Introduction
Peroxisome proliferator-activated receptor γ (PPARγ)
is a transcription factor that controls multiple cellular metabolic
processes and is a member of the nuclear hormone receptor protein
family involved in the regulation of multiple physiological and
pathological metabolic processes, such as lipid and glucose
metabolism regulation in vivo and inflammation regulation
(1). PPARγ is mainly expressed in
monocytes/macrophages and fat cells, and low levels of PPARγ can be
detected in the central nervous system. Recent studies have
revealed that brain ischemic injury promoted the expression and
activity of PPARγ (2) and that a
PPARγ agonist can protect against brain ischemic injury through
PPARγ activation (3,4).
12/15-lipoxygenase (12/15-LOX) is a fatty acid
dioxygenase that can oxidize arachidonic acid into
12-hydroxyeicosatetraenoic acid (12-HETE) in mammalian cells. In
vitro studies of non-neuronal cells revealed that 12-HETE can
activate PPARγ (5). However, no
studies have explored the influence of the 12/15-LOX pathway and
12-HETE on PPARγ activity in neurons damaged by ischemic injury.
The present study investigated the changes in 12/15-LOX and 12-HETE
levels and their impact on PPARγ activation in rat cortical neurons
treated with oxygen-glucose deprivation (OGD). Simultaneously,
antisense oligonucleotide technology was used to explore the
influence of 12/15-LOX inhibition on PPARγ expression and
activation to investigate the regulatory effect of 12/15-LOX
pathway on PPARγ. The aim of the present study was to understand
these mechanisms. The results may assist in the understanding of
brain ischemic injury and help develop treatments in the
future.
Materials and methods
Animal grouping and drug
administration
All the animal experiments were carried out
according to an institutionally approved protocol, in accordance
with the National Institutes of Health Guide for the Care and Use
of Laboratory Animals, and were approved by the Institutional
Animal Care and Use Committee of Tianjin Medical University
(Tianjin, China). Healthy adult male Sprague Dawley (SD) rats
weighing 280–330 g were purchased from the Laboratory Animal Center
at the Academy of Military Medical Sciences. All the rats consumed
water freely but were fasted for 12 h before surgery. The rats were
randomly allocated into three groups and were treated as follows:
i) 12 rats in the sham-operation group were used for brain tissue
homogenates for further analysis. ii) 6 rats in the
ischemia-reperfusion (I/R) group were used for tissue sections and
12 rats were used for brain tissue homogenates for further
analysis. iii) 12 rats in the I/R intervention group were used for
brain tissue homogenates. 12-HETE was purchased from Cayman
Chemical Co. (Ann Arbor, MI, USA) and dissolved in
phosphate-buffered saline (PBS) at 1 mg/ml. In the I/R intervention
group, rats received stereotactic injections of 15 ml 12-HETE 30
min before ischemia induction, whereas the rats in the
sham-operation group or I/R group received equal volumes of
PBS.
Stereotactic intracerebroventricular
injection
Rats were anesthetized with 10% chloral hydrate 30
min before the establishment of the middle cerebral artery
occlusion/reperfusion (MCAO/R) model. Subsequently, 15 ml 12-HETE
or an equal volume of PBS was injected into the right lateral
ventricle at 0.8 mm posterior to bregma, 1.3 mm right lateral to
midline and 3.5 mm deep into the subdural surface. Injection was
performed at a rate of 1 μl/min and finished in 15 min. The needle
was retained for 15 min and withdrawn at a rate of 1 mm/min to
prevent overflowing of 12-HETE or PBS.
Establishment of MCAO/R model
The rat cerebral artery was occluded with thread for
60 min and reperfusion was performed for 24 h. In the
sham-operation group the common carotid, external carotid and
internal carotid arteries were exposed and separated by surgery
without occlusion.
Primary culture of fetal rat cortical
neurons and neuronal cell identification
Day 18–21 embryonic Sprague Dawley (SD) rats were
sacrificed following anesthesia with ice and 75% ethanol and the
cerebral cortex was isolated under sterile conditions. Pia mater
and blood vessels were removed under a dissecting microscope. The
cerebral cortex were cut into pieces and digested with 0.125%
trypsin at 37°C for 20 min. Subsequently, 20% fetal bovine serum
(FBS) was added to stop digestion and the cells were triturated
with a Pasteur pipette and filtered with a 200-mesh cell strainer
to collect single-cell suspensions. The cells were counted with a
hemocytometer. Cells were seeded into Petri dishes were pre-coated
with 0.1 mg/ml L-polylysine at a final density of
105–106/ml. Primary cortical neurons were
incubated at 37°C in a 5% CO2 humidified atmosphere and
high glucose Dulbecco’s modified Eagle’s medium/nutrient mixture
F-12 supplemented with 10% FBS, 10% horse serum, 1% glutamine and
1% penicillin-streptomycin was used as culture medium. The medium
was changed to neurobasal medium (Invitrogen Life Technologies,
Carlsbad, CA, USA) supplemented with 2% B27 following cell
attachment (3 to 6 h after seeding). Half the volume of neurobasal
medium was refreshed every third day. Neuronal-specific marker
microtubule-associated protein 2 (MAP2) was stained at day 9 and
>95% cells were MAP2 positive.
Preparation of the neuronal model of
oxygen-glucose deprivation (OGD) and 12-HETE or 12/15-LOX antisense
oligonucleotide (asON-12/15-LOX) treatment
Day 9 cortical neurons were randomly divided into
the control, OGD-treated and intervention groups. The medium of the
OGD-treated neurons was changed to neurobasal medium without
glucose (Invitrogen Life Technologies) and the neurons were
incubated in a pre-adjusted tri-gas incubator (37°C, 95%
N2, 5% CO2, <1% O2). After 3 h
incubation, neurons were cultured in the normal medium at the
normal conditions for 24 h and were collected for further analysis.
