Introduction
Acute lung injury (ALI) is a major consequence of
septic shock and contributes to the high morbidity and mortality of
sepsis (1). It has been
demonstrated that, following severe injury or infection, some
patients undergo certain responses which involve the activation of
pro-inflammatory signaling pathways and the overexpression of
inflammatory mediators that result in systemic inflammatory
responses that culminate in severe shock, multi-organ failure and
death (2). Despite extensive
investigations, the cellular and molecular mechanisms that mediate
ALI during septic shock remain largely unknown. Furthermore,
developing effective methods for preventing and/or treating
sepsis-induced ALI has proven to be difficult. A growing body of
evidence suggests that there is a link between the innate immune
response and ALI in several important disease states, including
ischemia-reperfusion injury (3),
traumatic brain injury (4) and
septic shock (5).
High mobility group box 1 (HMGB1) is an
evolutionarily conserved protein present in virtually all types of
cells where it functions to stabilize nucleosomal structure and
regulate gene expression (6).
There is recent evidence suggesting that extracellular HMGB1
functions as a danger-associated molecular pattern (7) and is actively secreted by
immunologically activated immune cells or is passively released
from pathologically damaged cells (8). The in vivo neutralization of
HMGB1 by specific antibodies has been shown to protect mice against
lethal sepsis (9), as well as
lipopolysaccharide (LPS)-induced ALI (10). In a more clinically relevant
animal model of sepsis [induced by cecal ligation puncture (CLP)],
the delayed administration of HMGB1-specific neutralizing
antibodies, beginning 24 h after CLP, was shown to dose-dependently
protect rodents from lethal sepsis (11). Moreover, the targeted inhibition
of HMGB1 expression in innate immune cells (e.g., macrophages and
dendritic cells) has been shown to attenuate systemic HMGB1
accumulation, and similarly to protect mice from sepsis (12). Taken together, these experimental
data establish extracellular HMGB1 as a critical late mediator of
experimental sepsis. In vitro studies have demonstrated that
the HMGB1-stimulated inflammatory responses may be mediated through
several pattern-recognition receptors, including the receptors for
advanced glycation end products (13), Toll-like receptor 2 (TLR2)
(14), TLR4 (15) and TLR9 (16).
Ulinastatin (UTI) is a serine protease inhibitor
that modulates innate immunity and pro-inflammatory signaling in
sepsis (17,18). The administration of UTI has been
shown to decrease the LPS-induced increase in TLR4 expression
(19), and to attenuate
sepsis-induced nuclear factor-κB (NF-κB) activity (20). Previous studies have demonstrated
that UTI treatment improves the survival of mice with septis mice
(21), and inhibits LPS-induced
ALI in mice (19,20). Therefore, we hypothesized that UTI
may downregulate HMGB1 expression and that the inhibition of HMGB1
expression may be associated with the inhibition of TLR2/4 and
NF-κB activation by UTI during sepsis. Thus, the aim of the present
study was to determine whether UTI post-treatment attenuates ALI by
the inhibition of HMGB1 expression in rats and human alveolar
epithelial cells.
Materials and methods
Materials
LPS (Escherichia coli 055:B5) was obtained
from Sigma (St. Louis, MO, USA). The HMGB1, tumor necrosis factor-α
(TNF-α), interleukin-6 (IL-6) and myeloperoxidase (MPO)
enzyme-linked immunosorbent assay (ELISA) kits were obtained from
Invitrogen (Carlsbad, CA, USA). Anti-TLR2 (D-17, sc-12504),
anti-TLR4 (M-16, sc-12511), anti-p-NF-κB p65 (A-8, sc-166748) and
anti-NF-κB p65 (F-6, sc-8008) antibodies were obtained from Santa
Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Anti-IκB-α and
anti-p-IκB-α antibodies were obtained from Cell Signaling
Technology, Inc. (Beverly, MA, USA).
Animals
Adult male Sprague-Dawley rats (8–10 weeks of age,
weighing 250–300 g) were provided by the Experimental Animal Center
of Harbin Medical University, kept in a 12 h dark/12 h light cycle
in a temperature- and humidity-controlled room and fed standard
laboratory diet and provided with water. All procedures were
performed in accordance with the Declaration of Helsinki of the
World Medical Association. The study was approved by the Ethics
Committee of the First Affiliated Hospital of Harbin Medical
University, Harbin, China.
Animal experimental design
The animals were randomly divided into 6 groups and
each group contained 20 rats: i) the control group [given normal
saline (NS)]; ii) the UTI (20,000 U/kg) group (administered 20,000
U/kg UTI); iii) the LPS group (rats received 5 mg/kg LPS by
intratracheal instillation); iv) the LPS + UTI (5,000 U/kg) group
(rats received LPS plus 5,000 U/kg UTI); v) the LPS + UTI (10,000
U/kg) group (rats received LPS plus 10,000 U/kg UTI) group; vi) LPS
+ UTI (20,000 U/kg) group (rats received LPS plus 20,000 U/kg UTI).
LPS (5 mg/kg; to induce ALI), or the vehicle (NS) were
intratracheally administered, as previously described (22). UTI (5,000, 10,000 or 20,000 U/kg)
was intraperitoneally injected 30 min after the LPS administration.
The doses of these drugs were used based on previous studies
(17,19) and our preliminary experiments
(data not shown). At 24 h after the LPS administration, the rats
were sacrificed under sodium pentobarbitone (45 mg/kg body weight
ip, Sigma) anaesthesia according to the guidelines for euthanasia
in the Guide for Care and Use of Laboratory Animals, and the
bronchoalveolar lavage fluid (BALF) samples were collected for
counting and classification. Lung tissues were snap-frozen in
liquid nitrogen, and stored at −80°C for later analysis. To clarify
the role of HMGB1, TLR2/4 and NF-κB in LPS-induced pulmonary
inflammation, the rats were administered an intraperitoneal
injection of UTI (20,000 U/kg; Sigma), anti-HMGB1 antibody
(MABE148; 10 mg/kg), anti-TLR2/4 antibody (MABF84, MABF85; 10
mg/kg) (both from Calbiochem), or the NF-κB inhibitor, PDTC (10
mg/kg; Sigma), 30 min after the LPS administration, as previously
described (23). BALF was
collected at 24 h after the LPS administration to measure the
inflammatory markers.
Cell culture and treatment
The A549 human alveolar epithelial cells were
obtained from the Typical Species Preservation Center of Wuhan
University (Wuhan, Hubei, China). The A549 cells were seeded into
6-well plates and were cultured in Dulbecco's modified Eagle's
medium (DMEM) supplemented with 10% FBS, 100 U/ml penicillin and
100 µg/ml streptomycin at 37°C in a humidified atmosphere
containing 5% CO2. The cells were grown until 70%
confluent before being subjected to the different treatments. The
cells were divided into 4 groups as follows: i) the control group
(NS); ii) the UTI (100 U/ml) group; iii) the LPS group (stimulated
with 1 µg/ml LPS); and iv) the LPS + UTI (100 U/ml) group. The A549
cells were treated with UTI (100 U/ml) 24 h after the LPS (1 µg/ml)
stimulation. The doses of these drugs used were based on a previous
study (24) and our preliminary
experiments (data not shown). The cells were harvested at 24 h
after the addition of LPS to analyze the expression levels of TLR2,
TLR4, p-NF-κB p65 and p-IκB-α.
Lung wet/dry weight ratio in vivo
The water content of the lungs was determined by
calculating the wet/dry weight ratio of the lung tissues. The
inferior lobe of the right lung was excised, rinsed briefly in
phosphate-buffered saline (PBS), blotted and then weighed to obtain
the 'wet' weight. The lung was then dried at 80°C for 72 h to
obtain the 'dry' weight. The wet/dry ratio was calculated by
dividing the wet weight by the dry weight.
Determination of bronchoalveolar lavage
proteins and cell counts
Bronchoalveolar lavage (BAL) was performed by
intratracheal injection of 5 ml ice-cold PBS followed by gentle
aspiration. The recovery ratio of the fluid was ~90%. Then the
recovered fluid was pooled and centrifuged at 1,200 × g for 10 min
at 4°C. Supernatants were preserved for the measurement of total
protein concentration by the Bradford method with bovine serum
albumin (BSA) as a standard. The cell pellet was re-suspended in 50
μl PBS, and total cells recovered in BALF were counted. The
cell differentiation was determined for 200 cells by examination of
the hematoxylin and eosin (H&E)-stained smears.
