). Chemotherapy remains the
major strategy for the treatment of leukemia; however, traditional
therapy has lower efficacy and severe side-effects. Therefore, it
is urgent to develop an alternative medicine or alternative
therapeutic approaches for the treatment of leukemia. It has been
demonstrated that the trace element, selenium, has potential
effects on leukemia (2).
Selenium plays important roles in different
physiological functions of the human body. Epidemiological and
clinical studies have reported that the inadequate status of
selenium increases the risk of cancer (3). Basic research and clinical trials
have demonstrated the protective effects of selenium against
prostate, colorectal and other solid cancers (4–9).
The anticancer effects of selenium have been postulated to be
associated with the inhibition of cell proliferation and the
induction of apoptosis through different signaling pathways,
particularly its antioxidant and anti-inflammatory effects which
are mediated through the activity of selenoenzymes (9). Selenium compounds have also been
shown to be involved in the mitochondrial pathway, protein kinases,
tumor necrosis factor, the activation of caspases and reactive
oxygen species (3,7,8).
We have previously demonstrated that the anticancer
effects of selenium are mediated through the activation of c-Jun
NH2-ternimal kinase 1 (JNK1) and the inhibition of the
Wnt/β-catenin signaling pathway in colorectal cancer in mouse
models and colorectal cancer cell lines (10). JNK1 is a member of the
mitogen-activated protein kinase (MAPK) family, and plays a
critical role in the regulation of cell proliferation,
differentiation and apoptosis (11–14). In previous studies, we
demonstrated that JNK1 plays synergistic role with the cell cycle
regulator, p21 (12), and that
activated JNK1 (JNK1 phosphorylation) interacts with and
downregulates β-catenin signaling (15). In fact, p21 and Wnt/β-catenin are
negatively interacted through c-myc and cyclin D1 (16–18). Whether selenium also exerts
anticancer effects by promoting apoptosis and inhibiting cell
proliferation in leukemia cells, and the underlying mechanisms
involved, remains unclear.
In the present study, using HL-60 human
promyelocytic leukemia cells, we found that a higher concentration
of selenium significantly inhibited cell proliferation, induced
apoptosis, as well as changes in the cell cycle, which were
associasted with the enhanced phosphorylation of JNK and the
increased expression of p21 and p27, and with the deceased
expression of cyclin D1.
Materials and methods
Cell lines and chemicals
HL-60 human promyelocytic leukemia cells obtained
from the American Type Culture Collection (ATCC; Manassas, VA, USA)
were maintained in RPMI-1640 medium (Life Technologies, Inc.,
Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS)
and antibiotics (10,000 U/ml penicillin and 10 μg/ml streptomycin).
The cells were cultured at 37°C in a humidified atmosphere
containing 5% CO2. Sodium selenite (Se) was purchased
from Sigma-Aldrich (St. Louis, MO, USA).
Cell proliferation assay
A total of 1×104 HL-60 cells was seeded
in each well of a 96-well plate and incubated overnight. The medium
was replaced with fresh medium with a final concentration of Se
(100 or 250 nM). Phosphate-buffered saline (PBS) (0 nM of Se) was
used as a control. Three independent experiments were performed.
Followign 24 and 48 h of exposure to Se, cell proliferation was
determined by
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
(MTS) assay using the CellTiter 96 Non-Radioactive Cell
Proliferation Assay kit according to the manufacturer’s
instructions (Promega Corp., Madison, WI, USA).
Apoptosis and cell cycle analysis
To detect apoptosis, the HL-60 cells were treated
with various concentrations of Se (0, 100 and 250 nM) After 48 h of
treatment, the cells were harvested and fixed with 70% ethanol
followed by propidium iodide (PI) staining. The cells were then
counted using a flow cytometer (FACScan; BD Biosciences, San Jose,
CA, USA) to detect apoptosis and for analysis of the cell cycle.