The 12-HETE intervention group was treated with 1 μM 12-HETE 30 min
before OGD treatment and the 12-HETE concentration was maintained
during the whole culture process. The asON-12/15-LOX intervention
group was treated with 4 μM asON 48 h before OGD treatment and
maintained during the whole culture process. The same volume of
dimethyl sulfoxide (DMSO) was added into the OGD-treated group. The
sequences of the antisense oligonucleotides used in the study are
as follows: Antisense, 5′-CTC-AGG-AGG-GTG-TAA-ACA-3′ (6); sense, 5′-TGT-TTA-CAC-CCT-CCT-GAG-3′;
and scramble, 5′-AAG-ATT-GCG-GCG-CGA-CGA-TGA-3′.
Analysis of relative OGD-treated neuron
survival rate by 3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl
tetrazolium bromide (MTT) assay
Cortical neurons at day 9 were divided into 8
replicate blank groups (culture medium without neurons), control
groups and OGD-treated groups. After treatment with OGD for 24 h,
neurons were further cultured in normal medium and medium only was
added into blank wells. Subsequently, 20 μl 5 mg/ml MTT was added
into each well and incubated for 4 h. After discarding the medium,
100 μl DMSO was added, mixed thoroughly and incubated at 37°C for
15 min. The optical density (OD) of each well was analyzed with an
automatic plate reader and the absorbance at 570 nm was measured.
The relative cell survival rate was calculated as follows: Relative
cell survival rate = (OD OGD-treated group − OD blank group)/(OD
control group − OD blank group). The median concentration was used
for the model set.
Nuclear protein extraction
Nuclear proteins were isolated with a nuclear
protein extraction kit (Active Motif, Carlsbad, CA, USA) according
to the manufacturer’s instructions.
Western blotting
The primary antibodies were as follows: Mouse
anti-rat PPARγ monoclonal antibody (1:200; Santa Cruz, Dallas,
USA), rabbit anti-rat 12/15-LOX monoclonal antibody (1:1000; Cayman
Chemical Co.); and related horseradish peroxidase (HRP)-conjugated
secondary antibodies (Santa Cruz Biotechnology, Inc., Dallas, TX,
USA; 1:5000) were used as previously described (2).
ELISA analysis
The level of 12-HETE in neurons was determined with
the 12-HETE ELISA kit (Assay Design, Ann Arbor, MI, USA) and
performed according to the manufacturer’s instructions.
Total RNA isolation and reverse
transcription-polymerase chain reaction (RT-PCR) amplification
Total RNA was extracted from the cells with TRIzol
(Sigma, St. Louis, MO, USA), precipitated with
chloroform-isopropanol and quantified with absorbance at
OD260. cDNA was generated from 1.5 μg total RNA. PCR
amplification was performed using a 20 μl reaction system,
including 1 μl cDNA, 2 μl forward and reverse primers (5 μM), 200
μM dNTPs and 0.8 U Taq polymerase. Primers for lipoprotein lipase
(LPL) were as follows: Forward,
5′-TTCCATTACCAAGTCAAGATTCAC-3′; and reverse,
5′-TCAGCCCGACTTCTTCAGAGACTT-3′. PCR amplification products were
analyzed with 1.2% agarose gel electrophoresis and a gel imaging
system (Tanon Science & Technology Co., Ltd., Shanghai, China)
was used to quantify the band density. Glyceraldehyde 3-phosphate
dehydrogenase (GAPDH) was used as a loading control.
Construction of PGL3-PPRE and
determination of luciferase activity
pGL3-basic (Promega, Madison, WI, USA) was used as
the vector, which contains the luciferase mRNAs without promoter.
Three peroxisome proliferator responsive element (PPRE) fragments
were inserted upstream of the luciferase gene in the pGL3-basic
vector to construct the pGL3-PPRE vector. Neurons were transfected
with Lipofectamine 2000 (Invitrogen Life Technologies) and 24 h
later, 1 μM 12-HETE was added, whereas in the control group, the
same volume of DMSO was added. The cells were lysed 24 h later and
luciferase analysis was performed according to the instructions of
the luciferase assay system kit (Promega). A Safire2 basic plate
reader from Tecan Australia Pty Ltd. (Melbourne, Australia) was
used to measure the ODs of the cell lysates. The β-galactosidase
enzyme assay system kit (Promega) was used to measure the β-gal
activity to adjust the luciferase value for transfection
efficiency. Adjusted fluorescence value = measured fluorescence
value/β-gal value.
Immunofluorescence staining
Neurons were cultured on sterile slides precoated
with poly-L-lysine. To perform immunofluorescence staining, neurons
were fixed with 4% paraformaldehyde and blocked with serum for 30
min. Neurons were then incubated with a rabbit anti-rat 12/15-LOX
polyclonal antibody (1:100; Cayman Chemical Co.) or anti-PPARγ
antibody (1:50; Santa Cruz Biotechnology, Inc.) at 4°C overnight. A
tetraethyl rhodamine isothiocyanate-conjugated secondary antibody
was added the next day and incubated in the dark at 37°C for 1 h.
Neurons were washed with PBS-Tween 20 and finally incubated with
4′,6-diamidino-2-phenylindole at room temperature for 10 min to
stain the cell nuclei. Slides were treated with mounting medium and
analyzed with a Nikon Eclipse 80i microscope and Nikon DS-Ril
camera (Nikon, Toyko, Japan).