Cytokine measurements
The levels of HMGB1, TNF-α and IL-6 in the
supernatants of BALF were measured using commercially available
ELISA kits (Invitrogen) according to the manufacturer's
instructions. After incubation for 1 h at 37°C, the mixture of
100-ml sample and 100-ml rat HMGB1 (or TNF-α, IL-6) biotin
conjugate was added with 100 ml streptavidin–horseradish peroxidase
and incubated for another 30 min. Then, 100 ml stabilized chromogen
was added to 100 ml stop buffer, and the adsorption value of the
mixture at 450 nm was measured.
MPO activity assay
The lung tissues were homogenized in
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES; pH 8.0)
containing 0.5% cetyltrimethyl ammonium bromide and subjected to
three freeze-thaw cycles. The homogenate was then centrifuged (4°C,
12,000 × g for 30 min). The MPO activity was assayed using a
commercially available ELISA kit (Invitrogen). The samples were
diluted in phosphate citrate buffer (pH 5.0) and the absorbance of
the sample was measured at 460 nm using a microplate reader (Model
550; Bio-Rad Laboratories, Hercules, CA, USA). The specific of MPO
activity in the lungs is expressed as unit/gram of the tissue.
Histological examination
The right lobes were excised and fixed with 10%
neutral phosphate-buffered formalin, imbedded in paraffin and
sliced. Following H&E staining, pathological changes of lung
tissues were observed under a light microscope (BXFM; Olympus,
Tokyo, Japan). The standard lung injury score performed by a
blinded pathologist to objectively quantify the lung injury.
Western blot analysis
Protein concentrations were determined using a BCA
protein assay kit, and 20 μg proteins were loaded per well
on a 10% sodium dodecylsulfate-polyacrylamide gel and transferred
onto polyvinylidene difluoride membranes (Millipore, Bedford, MA,
USA). After being blocked for 3 h in Tris-buffered saline with 0.1%
Tween-20 (TBST) and 3% BSA, the membranes were incubated overnight
at 4°C with primary antibodies in TBST containing 3% BSA. The
membranes were then washed and incubated with horseradish
peroxidase-conjugated secondary antibodies (1662408; Bio-Rad
Laboratories) in TBST for 2 h and developed using an ECL detection
system (Amersham, Buckinghamshire, UK). The density of the bands on
the membranes was scanned and analyzed using an image analyzer (Lab
Works Software; UVP Bioimaging Systems Upland, CA, USA).
Protein extraction
For preparing whole cell lysates, the cells were
lysed in radioimmune precipitation assay (RIPA) buffer supplemented
with protease inhibitor cocktail (Roche Diagnostics). Nuclear and
cytoplasmic fractionations were created using the Proteo JET™
Cytoplasmic and Nuclear Protein Extraction kit (Fermentas Life
Science, St. Leon-Rot, Germany) according to the instructions
provided by the manufacturer.
Electrophoretic mobility shift assay
(EMSA)
Nuclear extracts were prepared as described above.
Oligonucleotides corresponding to the NF-κB (5′-AGTTGAGGGGACTTTCCCA
GGC-3′) binding site consensus sequences were synthesized and
end-labeled with biotin by Invitrogen. EMSAs were performed using
the LightShift chemiluminescent EMSA kit (Pierce, Rockford, IL,
USA).
Statistical analysis
Data are presented as the means ± standard deviation
(SD) of results obtained from 20 rats in each group in vivo
and 3 replicates in vitro. Statistical analysis of the
results was carried out by one-way analysis of variance (ANOVA)
followed by Tukey's post hoc test with SPSS 11.0 software (SPSS,
Inc., Chicago, IL, USA). Differences between each group were
assessed by two-way analysis of variance followed by Newman-Keuls
tests. P-values <0.05 were considered to indicate statistically
significant differences.
Results
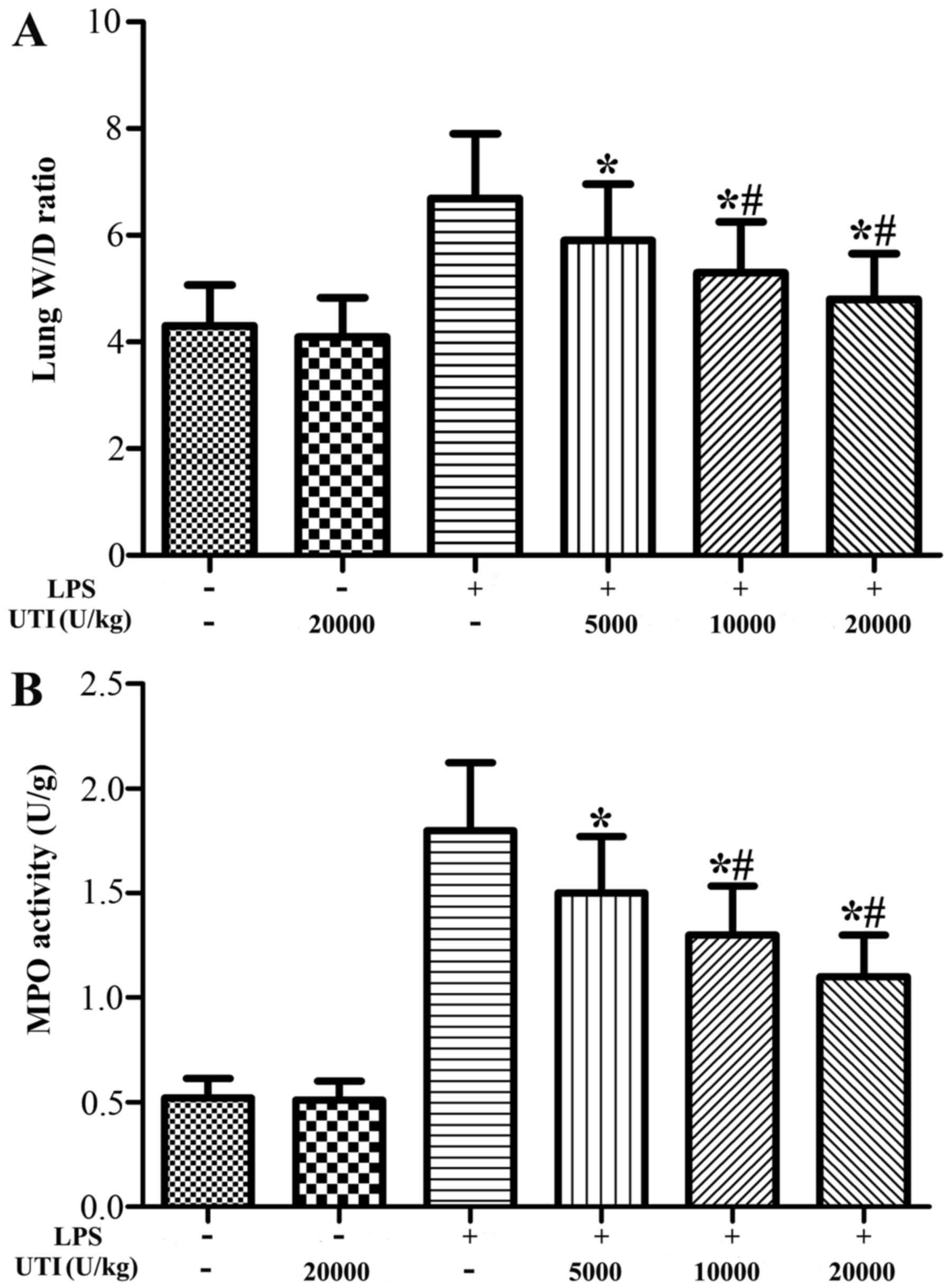
Effects of UTI on the lung wet/dry weight
ratio and MPO activity in lung tissues
To evaluate the LPS-induced changes in pulmonary
vascular permeability and neutrophil infiltration, the lung wet/dry
weight ratio and MPO activity in the lung tissues were analyzed.
The lung wet/dry ratio and MPO activity in the lung tissues were
significantly increased after the LPS challenge compared with the
control group (Fig. 1). However,
UTI post-treatment obviously attenuated the wet/dry ratio and MPO
activity in lung tissues in a dose-dependent manner (P<0.05)
(Fig. 1). There were no
significant differences in the lung wet/dry ratio and MPO activity
in lung tissues between the control and UTI group (UTI only).