Usually, approximately 10,000 cells were counted. The percentage of
apoptotic cells was calculated by dividing the total number of
cells by the number of apoptotic cells (e.g., the number of
apoptotic cells/the total number of harvested cells). Cell cycle
changes were assayed by dividing the total number of analyzed cells
by the number of cells at individual cell cycle phases.
Effects of a decrease in JNK1 expression
on cell proliferation and apoptosis
Small interfering RNA (siRNA) targeting human JNK1
(si-JNK1) (Sigma-Aldrich; sequence of si-JNK1 was
5′-GGGAUUUGUUAUCCAAAAU-3′) was transfected into the HL-60 cells.
Twenty-four hours after transfection, the cells were treated with
250 nM of Se for 48 h. siRNA for human green fluorescent protein
(GFP; si-Control) (Sigma-Aldrich) was used as a control. Cell
proliferation was determined by MTS assay, and apoptosis was
analyzed by flow cytometry, as described above.
Immunoblotting
For immunoblotting, the cells were collected 48 h
following treatment with Se. The cells were lysed using 1× RIPA
buffer (Upstate Biotechnology, Lake Placid, NY, USA) containing a
protease inhibitor cocktail (Sigma-Aldrich). Following cell lysis,
30 μg of protein were loaded on a 10% SDS gel followed by transfer
onto PVDF membranes. Antibodies against JNK1 and phosphorylated
JNK1 (Cell Signaling Technology, Danvers, MA, USA), p21, p27 and
cyclin D1 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA),
and β-actin (1:10,000; Sigma-Aldrich) were used. The anti-mouse
secondary antibody was purchased from Santa Cruz Biotechnology,
Inc. The detected signals were visualized by an enhanced
chemiluminescence reaction system, as recommended by the