Statistical analysis
Experimental data was exhibited as mean ± standard
deviation. Statistical analysis was performed with SPSS 15.0
statistical package (SPSS Inc, Chicago, IL, USA) and an independent
samples t test was performed for comparison between groups.
A χ2 test was used to analyze the difference in neuron
survival rates between groups. P<0.05 was considered to indicate
a statistically significant difference.
Results
Elevation of 12/15-LOX expression and
activity induced by I/R injury
Western blots were performed to analyze the
expression of 12/15-LOX in rat brain tissues from the I/R injury
model. Significant upregulation of 12/15-LOX expression was induced
by I/R injury (Fig. 1A). To
explore the activity changes of 12/15-LOX, the level of its
product, 12-HETE, was determined by ELISA and the results showed
that I/R injury clearly induced the production of 12-HETE (Fig. 1B).
Induction of PPARγ expression by I/R
injury and further upregulation of PPARγ expression with 12-HETE
intervention
Western blotting showed that compared to the I/R
group, I/R injury plus 12-HETE intervention markedly upregulated
the expression of PPARγ (Fig.
2).
Suppression of inducible nitric oxide
synthase (iNOS) expression by 12-HETE in rat brain tissues with I/R
injury
Western blots were performed to explore the
influence of 12-HETE intervention on iNOS expression and revealed
an evident inhibition of iNOS expression (Fig. 3).
Upregulation of 12/15-LOX expression and
12-HETE generation by OGD treatment in neurons
The MTT assay revealed that the relative neuron
survival rate decreased by 43.84±2.07% with OGD treatment for 24 h
compared to the control group. The difference between the two
groups was significant (P<0.05).
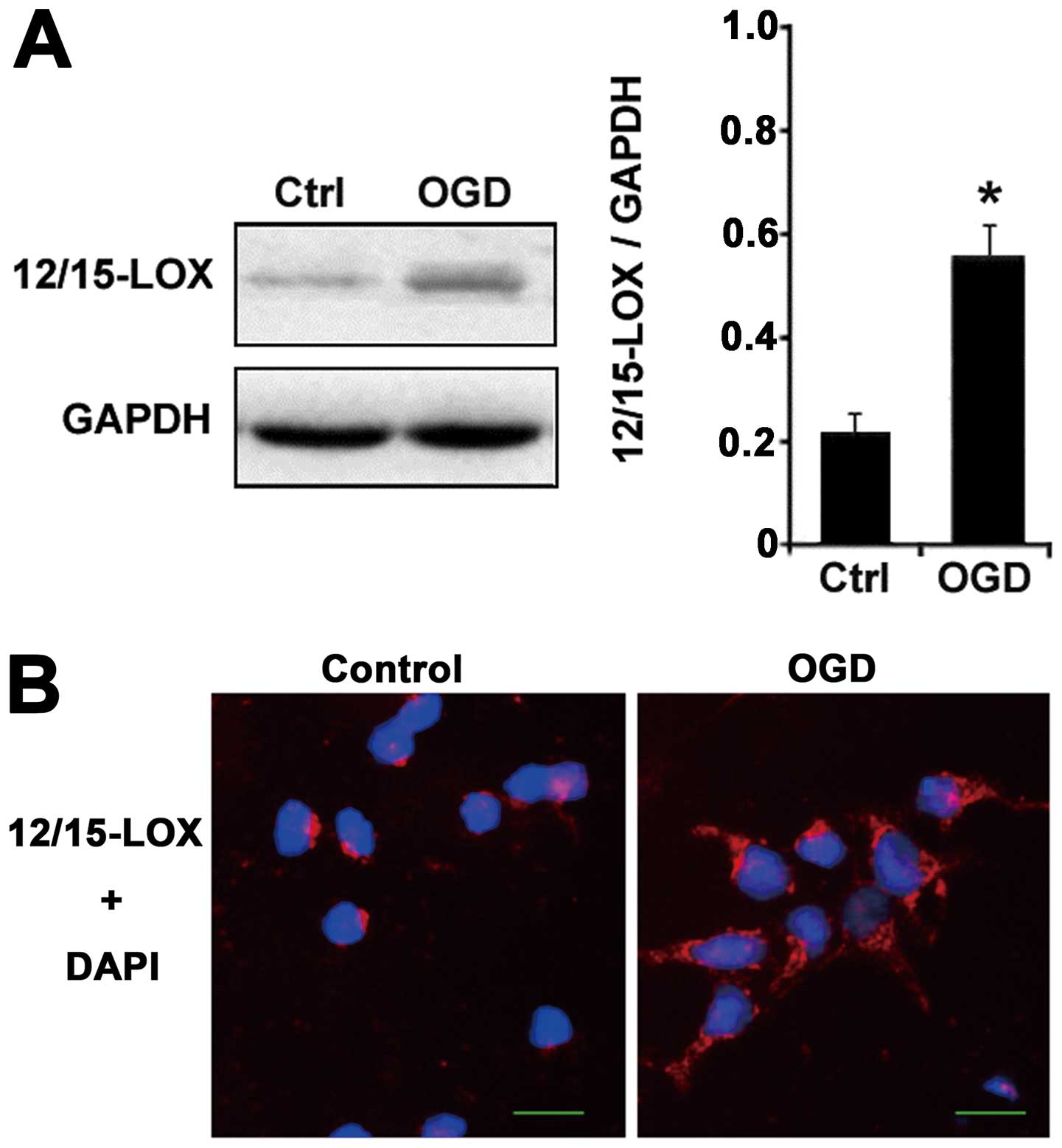
Western blots showed that the 12/15-LOX protein, the
critical enzyme generating 12-HETE, increased significantly with
OGD treatment compared with the control group (Fig. 4A) and immunofluorescence
experiments confirmed that the upregulation of 12/15-LOX expression
was induced by OGD (Fig. 4B).