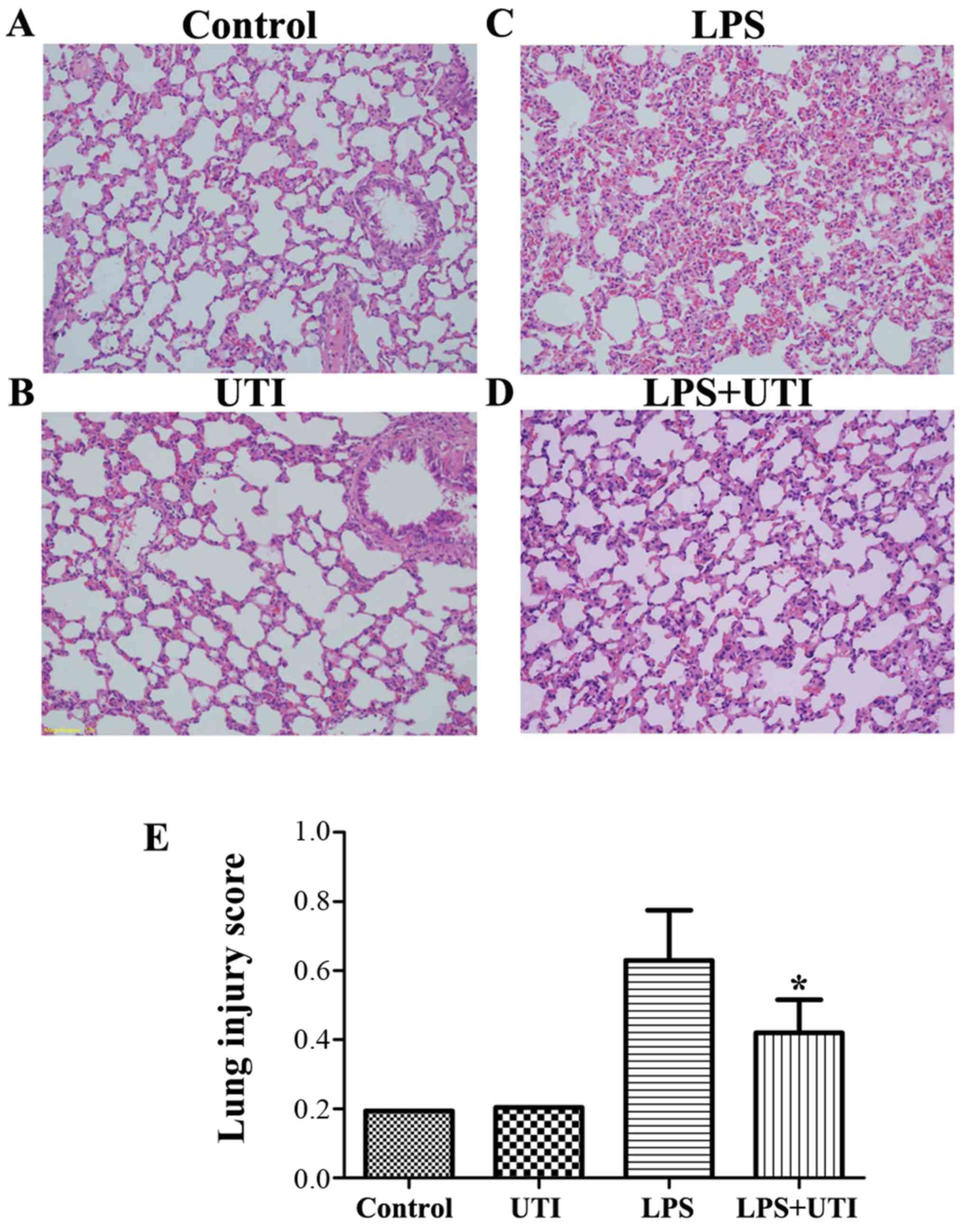
Effects of UTI on LPS-mediated lung
histopathological changes
To evaluate the histological changes following UTI
post-treatment in LPS-challenged rats, the lung tissues were
harvested at 24 h after the administration of LPS. The lung tissues
from the control group exhibited a normal structure and no
histopathological changes were observed (Fig. 2A). In the LPS group, histological
examination revealed severe lung destruction, as indicated by
H&E staining, which manifested as severe pulmonary edema,
hemorrhagia in the stroma, alveolar collapse and mass inflammatory
cell infiltration (Fig. 2C).
However, UTI post-treatment effectively alleviated the destruction
of lung structure (Fig. 2D).
Furthermore, the UTI post-treatment group had a significantly lower
lung injury score than the control group (P<0.05) (Fig. 2E).
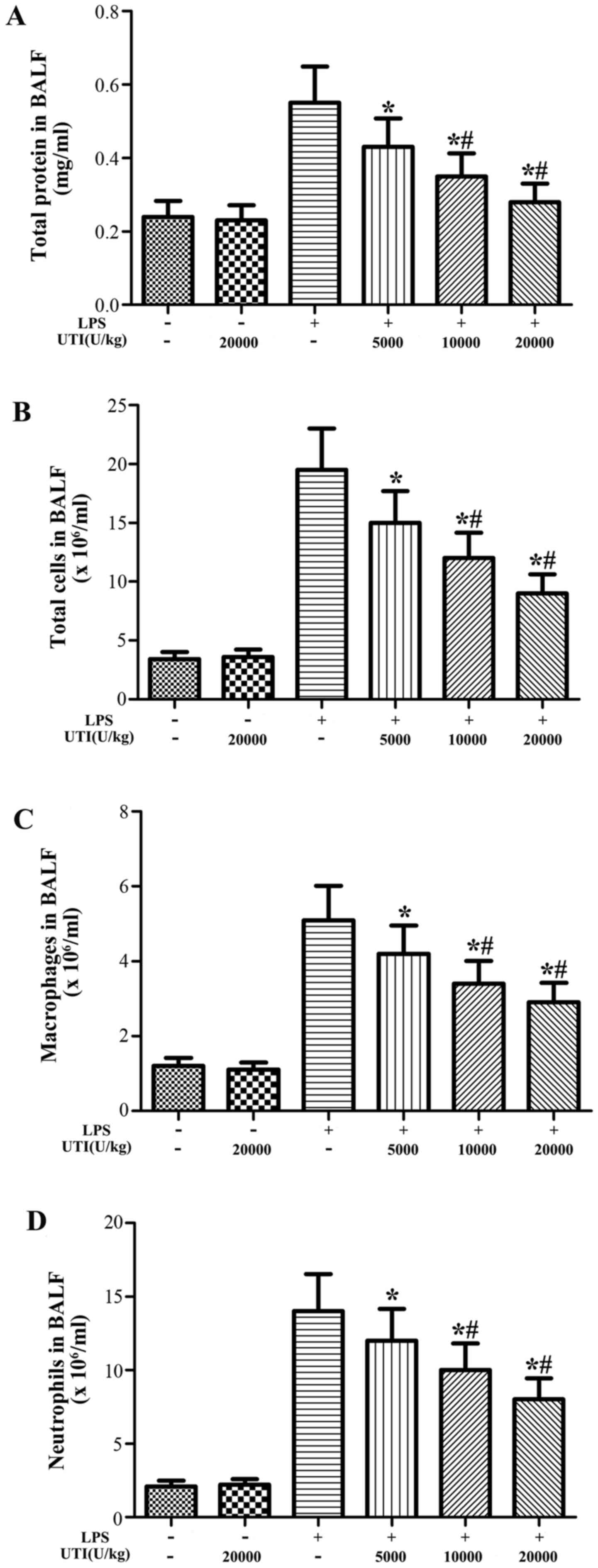
Effects of UTI on the concentration of
total protein and the inflammatory cell counts in BALF
To examine the effects of UTI on LPS-induced
pulmonary inflammation, the concentration of total protein and the
numbers of inflammatory cells, such as neutrophils and macrophages,
in BALF were analyzed at 24 h after the LPS injection. After the
LPS challenge, the concentration of total protein and the numbers
of total cells, neutrophils and macrophages significantly increased
compared with the control group (Fig.
3). However, this increase was apparently attenuated by UTI
post-treatment in a dose-dependent manner (P<0.05) (Fig. 3).