manufacturer (ECL-Plus; Amersham, Piscataway, NJ, USA). The
immunoblotting intensities were quantified using Quantity One
Software (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Statistical analysis
The significance of the differences between the
means of the various subgroups was assessed by an unpaired
two-tailed Student’s t-test. Data are presented as the means ± SD.
A value of P<0.05 was considered to indicate a statistically
significant difference.
Results
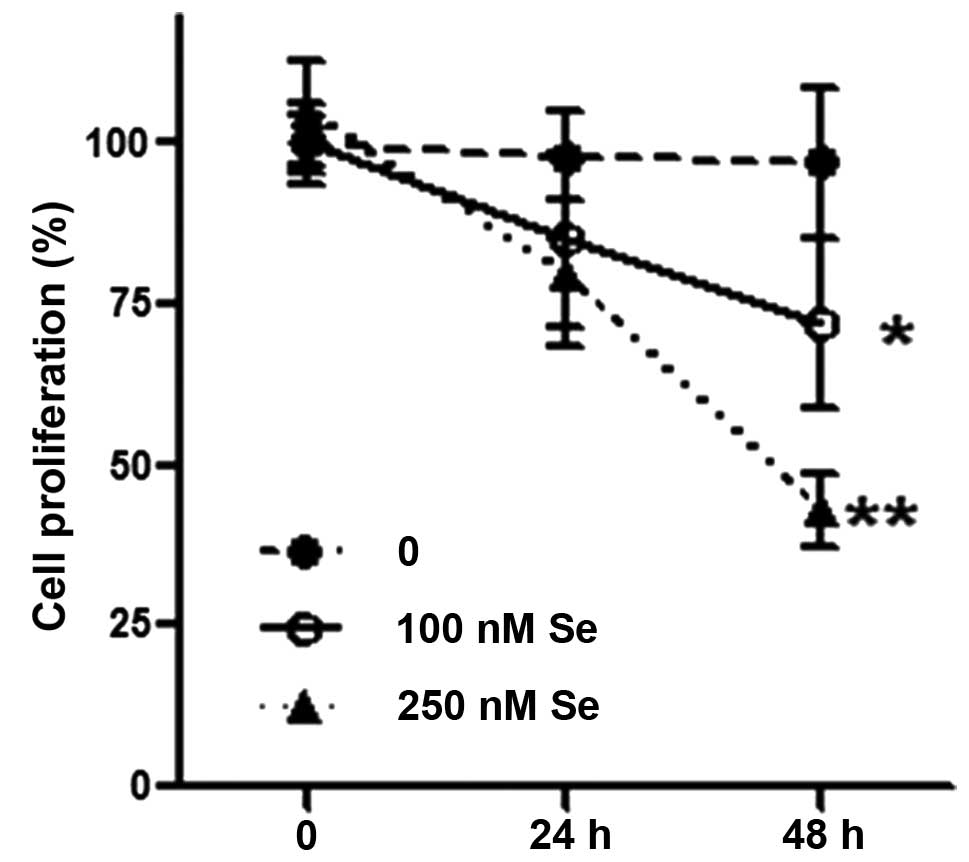
Se inhibits HL-60 cell proliferation
Se (250 nM) significantly inhibited HL-60 cell
proliferation after 48 h of treatment (P<0.01, compared to the
cells treated with 0 and 100 nM Se), although 100 nM of Se also
exerted slight inhibitory effects on cell proliferation (P<0.05,
compared to the cells treated with 0 and 100 nM Se) (Fig. 1).
Se promotes the apoptosis of HL-60
cells
The HL-60 cells were treated with 0, 100 or 250 nM
of Se for 48 h, and the cells were then collected for the analysis
of apoptosis. Se (100 nM) induced HL-60 cell apoptosis, although
the induction was not significant (Fig. 2). However, compared to the cells
treated with 0 and 100 nM Se, the higher concentration of Se (250
nM) significantly induced apoptosis (P<0.05).
Se causes changes in the cell cycle in
HL-60 cells
Following treatment with Se for 48 h, 100 nM of Se
caused changes in the cell cycle; however, these changes were not
significant, although the percentage of cells in the M phase
decreased and that of cells in the S phase increased compared to
the untreated cells (Fig. 3). The
higher concentration of Se (250 nM) significantly caused changes in