Stimulation of PPARγ nuclear protein
expression by OGD treatment in neurons and upregulation of PPARγ
nuclear protein expression by 12-HETE in OGD-treated neurons
Western blotting was used to determine the
expression levels of PPARγ in the cell nucleus, and the results
showed that PPARγ nuclear protein increased significantly in
neurons with OGD treatment. PPARγ nuclear protein was also
significantly elevated in the 12-HETE intervention plus OGD-treated
group compared to the OGD-treated group (Fig. 5).
Blockade of 12/15-LOX expression by
asON-12/15-LOX treatment in OGD-treated neurons
Compared to the OGD-treated group, asON-12/15-LOX
treatment reduced the expression of 12/15-LOX significantly
(Fig. 6). Whereas sense
oligonucleotides or scramble oligonucleotides of 12/15-LOX did not
change the expression of 12/15-LOX (data not shown).
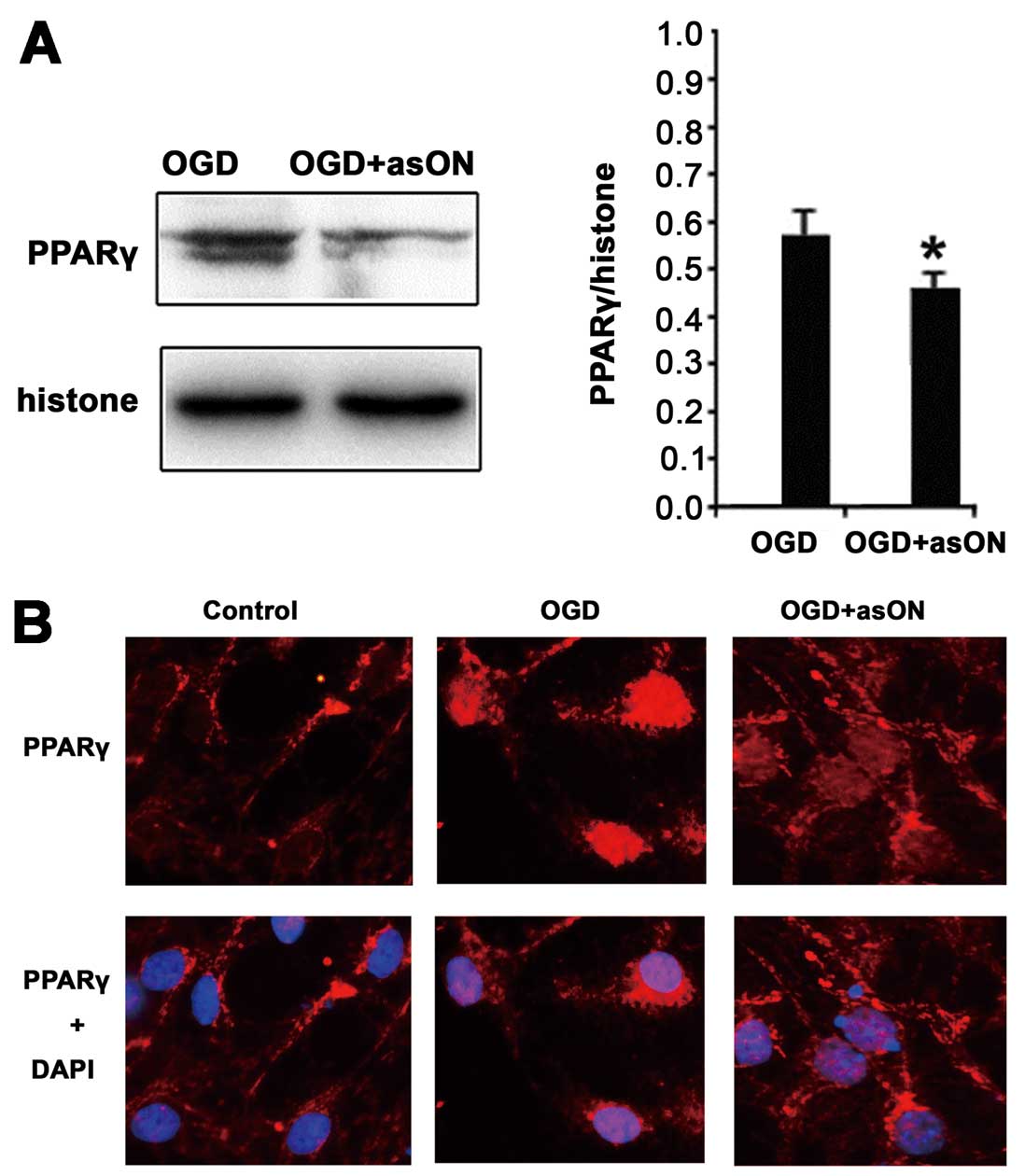
Inhibition of PPARγ nuclear protein
expression by asON-12/15-LOX treatment in OGD-treated neurons
asON-12/15-LOX treatment inhibited the expression of
PPARγ nuclear protein significantly compared to the OGD-treated
group (Fig. 7A).
Immunofluorescence analysis confirmed the results of the western
blot, showing that the expression of PPARγ nuclear protein was
upregulated with OGD treatment and the upregulation was inhibited
by asON-12/15-LOX treatment (Fig.
7B).