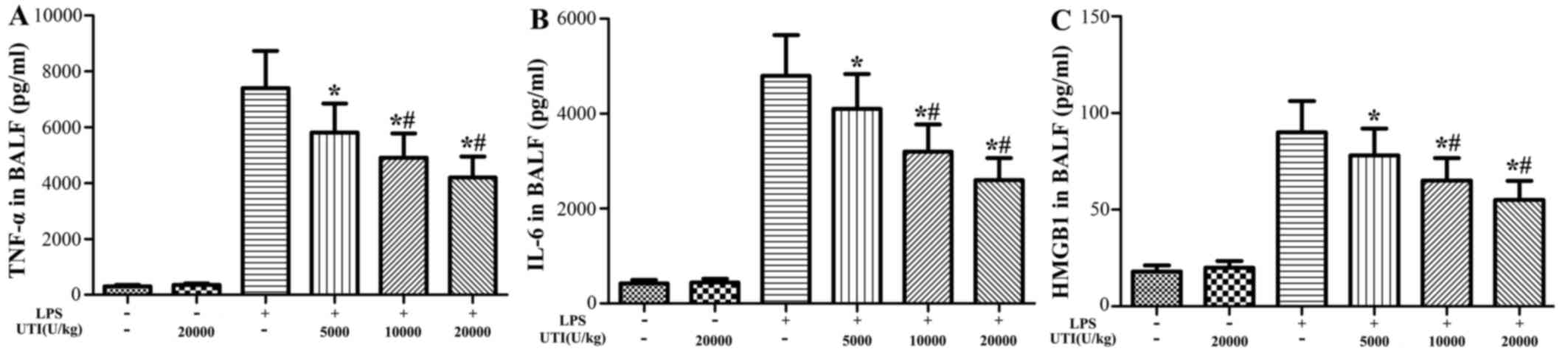
Effects of UTI on the concentrations of
HMGB1, TNF-α and IL-6 in BALF
To further evaluate the anti-inflammatory effects of
UTI, the concentrations of the pro-inflammatory cytokines, TNF-α,
IL-6 and HMGB1, in BALF were analyzed at 24 h after the LPS
administration by ELISA. The concentrations of TNF-α, IL-6 and
HMGB1 in BALF significantly increased in the LPS group (Fig. 4). However, UTI post-treatment
markedly decreased the levels of TNF-α, IL-6 and HMGB1 compared to
those in the LPS group in a dose-dependent manner (P<0.05)
(Fig. 4).
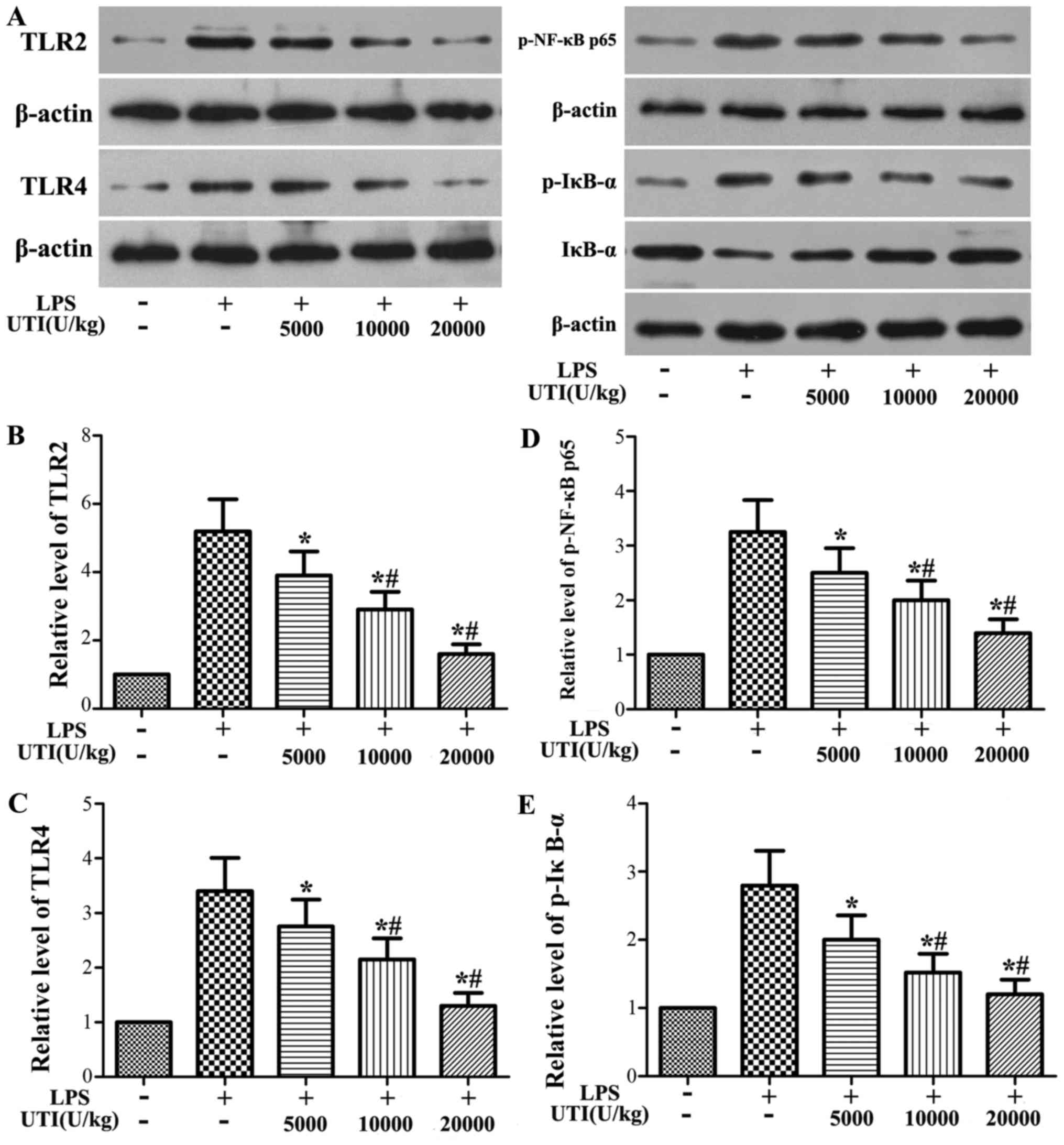
Effects of UTI on the expression levels
of TLR2/4 and the activation of NF-κB in lung tissues
Western blot analysis was used to determine the
expression levels of TLR2/4 and the phosphorylation of NF-κB p65
and IκB-α. The protein levels of TLR2/4 and the phosphorylation
levels of NF-κB p65 and IκB-α significantly increased in the LPS
group at 24 h after the LPS administration (Fig. 5). However, this increase was
apparently attenuated by UTI post-treatment in a dose-dependent
manner (P<0.05) (Fig. 5).
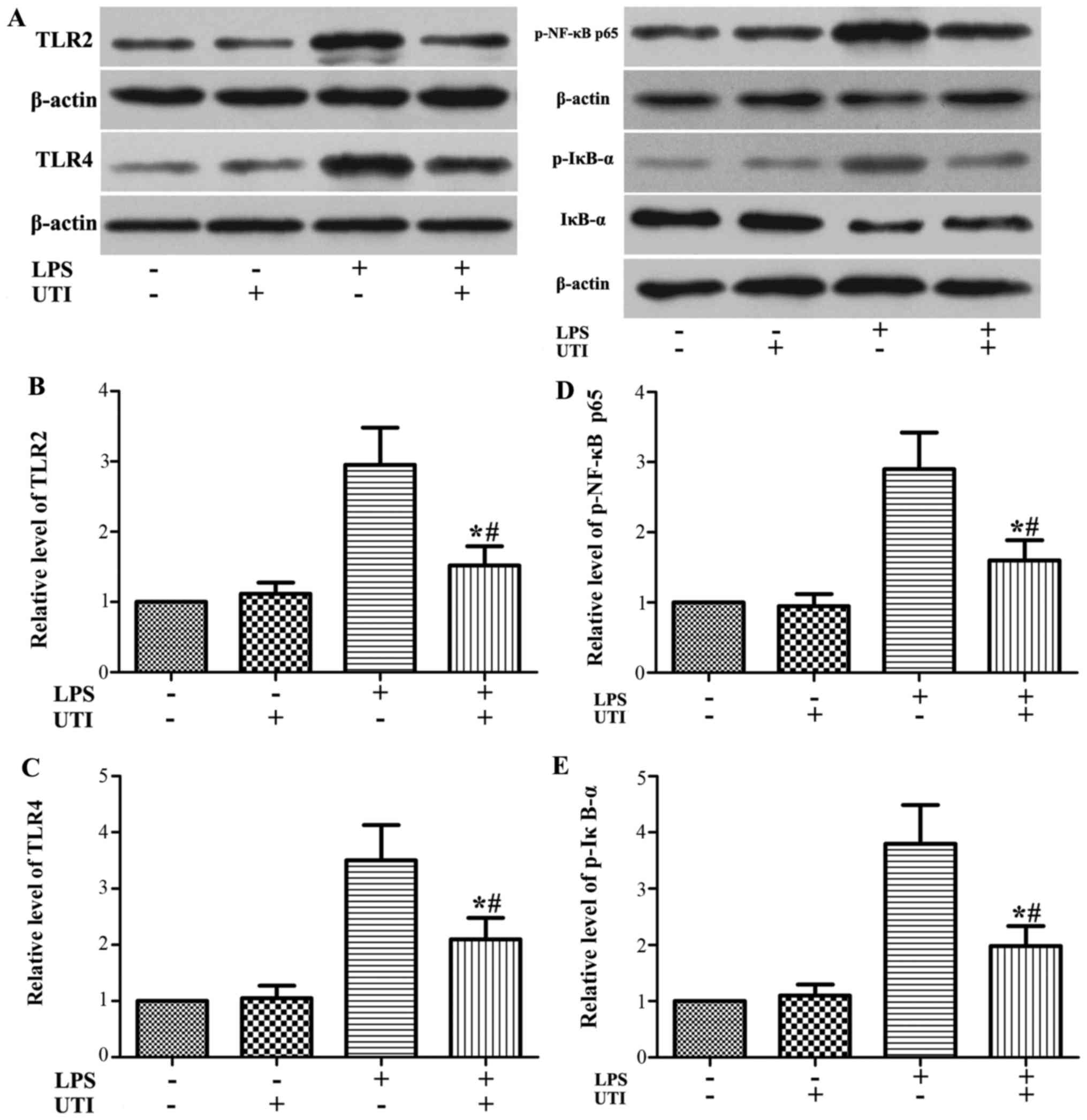
Effects of UTI on the expression levels
of TLR2/4 and the activation of NF-κB in A549 alveolar epithelial
cells
To further examine the anti-inflammatory mechanistic
basis of UTI, we examined the effects of UTI on TLR2/4 expression
and NF-κB activation in A549 alveolar epithelial cells. Similar to
what we observed in the animal experiments, the expression levels
of TLR2/4 and the phosphorylation of NF-κB p65 and IκB-α were
markedly increased in the cells stimulated with LPS. However, these
increases were markedly inhibited by UTI post-treatment (P<0.05)
(Fig. 6).