the cell cycle in HL-60 cells; in particular, Se induced cell cycle
arrest at the S phase (P<0.05, compared to the cells treated
with 0 and 100 nM Se).
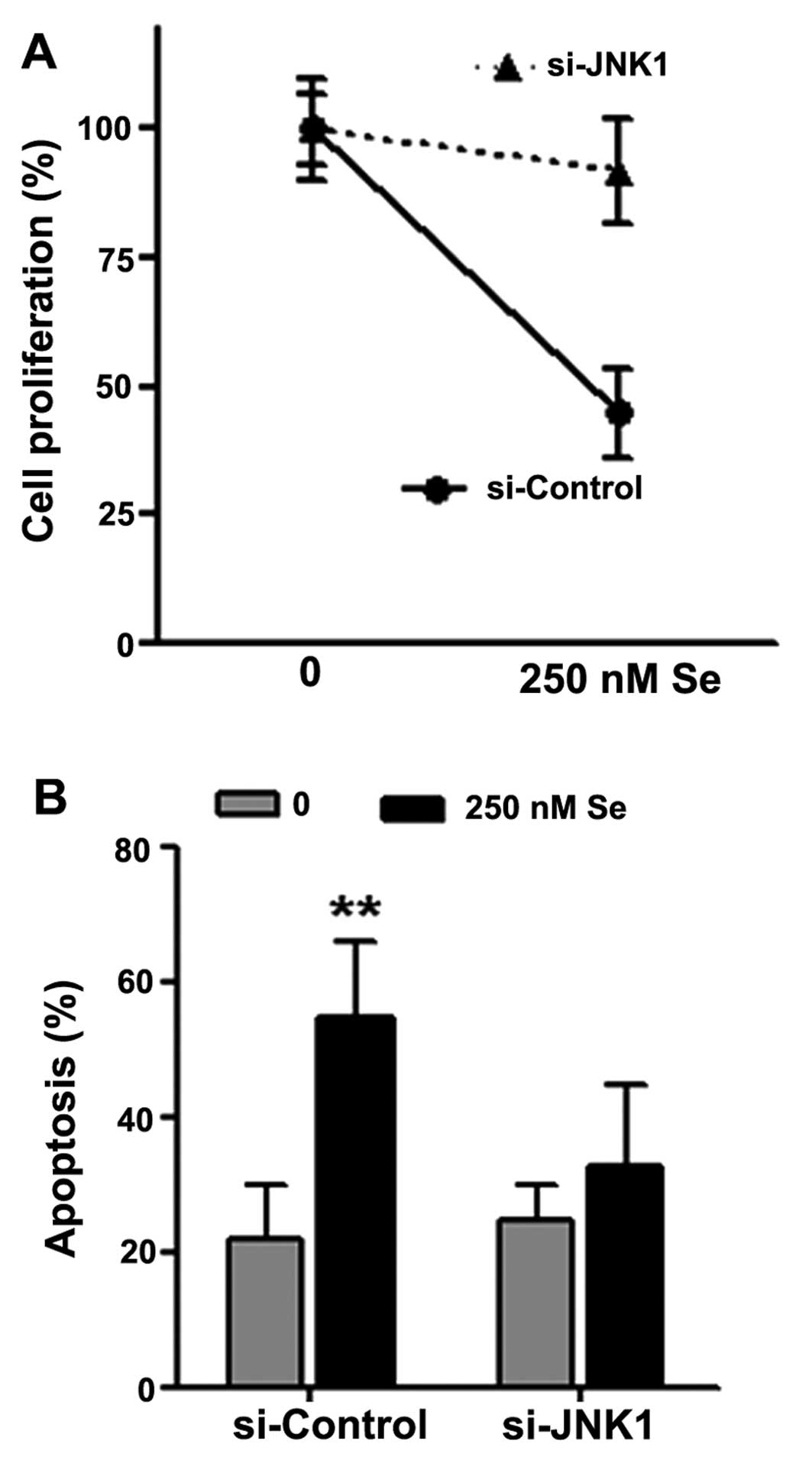
Decrease in JNK1 expression attenuates
the effects of Se on cell proliferation and apoptosis
In order to determine the importance of JNK1 in the
inhibitory effects of Se on cell proliferation and its
apoptosis-promoting effects, we transfected the HL-60 cells with
siRNA targeting human JNK1, and subsequently treated the cells with
250 nM Se for 48 h. We found that the knockdown of JNK1 abrogated
the inhibitory effects of Se on cell proliferation (Fig. 4A) and the induction of apoptosis
(Fig. 4B).
Se induces changes in protein expression
in HL-60 cells
To determine the potential mechanisms through which
Se induces apoptosis and cell cycle arrest, we assayed the changes
in the expression of the apoptosis-associated protein, JNK1, and
the cell cycle regulators, p21, p27 and cyclin D1. Se (250 nM)
significantly induced JNK1 phosphorylation even though the levels
of total JNK1 were not altered (Fig.
5). In addition, the expression of p21 and p27 was increased
(by approximately 100- and 300-fold, respectively) and that of
cyclin D1 was decreased by approximately 60-fold, compared to the
untreated cells and those treated with 100 nM Se.
Discussion
In this study, using an in vitro cell culture
model, found that a higher concentration of Se inhibited HL-60 cell
proliferation, induced HL-60 cell apoptosis and caused cell cycle
arrest, and these effects of selenium were associated with enhanced
JNK1 phosphorylation and increased p21/p27 expression (Fig. 6).