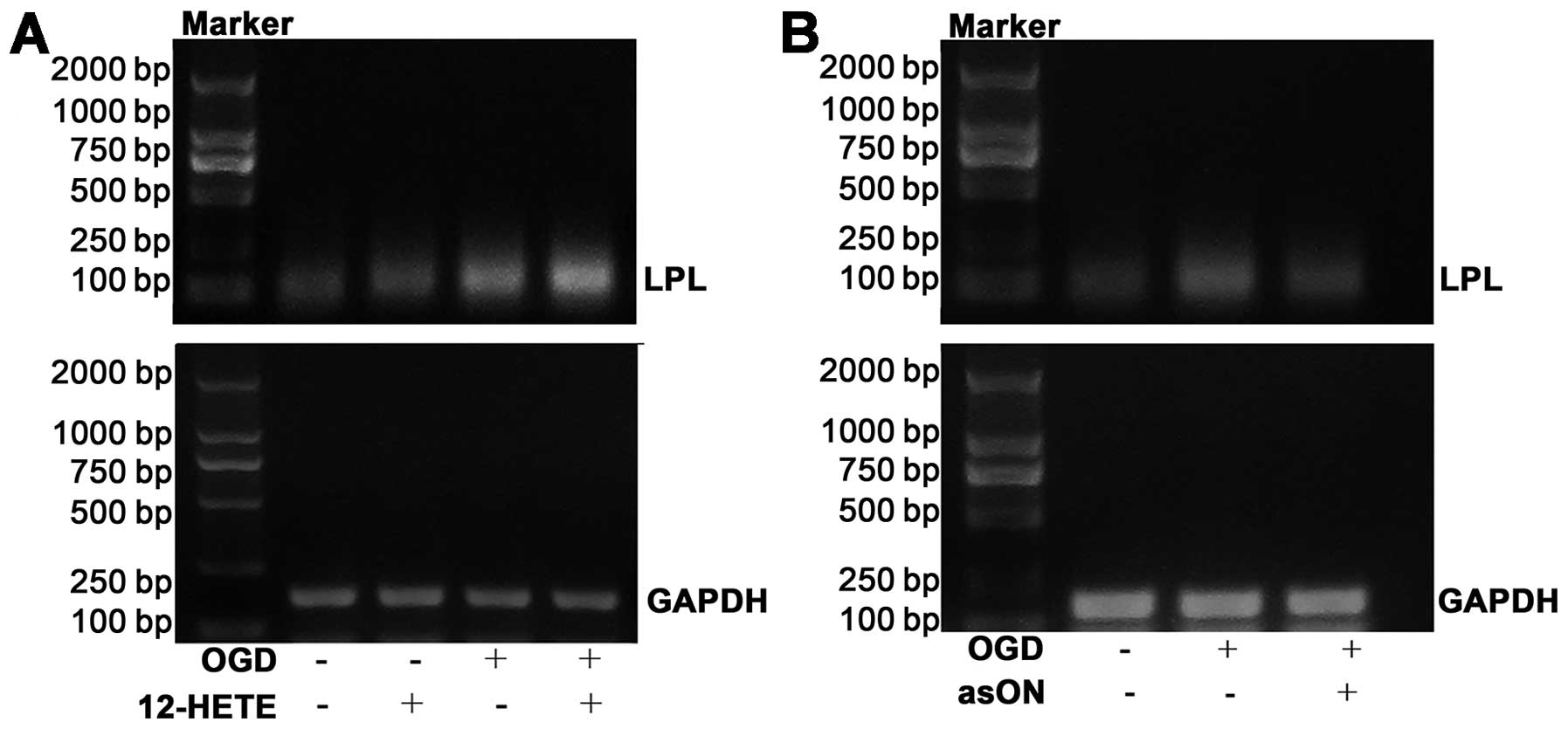
Expression of PPARγ target genes by
12-HETE treatment and suppression of PPARγ target genes by
asON-12/15-LOX treatment in OGD-treated neurons
RT-PCR was employed to analyze the expression of
LPL, a PPARγ target gene, in OGD-treated neurons and to
explore the impact of 12-HETE intervention on its expression. The
results showed that 12-HETE intervention markedly increased the
expression of LPL mRNA compared to OGD-treated neurons
(Fig. 8A). However, treatment
with asON-12/15-LOX clearly inhibited the expression of LPL
in OGD-treated neurons (Fig.
8B).