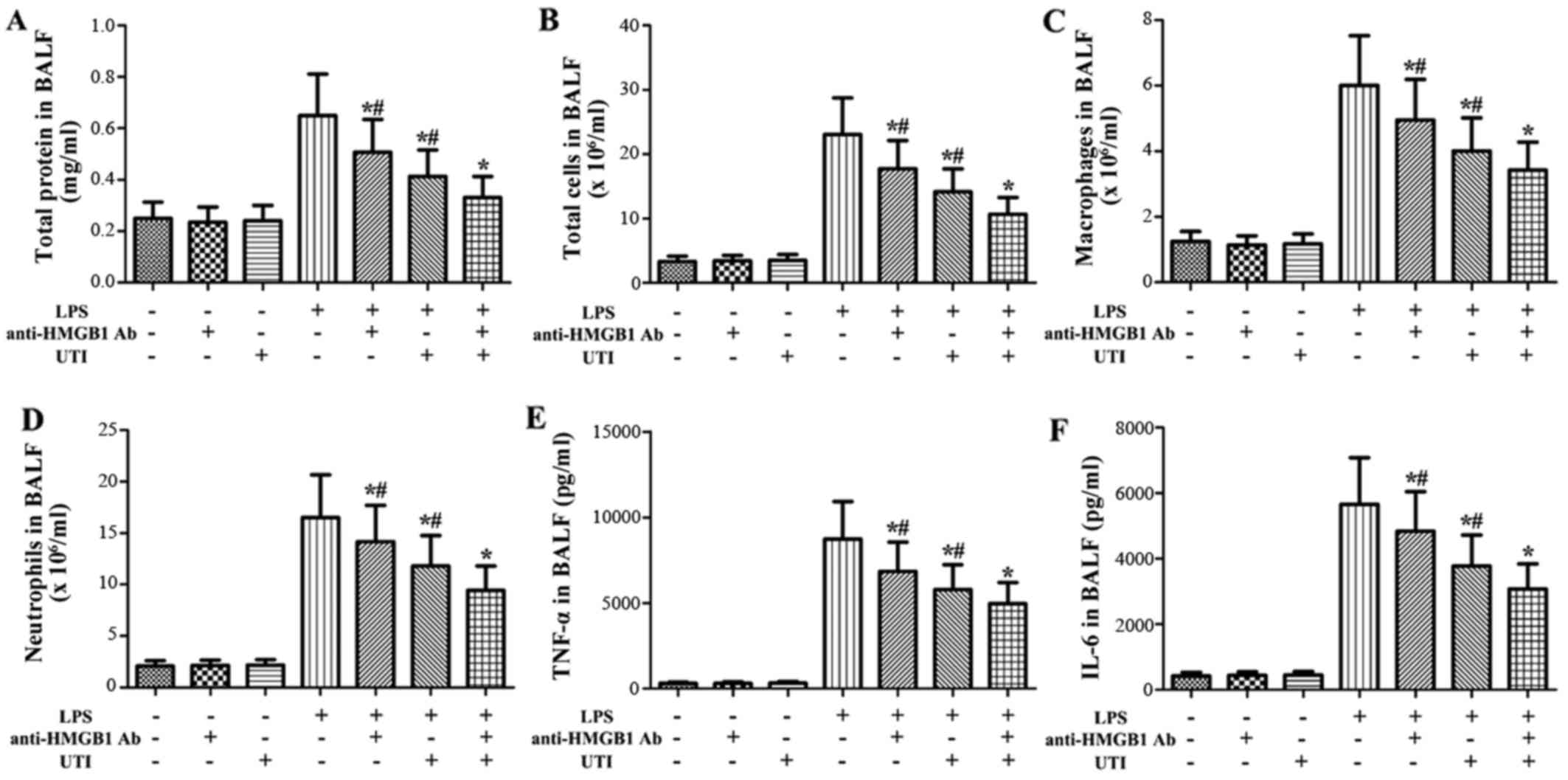
Effects of anti-HMGB1 antibody and UTI on
inflammatory markers in BALF of LPS-induced ALI
To clarify the role of HMGB1 in LPS-induced
pulmonary inflammation, the rats were administered an
intraperitoneal injection of UTI (20,000 U/kg) or anti-HMGB1
antibody (10 mg/kg) 30 min after the LPS administration. BALF was
collected at 24 h after the LPS administration to measure the
inflammatory markers. After the LPS challenge, the levels of
inflammatory markers significantly increased compared with the
control group (Fig. 7). However,
this increase was markedly attenuated by anti-HMGB1 anti body
(P<0.05) (Fig. 7).
Furthermore, anti-HMGB1 antibody significantly enhanced the
anti-inflammatory effects of UTI on inflammatory markers in BALF
from rats with LPS-induced ALI (P<0.05) (Fig. 7).
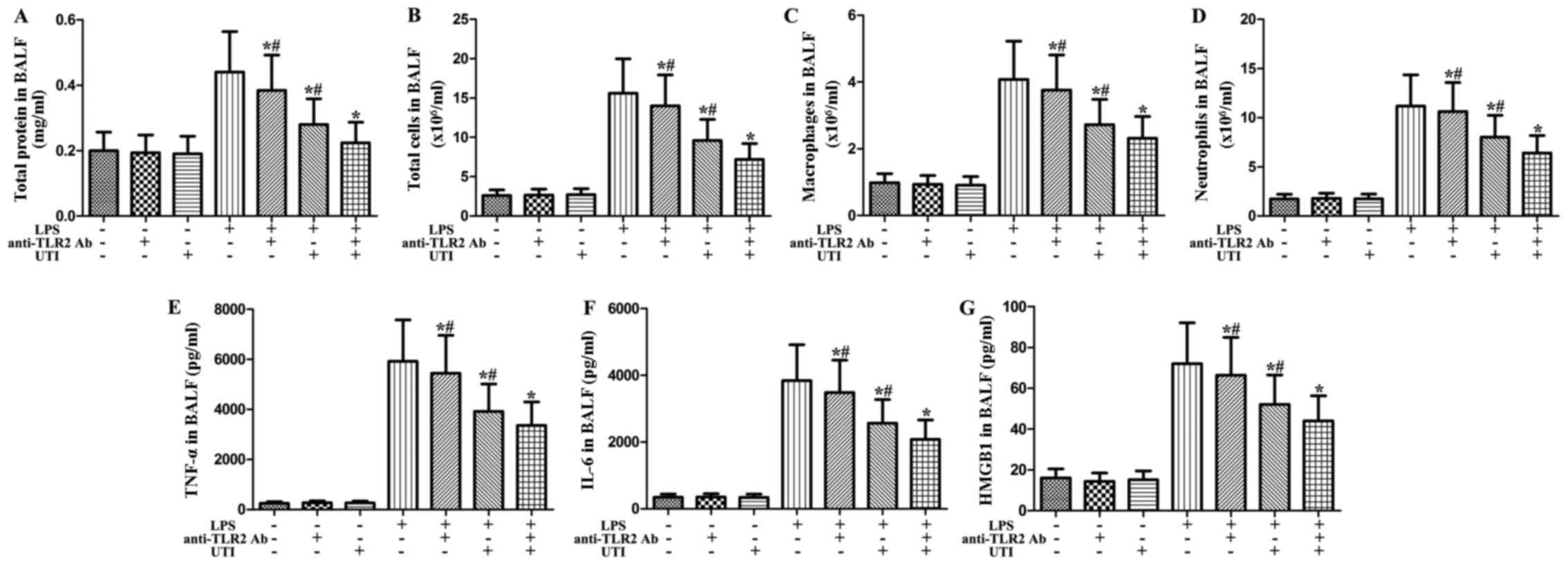
Effects of anti-TLR2 antibody and UTI on
inflammatory markers in BALF from rats with LPS-induced ALI
To clarify the role of TLR2 in LPS-induced pulmonary
inflammation, rats were administered an intraperitoneal injection
of UTI (20,000 U/kg) or anti-TLR2 antibody (10 mg/kg) 30 min after
the LPS administration. BALF was collected at 24 h after the LPS
administration to measure the inflammatory markers. After the LPS
challenge, the levels of inflammatory markers significantly
increased compared with the control group (Fig. 8). However, this increase was
markedly attenuated by anti-TLR2 antibody (P<0.05) (Fig. 8). Furthermore, anti-TLR2 antibody
significantly enhanced the anti-inflammatory effects of UTI on
inflammatory markers in BALF from rats with LPS-induced ALI
(P<0.05) (Fig. 8).