It has been reported that selenium exerts multiple
anticancer effects, such as antioxidant (19,20), anti-inflammatory and/or
suppressive effects by inhibiting β-catenin through 1,4-phenylene
bis (methylene) selenocyanate (9,21,22), and increasing the phosphorylation
of mitogen-activated protein kinase (MAPK) in prostate cancer cells
through methylseleninic acid (23,24). In our previous study, we
demonstrated that tumor inhibition by Se in colorectal cancer was
associated with the phosphorylation of JNK1 and the consequent
inhibition of β-catenin and its transcriptional targets, c-myc,
cyclin D1 and CDK4, leading to the induction of apoptosis and
inhibition of cell proliferation. In addition, Cox2 was almost
completed eliminated by Se in mouse intestinal epithelial cells
(10). In another recent study,
using an Apc/p21 complex mouse model of colorectal cancer, we found
that the combination of selenium and the non-steroidal
anti-inflammation drug, sulindac, led to the significant induction
of the expression of p27 and p53 and JNK1 phosphorylation, as well
as to the suppression of β-catenin and its downstream targets, also
leading to a demythelation on the p21 promoter (25), an additional mechanism of selenium
in cancer prevention, in which selenium showed a synergistic role
with sulindac in exerting maximal inhibitory effects on tumor
growth. This finding also provides an important chemopreventive
strategy using a combination of anticancer agents, which has a
great impact on cancer prevention and has a promising translational
potential.
JNK1 plays important roles in the regulation of cell
proliferation, differentiation and apoptosis in response to
cellular stress and chemopreventive agents (12,15,26,27). In the current study, we
demonstrated that the selenium-induced inhibition of leukemia HL-60
cell proliferation and the induction of apoptosis was through JNK1
activation. Experiments using siRNA targerting JNK1 further
demonstrated that the selenium-mediated effects on HL-60 cells
required JNK1. Our data strongly suggest that JNK1 plays a critical
role in the selenium-mediated chemoprevention of leukemia cells, in
which JNK1 phosphorylation or activation may be one of the key
factors through which selenium induces apoptosis.
Basic research and clinical trials have shown the
strong tumor preventive effects of selenium (6), whereas the outcome of the Selenium
and Vitamin E Cancer Prevention Trial (SELECT) showed that selenium
or vitamin E, alone or in combination did not prevent cancer but
caused an increased risk of cancer and metabolism-associated
diseases (28). This failure may
be due to the different form of selenium used, as well as the
dosage, and the baseline selenium status and genotypes (e.g.,
polymorphims) of selenium-containing proteins, such as glutathione
peroxidase 1 (GPx1) and selenium-binding protein 1 (SBP1). Both
proteins are selenium-contain proteins and are negatively regulated
by each other, and this interaction plays very important roles in
both cancer prevention and carcinogenesis (29).
The promotion of apoptosis and the induction of cell
cycle arrest are major mechanisms of action of chemotherapeutic
agents, in which cell cycle regulators, such as cyclins (e.g.,
cyclin D1 and cyclin E), cyclin-dependent kinases (e.g., cdk2, cdk4
and cdk6), and cyclin-dependent kinase inhibitors (e.g., p21, p27
and p16) (30) play crucial
roles. In this study, we found that apart from the induction of
apoptosis, selenium induced cell cycle arrest at the S phase. This
effect was associated with the increased expression of p21 and p27
and the decreased expression of cyclin D1.
In conclusion, our data demonstrate that Se is
effective in inhibiting HL-60 cell proliferation, promoting
apoptosis and inducing cell cycle arrest in leukemia cells by
targeting JNK1 and the cell cycle signaling pathway, which provides
further evidence of the anticancer bioactivity of selenium and
suggests that selenium may have additional usage beyond solid
tumors. In addition, our data have improved our understanding of
the mechanisms responsible for the selenium-induced anticancer
effects and suggest a novel use of selenium in leukemia.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (81272251), the Doctor Start-up
Research Fund (100820 and 505011) and the Key Research Fund
(ZD2011-18) from Xinxiang Medical University.