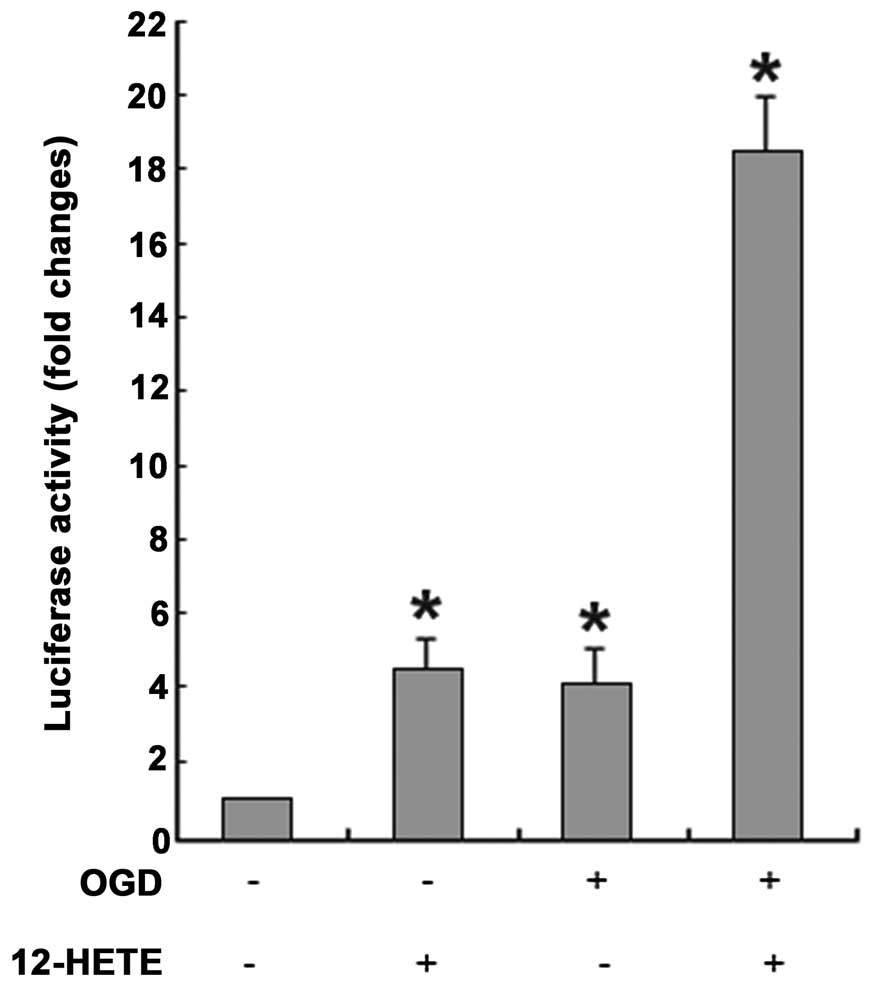
Enhancement of PPARγ binding ability to
DNA by 12-HETE treatment
Normal cultured cortical neurons were transfected
with pGL3-PPRE and were subsequently treated with 12-HETE, OGD or
OGD plus 12-HETE. After 24 h, luciferase activities in the treated
neurons clearly increased compared to control neurons treated with
DMSO (t=−10.753, −9.679 and −21.978, respectively,
P<0.05). Luciferase activity increased similarly in 12-HETE- and
OGD-treated neurons with 4-fold changes, but luciferase activity
increased by 18-fold in neurons treated with OGD plus 12-HETE
(Fig. 9). Furthermore, relative
survival rates of neurons were measured with the MTT assay and the
possibility that luciferase activity was enhanced by cell survival
changes derived from 12-HETE or OGD treatment was excluded (data
not shown). Only minor luciferase activity was detected in
12-HETE-treated neurons transfected with pGL3-Basic, whereas
luciferase activity could not be detected in neurons transfected
with mixtures without any plasmids or in neuron lysates.
Discussion
The aim of the present study was to investigate the
influence of 12/15-LOX on the activity of PPARγ in ischemia
reperfusion. This was of interest as this information may assist
with future treatment of brain ischemic injury. PPARγ is a member
of the nuclear receptor superfamily and a ligand-dependent
transcriptional factor. PPARγ activation leads to its nuclear
translocation in order to regulate the transcription of target
genes. Numerous studies have reported that PPARγ expression and
activity in brain tissues were induced by I/R injury (7–10),
and that PPARγ activation reduced infarct volume and inhibition of
inflammation mediators including intercellular adhesion molecule 1,
interleukin-β, cyclooxygenase-2 and iNOS (3,4,11,12). Therefore, it has been proposed
that induction of PPARγ expression and activation by brain ischemia
is a protective response to damage. This protective response may be
attributed to PPARγ activation induced by endogenous agonists
following ischemia, but the detailed mechanisms remain elusive.
12/15-LOX, a member of lipoxygenase family, is a
lipid peroxidase encoded by the ALOX15 gene that can oxidize
free polyunsaturated fatty acids and phospholipids in biological
membranes to generate oxidative products. 12-HETE is the oxidative
derivative of arachidonic acid catalyzed by 12/15-LOX. In brain
tissues, 12/15-LOX is mainly expressed in neurons or certain
astrocytes in the cerebral hemisphere, basal ganglia and
hippocampus. 12-HETE is the major product of 12/15-LOX in the
central nervous system (3).
Previous studies revealed that in brain tissues, 12/15-LOX may
exert its physiological functions through oxidative modifications
of membrane structures and generation of mediators or signaling
molecules with biological activities involved in synaptic
transmission (13,14). In vitro studies confirmed
that 12/15-LOX products, such as 13-hydroxyoctadecadienoic acid
(13-HODE), 12-HETE and 15--HETE served as endogenous PPARγ
agonists. Furthermore, higher expression levels of 12/15-LOX
enhanced the transcriptional activation effect of PPARγ in specific
cell types. For example, in monocytes, 12/15-LOX products
interacted with PPARγ directly to stimulate the expression of
cluster of differentiation 36 and upregulate PPARγ expression
simultaneously (15). The
induction of PPARγ and its target genes by IL4 in macrophages
through the 12/15-LOX pathway has already been shown. Similarly,
13-HODE and 15-HETE PPARγ and increase the expression of
PPARγ mRNA in human vascular smooth muscle cells (16) and colon tumor cell line (17). Furthermore, in vivo
experiments have confirmed the regulatory effect of 12/15-LOX on
PPARγ and in the mouse uterus, PPARγ activation can occur through
the 12/15-LOX pathway to mediate the impact of 12/15-LOX on the
pregnant uterus (5). These
results indicate that elevation of PPARγ expression and activation
could be induced by 12/15-LOX in multiple tissues and cells.
Therefore, we speculate that 12/15-LOX has similar roles in the
central nervous system.