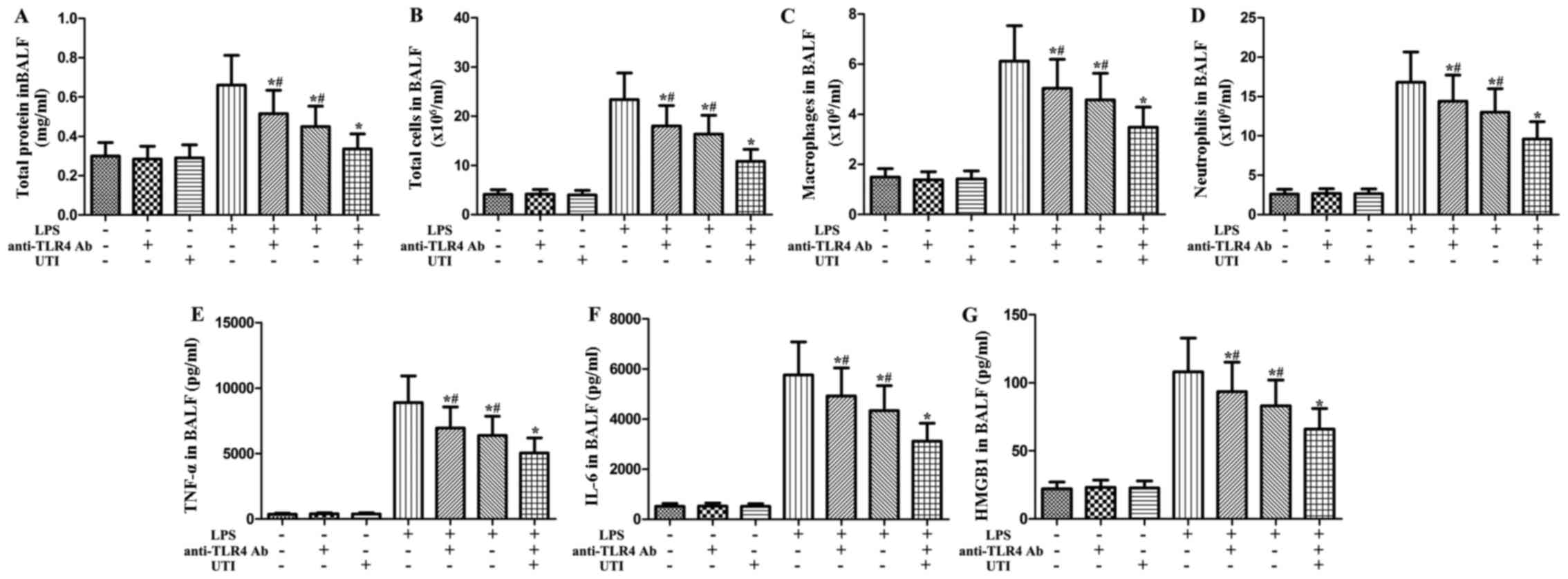
Effects of anti-TLR4 antibody and UTI on
inflammatory markers in BALF from rats with LPS-induced ALI
To clarify the role of TLR4 in LPS-induced pulmonary
inflammation, rats were administered an intraperitoneal injection
of UTI (20,000 U/kg) or anti-TLR4 antibody (10 mg/kg) 30 min after
the LPS administration. BALF was collected at 24 h after the LPS
administration to measure the levels of inflammatory markers. As
shown in Fig. 9, after the LPS
challenge, the levels of inflammatory markers significantly
increased compared with the control group. However, this increase
was markedly attenuated by anti-TLR4 antibody (P<0.05) (Fig. 9). Furthermore, anti-TLR4 antibody
significantly enhanced the anti-inflammatory effects of UTI on
inflammatory markers in BALF from rats with LPS-induced ALI
(P<0.05) (Fig. 9).
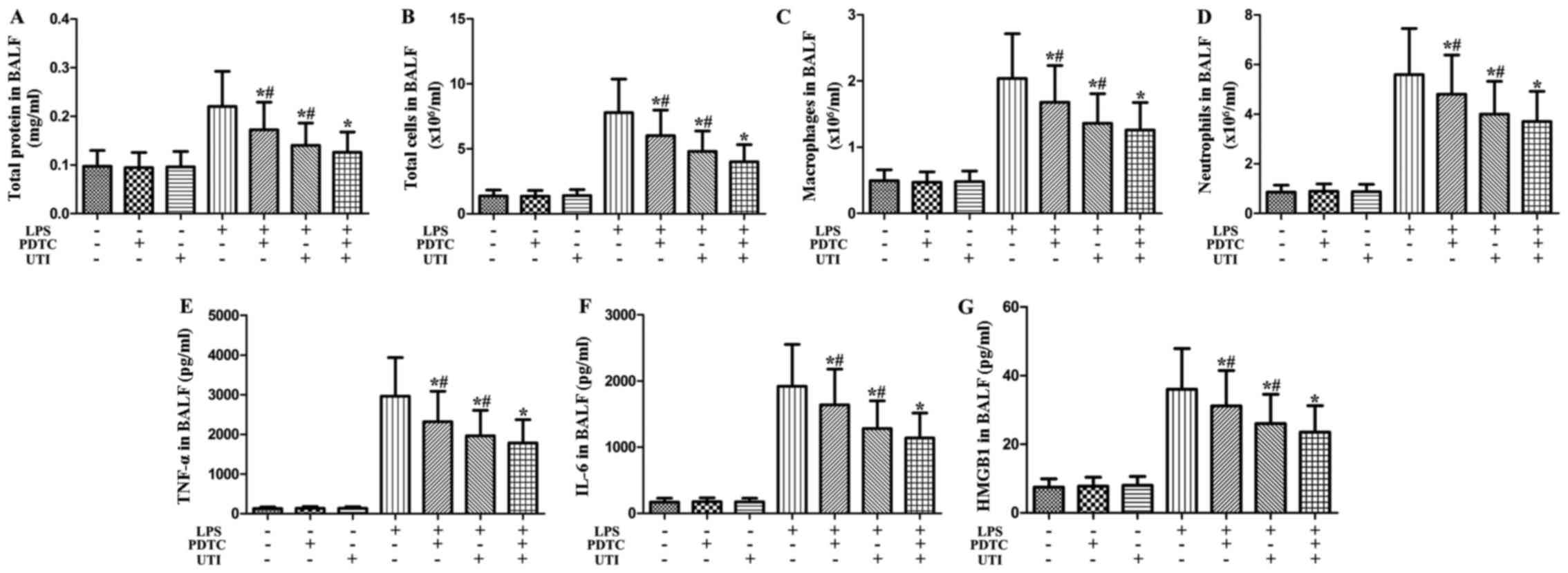
Effects of NF-κB inhibitor and UTI on
inflammatory markers in BALF from rats with LPS-induced ALI
To clarify the role of NF-κB in LPS-induced
pulmonary inflammation, rats were administered an intraperitoneal
injection of UTI (20,000 U/kg) or NF-κB inhibitor PDTC (10 mg/kg)
30 min after the LPS administration. BALF was collected at 24 h
after the LPS administration to measure the inflammatory markers.