Abbreviations:
|
Se
|
selenium
|
|
JNK1
|
c-Jun NH2 terminal kinase 1
|
References
|
1
|
Siegel R, Ward E, Brawley O and Jemal A:
Cancer statistics, 2011: the impact of eliminating socioeconomic
and racial disparities on premature cancer deaths. CA Cancer J
Clin. 61:212–236. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Uğuz AC, Naziroğlu M, Espino J, Bejarano
I, Gonzalez D, Rodriguez AB and Pariente JA: Selenium modulates
oxidative stress-induced cell apoptosis in human myeloid HL-60
cells through regulation of calcium release and caspase-3 and -9
activities. J Membr Biol. 232:15–23. 2009.
|
|
3
|
Ip C: Lessons from basic research in
selenium and cancer prevention. J Nutr. 128:1845–1854.
1998.PubMed/NCBI
|
|
4
|
Wang L, Bonorden MJ, Li GX, Lee HJ, Hu H,
Zhang Y, Liao JD, Cleary MP and Lü J: Methyl-selenium compounds
inhibit prostate carcinogenesis in the transgenic adenocarcinoma of
mouse prostate model with survival benefit. Cancer Prev Res
(Phila). 2:484–495. 2009. View Article : Google Scholar
|
|
5
|
Facompre N and El-Bayoumy K: Potential
stages for prostate cancer prevention with selenium: implications
for cancer survivors. Cancer Res. 69:2699–2703. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Clark LC, Combs GF Jr, Turnbull BW, Slate
EH, Chalker DK, Chow J, Davis LS, Glover RA, Graham GF, Gross EG,
et al: Effects of selenium supplementation for cancer prevention in
patients with carcinoma of the skin. A randomized controlled trial
Nutritional Prevention of Cancer Study Group. JAMA. 276:1957–1963.
1996. View Article : Google Scholar
|
|
7
|
Greenwald P, Anderson D, Nelson SA and
Taylor PR: Clinical trials of vitamin and mineral supplements for
cancer prevention. Am J Clin Nutr. 85:314S–317S. 2007.PubMed/NCBI
|
|
8
|
Hawk ET and Levin B: Colorectal cancer
prevention. J Clin Oncol. 23:378–391. 2005. View Article : Google Scholar
|
|
9
|
Peters U and Takata Y: Selenium and the
prevention of prostate and colorectal cancer. Mol Nutr Food Res.
52:1261–1272. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fang W, Han A, Bi X, Xiong B and Yang W:
Tumor inhibition by sodium selenite is associated with activation
of c-Jun NH2-terminal kinase 1 and suppression of beta-catenin
signaling. Int J Cancer. 127:32–42. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Song Z, Tong C, Liang J, Dockendorff A,
Huang C, Augenlicht LH and Yang W: JNK1 is required for
sulindac-mediated inhibition of cell proliferation and induction of
apoptosis in vitro and in vivo. Eur J Pharmacol. 560:95–100. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tong C, Yin Z, Song Z, Dockendorff A,
Huang C, Mariadason J, Flavell RA, Davis RJ, Augenlicht LH and Yang
W: c-Jun NH2-terminal kinase 1 plays a critical role in intestinal
homeostasis and tumor suppression. Am J Pathol. 171:297–303. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Davis RJ: Signal transduction by the JNK
group of MAP kinases. Cell. 103:239–252. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Johnson GL and Lapadat R:
Mitogen-activated protein kinase pathways mediated by ERK, JNK, and
p38 protein kinases. Science. 298:1911–1912. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hu D, Fang W, Han A, Gallagher L, Davis
RJ, Xiong B and Yang W: c-Jun N-terminal kinase 1 interacts with
and negatively regulates Wnt/beta-catenin signaling through
GSK3beta pathway. Carcinogenesis. 29:2317–2324. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang WC, Mathew J, Velcich A, Edelmann W,
Kucherlapati R, Lipkin M, Yang K and Augenlicht LH: Targeted
inactivation of the p21(WAF1/cip1) gene enhances Apc-initiated
tumor formation and the tumor-promoting activity of a Western-style
high-risk diet by altering cell maturation in the intestinal
mucosal. Cancer Res. 61:565–569. 2001.
|
|
17
|
van de Wetering M, Sancho E, Verweij C, de
Lau W, Oving I, Hurlstone A, van der Horn K, Batlle E, Coudreuse D,
Haramis AP, et al: The beta-catenin/TCF-4 complex imposes a crypt
progenitor phenotype on colorectal cancer cells. Cell. 111:241–250.