In the present study, I/R injury has been
demonstrated to induce the expression of 12/15-LOX and its product
12-HETE. A previous study implicated the upregulation of 12-HETE
level with oxidative stress caused by I/R injury in brain tissues
(18). However, other studies
have shown that 12-HETE is involved in synaptic transmission as a
second messenger in the central nervous system and that it also
participates in a variety of physiological activities, including
learning and memory (19,20). Notably, 12-HETE served as an
endogenous agonist of PPARγ to modulate its activity (5).
PPARγ is a nuclear transcription factor and
following activation it transports into the cell nucleus from the
cytoplasm to regulate the expression of target genes. Therefore,
the expression of PPARγ protein in the nucleus is associated with
its activity status. Regulation of target gene expression by PPARγ
is through PPARγ binding to a specific DNA element in the promoters
of the target genes (peroxisome proliferator responsive element,
termed PPRE) to promote target gene expression (21–23).
The present study revealed that 12-HETE intervention
in rats with I/R injury elevated the expression of PPARγ total
protein. Furthermore, treatment of OGD-damaged rat cortical neurons
with 12-HETE induced the expression of PPARγ, enhanced its binding
ability to DNA and promoted the expression of target genes,
suggesting the stimulatory effect of 12-HETE on PPARγ activity.
Several associated studies have reported an inhibitory effect of
PPARγ on inflammation following ischemia. For instance, PPARγ
agonists, including pioglitazone, rosiglitazone and troglitazone,
reduced infarct volume, improved neuron functions, suppressed the
expression of variant inflammatory mediators, reduced neutrophil
infiltration, and inhibited the activation of microglias,
macrophages and the inflammation-associated NF-κB pathway (4,10–12). Furthermore, other studies have
reported that the neuroprotective effect of PPARγ is partially
achieved through suppression of iNOS expression and activation
(24–27). In accordance with these results,
it was observed that elevated expression of iNOS in brain tissues
with I/R injury was inhibited by 12-HETE intervention and PPARγ
activity was stimulated simultaneously, indicating that
12-HETE-induced PPARγ activation inhibited inflammation responses,
to achieve a neuroprotective effect.
Antisense oligonucleotides were also used to inhibit
the expression of 12/15-LOX and revealed that PPARγ nuclear
expression was negatively regulated by asON-12/15-LOX, confirming
the regulatory effect of 12/15-LOX on PPARγ. Cell transfection
reagents are often used to promote the delivery of antisense
oligonucleotides into cells to enhance the inhibitory effect.
However, cell transfection reagents often damage cells. As primary
cultured neurons in vitro are vulnerable and prone to
injury, the oligonucleotides were dissolved into the culture medium
directly to treat neurons. This method inhibited the expression of
12/15-LOX significantly, indicating its feasibility. Previous
studies have also proved the efficiency of such methods in sensory
neuron treatment (6,28).
The present study has certain limitations. Rat-based
models of oxygen-glucose deprivation and ischemia reperfusion were
used, however, it would be noteworthy to observe if the role of
12/15-LOX can also be followed in human derived cells. A number of
details of the mechanism of PPARγ protection remain to be revealed
and therefore, further work is required prior to considering these
results in terms of clinical therapy.
In conclusion, the level of the 12/15-LOX-derived
product 12-HETE was significantly elevated in OGD-treated cortical
neurons and confirmed the agonistic effect of 12-HETE on PPARγ. The
expression of PPARγ nuclear expression could be blocked with
12/15-LOX inhibition in OGD-treated neurons. These results revealed
that PPARγ is activated by the 12/15-LOX pathway in OGD-treated
neurons and that PPARγ activation has a neuroprotective effect,
indicating that it is a neuronal self-protective response to damage
and injury.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 81401023).
References
|
1
|
Boyle PJ: Diabetes mellitus and
macrovascular disease: mechanisms and mediators. Am J Med. 120(9
Suppl): S12–S17. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Xu YW, Sun L, Liang H, Sun GM and Cheng Y:
12/15-lipoxygenase inhibitor baicalein suppresses PPAR gamma
expression and nuclear translocation induced by cerebral
ischemia/reperfusion. Brain Res. 1307:149–157. 2010. View Article : Google Scholar
|
|
3
|
Collino M, Aragno M, Mastrocola R, et al:
Modulation of the oxidative stress and inflammatory response by
PPAR-gamma agonists in the hippocampus of rats exposed to cerebral
ischemia/reperfusion. Eur J Pharmacol. 530:70–80. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhao Y, Patzer A, Herdegen T, Gohlke P and
Culman J: Activation of cerebral peroxisome proliferator-activated
receptors gamma promotes neuroprotection by attenuation of neuronal
cyclooxygenase-2 overexpression after focal cerebral ischemia in
rats. FASEB J. 20:1162–1175. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li Q, Cheon YP, Kannan A, Shanker S,
Bagchi IC and Bagchi MK: A novel pathway involving progesterone
receptor, 12/15-lipoxygenase-derived eicosanoids and peroxisome
proliferator-activated receptor gamma regulates implantation in
mice. J Biol Chem. 279:11570–11581. 2004. View Article : Google Scholar
|
|
6
|
Lebeau A, Terro F, Rostene W and Pelaprat
D: Blockade of 12-lipoxygenase expression protects cortical neurons
from apoptosis induced by beta-amyloid peptide. Cell Death Differ.
11:875–884. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ou Z, Zhao X, Labiche LA, et al: Neuronal
expression of peroxisome proliferator-activated receptor-gamma
(PPARgamma) and 15d-prostaglandin J2-mediated protection of brain
after experimental cerebral ischemia in rat. Brain Res.