After the LPS challenge, the levels of inflammatory markers
significantly increased compared with the control group (Fig. 10). However, this increase was
markedly attenuated by the NF-κB inhibitor, PDTC (P<0.05)
(Fig. 10). Furthermore, the
NF-κB inhibitor, PDTC, significantly enhanced the anti-inflammatory
effects of UTI on inflammatory markers in BALF from rats with
LPS-induced ALI (P<0.05) (Fig.
10).
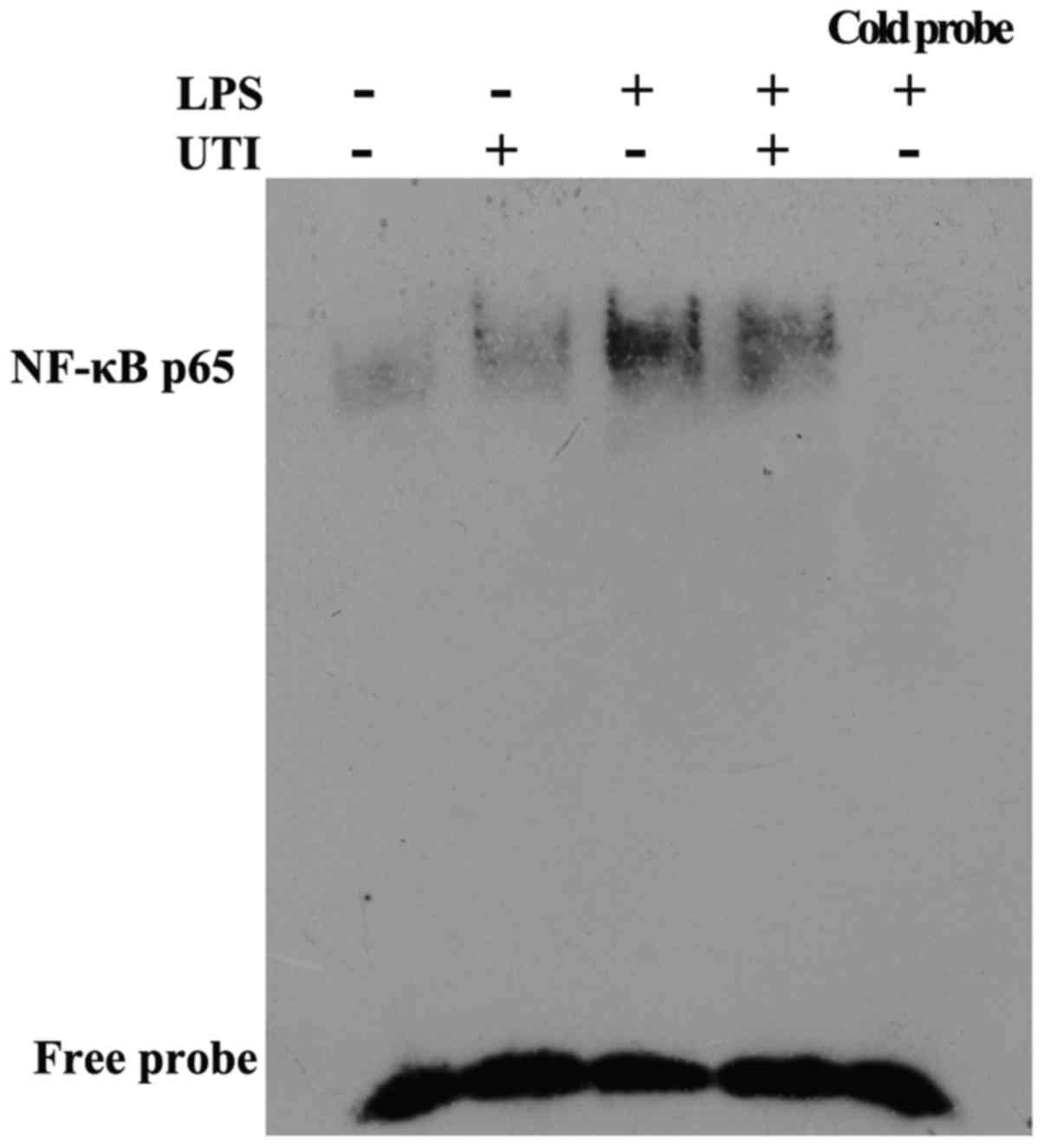
UTI post-treatment inhibits NF-κB DNA
binding activity in lung tissues
To determine the effects of UTI on transcription
factor signaling pathways that may mediate LPS-induced inflammatory
cytokine production, EMSA was performed. The rats were
admininstered an intraperitoneal injection of UTI (20,000 U/kg) 30
min after the LPS administration. At 24 h after the LPS
administration, nuclear extracts obtained from lung tissues were
collected. Post-treatment with UTI markedly reduced the LPS-induced
DNA-binding activity of NF-κB (Fig.
11).
Discussion
In this study, we evaluated the protective effects
of UTI in a model of LPS-induced ALI in rats and human alveolar
epithelial cells. Consistent with previously published data
(20), we demonstrated that UTI
post-treatment attenuated lung inflammatory injury following LPS
challenge, as revealed by the decreased elevation of the lung wet
to dry weight ratio, total cells, neutrophils, macrophages and MPO
activity, associated with reduced lung histological damage. In
addition, we also found that UTI post-treatment markedly inhibited
the release of HMGB1 and other pro-inflammatory cytokines in a rat
model of LPS-induced ALI. Furthermore, UTI significantly inhibited
the LPS-induced increase in TLR2/4 protein expression and NF-κB
activation in vivo and in vitro.
Previous studies have demonstrated that UTI inhibits
the inflammatory response and mitigates sepsis-induced ALI
(18,20). As is already known, the etiologies
of ALI are complex, and additional mechanisms through which UTI
exerts protective effects against LPS-induced lung injury need to
be further investigated. Evidence suggests that HMGB1 plays a
critical role in the progression of ALI. HMGB1-based therapeutic
strategies may be more effective in ALI, as HMGB1 is a late
inflammatory mediator, and this may provide a much broader
intervention window. Therefore, the current study aimed to examine
the hypothesis that UTI post-treatment alleviates LPS-induced ALI
by preventing the expression of HMGB1.
Clinical and experimental studies have indicated
that ALI is an early and fatal complication of septic shock
(1) and that the TLR2/4-mediated
NF-κB activation signaling pathway may be an early molecular event
leading to ALI during septic shock (25,26). Previous studies have demonstrated
that UTI significantly improved the survival of mice with septis
(21) and the mechanisms involved
the downregulation of the expression of TLR4 (19). To evaluate our hypothesis, we
examined pulmonary inflammation in LPS-induced ALI with or without
UTI post-treatment. We observed that the pulmonary inflammation was
significantly increased in a rat model of LPS-induced ALI. However,
UTI post-treatment significantly inhibited pulmonary inflammation
by inhibiting the expression of TLR2/4 in a rat model of
LPS-induced ALI. It has been demonstrated that the administration
of UTI significantly blunted NF-κB activation in rats with septis
(18). NF-κB is a critical
transcription factor in TLR-mediated signaling pathways (27) and plays a critical role in the
regulation of the expression of a number of genes, including
inflammatory cytokines, such as HMGB1, TNF-α and IL-1β (28–31). In this study, we demonstrated that
UTI post-treatment downregulated HMGB1 expression in a rat model of
LPS-induced ALI and that the inhibition of HMGB1 expression was
associated with the inhibition of TLR2/4 and NF-κB activation by
UTI post-treatment.
UTI is a synthetic glycoprotein with a molecular
weight of 67 kDa, first purified from human urine (32). It is frequently used clinically
for the treatment of shock (33)
and acute pancreatitis (34).
According to the pharmaceutical reference of UTI, side-effects have
been reported in 74/8,710 patients (0.8%) at doses up to of
3×105 U/day (32).
These included abnormalities in serum tests, such as elevations in
liver enzyme levels, abdominal symptoms, skin rashes and angialgia
after intravenous administrations (32). In this study, we found that UTI
post-treatment (5,000, 10,000 or 20,000 U/kg) attenuated
LPS-induced ALI in a dose-dependent manner. Due to the toxicity of
systemic UTI, we consider that UTI post-treatment at 10,000 U/kg
was effective and safe; however, this should be explored further in
larger animal studies and more relevant sepsis models, as well as
eventually, in human trials.
In conclusion, the present study provides evidence
that UTI post-treatment attenuates LPS-induced ALI accompanied with
a decreased HMGB1 expression in a model of LPS-induced ALI. The
mechanisms responsible for these effects involve, at least in part,
the downregulation of TLR2/4 expression and the inhibition of the
NF-κB pathway.
References
|
1
|
Su CF, Kao SJ and Chen HI: Acute
respiratory distress syndrome and lung injury: Pathogenetic
mechanism and therapeutic implication. World J Crit Care Med.