2002.PubMed/NCBI
|
|
18
|
Tetsu O and McCormick F: Beta-catenin
regulates expression of cyclin D1 in colon carcinoma cells. Nature.
398:422–426. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lü J and Jiang C: Selenium and cancer
chemoprevention: hypotheses integrating the actions of
selenoproteins and selenium metabolites in epithelial and
non-epithelial target cells. Antioxid Redox Signal. 7:1715–1727.
2005.
|
|
20
|
Diwadkar-Navsariwala V and Diamond AM: The
link between selenium and chemoprevention: a case for
selenoproteins. J Nutr. 134:2899–2902. 2004.PubMed/NCBI
|
|
21
|
Narayanan BA, Narayanan NK, Desai D,
Pittman B and Reddy BS: Effects of a combination of docosahexaenoic
acid and 1,4-phenylene bis(methylene) selenocyanate on
cyclooxygenase 2, inducible nitric oxide synthase and beta-catenin
pathways in colon cancer cells. Carcinogenesis. 25:2443–2449. 2004.
View Article : Google Scholar
|
|
22
|
Rao CV, Cooma I, Rodriguez JG, Simi B,
El-Bayoumy K and Reddy BS: Chemoprevention of familial adenomatous
polyposis development in the APC(min) mouse model by 1,4-phenylene
bis(methylene)selenocyanate. Carcinogenesis. 21:617–621. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hu H, Jiang C, Ip C, Rustum YM and Lü J:
Methylseleninic acid potentiates apoptosis induced by
chemotherapeutic drugs in androgen-independent prostate cancer
cells. Clin Cancer Res. 11:2379–2388. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jiang C, Wang Z, Ganther H and Lu J:
Distinct effects of methylseleninic acid versus selenite on
apoptosis, cell cycle, and protein kinase pathways in DU145 human
prostate cancer cells. Mol Cancer Ther. 1:1059–1066.
2002.PubMed/NCBI
|
|
25
|
Bi X, Pohl N, Dong H and Yang W: Selenium
and sulindac are synergistic to inhibit intestinal tumorigenesis in
Apc/p21 mice. J Hematol Oncol. 6:82013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hu D, Bi X, Fang W, Han A and Yang W:
GSK3beta is involved in JNK2-mediated beta-catenin inhibition. PLoS
One. 4:e66402009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bode AM and Dong Z: The functional
contrariety of JNK. Mol Carcinog. 46:591–598. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lippman SM, Klein EA, Goodman PJ, Lucia
MS, Thompson IM, Ford LG, Parnes HL, Minasian LM, Gaziano JM,
Hartline JA, et al: Effect of selenium and vitamin E on risk of
prostate cancer and other cancers: the Selenium and Vitamin E
Cancer Prevention Trial (SELECT). JAMA. 301:39–51. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fang W, Goldberg ML, Pohl NM, Bi X, Tong
C, Xiong B, Koh TJ, Diamond AM and Yang W: Functional and physical
interaction between the selenium-binding protein 1 (SBP1) and the
glutathione peroxidase 1 selenoprotein. Carcinogenesis.
31:1360–1366. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chu IM, Hengst L and Slingerland JM: The
Cdk inhibitor p27 in human cancer: prognostic potential and
relevance to anticancer therapy. Nat Rev Cancer. 8:253–267. 2008.
View Article : Google Scholar : PubMed/NCBI
|