1096:196–203. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sundararajan S, Gamboa JL, Victor NA,
Wanderi EW, Lust WD and Landreth GE: Peroxisome
proliferator-activated receptor-gamma ligands reduce inflammation
and infarction size in transient focal ischemia. Neuroscience.
130:685–696. 2005. View Article : Google Scholar
|
|
9
|
Pereira MP, Hurtado O, Cárdenas A, et al:
Rosiglitazone and 15-deoxy-delta12,14-prostaglandin J2 cause potent
neuroprotection after experimental stroke through noncompletely
overlapping mechanisms. J Cereb Blood Flow Metab. 26:218–229. 2006.
View Article : Google Scholar
|
|
10
|
Chu K, Lee ST, Koo JS, et al: Peroxisome
proliferator-activated receptor-gamma-agonist, rosiglitazone,
promotes angiogenesis after focal cerebral ischemia. Brain Res.
1093:208–218. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tureyen K, Kapadia R, Bowen KK, et al:
Peroxisome proliferator-activated receptor-gamma agonists induce
neuroprotection following transient focal ischemia in normotensive,
normoglycemic as well as hypertensive and type-2 diabetic rodents.
J Neurochem. 101:41–56. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhao X, Strong R, Zhang J, et al: Neuronal
PPARgamma deficiency increases susceptibility to brain damage after
cerebral ischemia. J Neurosci. 29:6186–6195. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Czapski GA, Czubowicz K and Strosznajder
RP: Evaluation of the antioxidative properties of lipoxygenase
inhibitors. Pharmacol Rep. 64:1179–1188. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Giannopoulos PF, Joshi YB, Chu J and
Praticò D: The 12–15-lipoxygenase is a modulator of
Alzheimer’s-related tau pathology in vivo. Aging Cell.
12:1082–1090. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Praticò D, Zhukareva V, Yao Y, et al:
12/15-lipoxygenase is increased in Alzheimer’s disease: possible
involvement in brain oxidative stress. Am J Pathol. 164:1655–1662.
2004. View Article : Google Scholar
|
|
16
|
Limor R, Sharon O, Knoll E, Many A,
Weisinger G and Stern N: Lipoxygenase-derived metabolites are
regulators of peroxisome proliferator-activated receptor gamma-2
expression in human vascular smooth muscle cells. Am J Hypertens.
21:219–223. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bull AW, Steffensen KR, Leers J and Rafter
JJ: Activation of PPAR gamma in colon tumor cell lines by oxidized
metabolites of linoleic acid, endogenous ligands for PPAR gamma.
Carcinogenesis. 24:1717–1722. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yao Y, Clark CM, Trojanowski JQ, Lee VM
and Pratico D: Elevation of 12/15 lipoxygenase products in AD and
mild cognitive impairment. Ann Neurol. 58:623–626. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Palluy O, Bendani M, Vallat JM and Rigaud
M: 12-lipoxygenase mRNA expression by cultured neurons. C R Acad
Sci III. 317:813–818. 1994.PubMed/NCBI
|
|
20
|
Pekcec A, Yigitkanli K, Jung JE, Pallast
S, Xing C, Antipenko A, Minchenko M, Nikolov DB, Holman TR, Lo EH
and van Leyen K: Following experimental stroke, the recovering
brain is vulnerable to lipoxygenase-dependent semaphorin signaling.
FASEB J. 27:437–445. 2013. View Article : Google Scholar :
|
|
21
|
Marcus SL, Miyata KS, Zhang B, Subramani
S, Rachubinski RA and Capone JP: Diverse peroxisome
proliferator-activated receptors bind to the peroxisome
proliferator-responsive elements of the rat hydratase/dehydrogenase
and fatty acyl-CoA oxidase genes but differentially induce
expression. Proc Natl Acad Sci USA. 90:5723–5727. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Okuno Y, Matsuda M, Miyata Y, et al: Human
catalase gene is regulated by peroxisome proliferator activated
receptor-gamma through a response element distinct from that of
mouse. Endocr J. 57:303–309. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Venkatachalam G, Kumar AP, Yue LS, Pervaiz
S, Clement MV and Sakharkar MK: Computational identification and
experimental validation of PPRE motifs in NHE1 and MnSOD genes of
human. BMC Genomics. 10(Suppl 3): S52009. View Article : Google Scholar :
|
|
24
|
Gresa-Arribas N, Viéitez C, Dentesano G,
Serratosa J, Saura J and Solà C: Modelling neuroinflammation in
vitro: a tool to test the potential neuroprotective effect of
anti-inflammatory agents. PLoS One. 7:e452272012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kapinya KJ, Löwl D, Fütterer C, et al:
Tolerance against ischemic neuronal injury can be induced by
volatile anesthetics and is inducible no synthase dependent.
Stroke. 33:1889–1898. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Choi SH, Lee DY, Kim SU and Jin BK:
Thrombin-induced oxidative stress contributes to the death of
hippocampal neurons in vivo: role of microglial NADPH oxidase. J
Neurosci. 25:4082–4090. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xing B, Xin T, Hunter RL and Bing G:
Pioglitazone inhibition of lipopolysaccharide-induced nitric oxide
synthase is associated with altered activity of p38 MAP kinase and
PI3K/Akt. J Neuroinflammation. 5:42008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mulderry PK and Dobson SP: Regulation of
VIP and other neuropeptides by c-Jun in sensory neurons:
implications for the neuropeptide response to axotomy. Eur J
Neurosci. 8:2479–2491. 1996. View Article : Google Scholar : PubMed/NCBI
|