1:50–60. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Castellheim A, Brekke OL, Espevik T,
Harboe M and Mollnes TE: Innate immune responses to danger signals
in systemic inflammatory response syndrome and sepsis. Scand J
Immunol. 69:479–491. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jing H, Yao J, Liu X, Fan H, Zhang F, Li
Z, Tian X and Zhou Y: Fish-oil emulsion (omega-3 polyunsaturated
fatty acids) attenuates acute lung injury induced by intestinal
ischemia-reperfusion through Adenosine 5′-monophosphate-activated
protein kinase-sirtuin1 pathway. J Surg Res. 187:252–261. 2014.
View Article : Google Scholar
|
|
4
|
Song Z, Zhao X, Gao Y, Liu M, Hou M, Jin H
and Cui Y: Recombinant human brain natriuretic peptide ameliorates
trauma-induced acute lung injury via inhibiting JAK/STAT signaling
pathway in rats. J Trauma Acute Care Surg. 78:980–987. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lin WC, Chen CW, Huang YW, Chao L, Chao J,
Lin YS and Lin CF: Kallistatin protects against sepsis-related
acute lung injury via inhibiting inflammation and apoptosis. Sci
Rep. 5:124632015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lu B, Wang C, Wang M, Li W, Chen F, Tracey
KJ and Wang H: Molecular mechanism and therapeutic modulation of
high mobility group box 1 release and action: An updated review.
Expert Rev Clin Immunol. 10:713–727. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Agalave NM and Svensson CI: Extracellular
high-mobility group box 1 protein (HMGB1) as a mediator of
persistent pain. Mol Med. 20:569–578. 2015.
|
|
8
|
Wang H, Ward MF and Sama AE: Targeting
HMGB1 in the treatment of sepsis. Expert Opin Ther Targets.
18:257–268. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang H, Bloom O, Zhang M, Vishnubhakat JM,
Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, et
al: HMG-1 as a late mediator of endotoxin lethality in mice.
Science. 285:248–251. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lutz W and Stetkiewicz J: High mobility
group box 1 protein as a late-acting mediator of acute lung
inflammation. Int J Occup Med Environ Health. 17:245–254.
2004.PubMed/NCBI
|
|
11
|
Yang H, Ochani M, Li J, Qiang X, Tanovic
M, Harris HE, Susarla SM, Ulloa L, Wang H, DiRaimo R, et al:
Reversing established sepsis with antagonists of endogenous
high-mobility group box 1. Proc Natl Acad Sci USA. 101:296–301.
2004. View Article : Google Scholar :
|
|
12
|
Ye C, Choi JG, Abraham S, Wu H, Diaz D,
Terreros D, Shankar P and Manjunath N: Human macrophage and
dendritic cell-specific silencing of high-mobility group protein B1
ameliorates sepsis in a humanized mouse model. Proc Natl Acad Sci
USA. 109:21052–21057. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
van Beijnum JR, Buurman WA and Griffioen
AW: Convergence and amplification of toll-like receptor (TLR) and
receptor for advanced glycation end products (RAGE) signaling
pathways via high mobility group B1 (HMGB1). Angiogenesis.
11:91–99. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Park JS, Gamboni-Robertson F, He Q,
Svetkauskaite D, Kim JY, Strassheim D, Sohn JW, Yamada S, Maruyama
I, Banerjee A, et al: High mobility group box 1 protein interacts
with multiple Toll-like receptors. Am J Physiol Cell Physiol.
290:C917–C924. 2006. View Article : Google Scholar
|
|
15
|
Kim S, Kim SY, Pribis JP, Lotze M, Mollen
KP, Shapiro R, Loughran P, Scott MJ and Billiar TR: Signaling of
high mobility group box 1 (HMGB1) through toll-like receptor 4 in
macrophages requires CD14. Mol Med. 19:88–98. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hirata Y, Kurobe H, Higashida M, Fukuda D,
Shimabukuro M, Tanaka K, Higashikuni Y, Kitagawa T and Sata M:
HMGB1 plays a critical role in vascular inflammation and lesion
formation via toll-like receptor 9. Atherosclerosis. 231:227–233.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cao YZ, Tu YY, Chen X, Wang BL, Zhong YX
and Liu MH: Protective effect of Ulinastatin against murine models
of sepsis: Inhibition of TNF-α and IL-6 and augmentation of IL-10
and IL-13. Exp Toxicol Pathol. 64:543–547. 2012. View Article : Google Scholar
|
|
18
|
Wang N, Liu X, Zheng X, Cao H, Wei G, Zhu
Y, Fan S, Zhou H and Zheng J: Ulinastatin is a novel candidate drug
for sepsis and secondary acute lung injury, evidence from an
optimized CLP rat model. Int Immunopharmacol. 17:799–807. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gao C, Li R and Wang S: Ulinastatin
protects pulmonary tissues from lipopolysaccharide-induced injury
as an immunomodulator. J Trauma Acute Care Surg. 72:169–176.
2012.
|
|
20
|
Li W, Qiu X, Jiang H, Zhi Y, Fu J and Liu
J: Ulinastatin inhibits the inflammation of LPS-induced acute lung
injury in mice via regulation of AMPK/NF-κB pathway. Int
Immunopharmacol. 29:560–567. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Huang N, Wang F, Wang Y, Hou J, Li J and
Deng X: Ulinastatin improves survival of septic mice by suppressing
inflammatory response and lymphocyte apoptosis. J Surg Res.
182:296–302. 2013. View Article : Google Scholar
|
|
22
|
Shen W, Gan J, Xu S, Jiang G and Wu H:
Penehyclidine hydrochloride attenuates LPS-induced acute lung
injury involvement of NF-kappaB pathway. Pharmacol Res. 60:296–302.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang F, Meng Y, Zhang Y, Zhao G, Zheng X,
Xiao Q and Yu Y: Ketamine reduces lipopolysaccharide-induced
high-mobility group box-1 through heme oxygenase-1 and nuclear
factor erythroid 2-related factor 2/p38 mitogen-activated protein
kinase. J Surg Res. 194:599–613. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sung YH, Shin MS, Ko IG, Kim SE, Kim CJ,
Ahn HJ, Yoon HS and Lee BJ: Ulinastatin suppresses
lipopolysaccharide-induced prostaglandin E2 synthesis and nitric
oxide production through the downregulation of nuclear factor-κB in
BV2 mouse microglial cells. Int J Mol Med. 31:1030–1036.
2013.PubMed/NCBI
|
|
25
|
Feng G, Sun B and Li TZ: Daidzein
attenuates lipopolysaccharide-induced acute lung injury via
toll-like receptor 4/NF-kappaB pathway. Int Immunopharmacol.
26:392–400. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tianzhu Z and Shumin W: Esculin inhibits
the inflammation of LPS-induced acute lung injury in mice via
regulation of TLR/NF-κB pathways. Inflammation. 38:1529–1536. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kawasaki T and Kawai T: Toll-like receptor
signaling pathways. Front Immunol. 5:4612014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chang Y, Huang X, Liu Z, Han G, Huang L,
Xiong YC and Wang Z: Dexmedetomidine inhibits the secretion of high
mobility group box 1 from lipopolysaccharide-activated macrophages
in vitro. J Surg Res. 181:308–314. 2013. View Article : Google Scholar
|
|
29
|
Liu Z, Zhang J, Huang X, Huang L, Li S and
Wang Z: Magnesium sulfate inhibits the secretion of high mobility
group box 1 from lipopolysaccharide-activated RAW264.7 macrophages
in vitro. J Surg Res. 179:e189–e195. 2013. View Article : Google Scholar
|
|
30
|
Liu Z, Chang Y, Zhang J, Huang X, Jiang J,
Li S and Wang Z: Magnesium deficiency promotes secretion of
high-mobility group box 1 protein from lipopolysaccharide-activated
macrophages in vitro. J Surg Res. 180:310–316. 2013. View Article : Google Scholar
|
|
31
|
Yang Q, Liu X, Yao Z, Mao S, Wei Q and
Chang Y: Penehyclidine hydrochloride inhibits the release of
high-mobility group box 1 in lipopolysaccharide-activated RAW264.7
cells and cecal ligation and puncture-induced septic mice. J Surg
Res. 186:310–317. 2014. View Article : Google Scholar
|
|
32
|
Yamauchi Y, Izumi Y, Inoue M, Sugiura H,
Goto T, Anraku M, Ohtsuka T, Kohno M, Soejima K and Nomori H:
Safety of postoperative administration of human urinary trypsin
inhibitor in lung cancer patients with idiopathic pulmonary
fibrosis. PLoS One. 6:e290532011. View Article : Google Scholar
|
|
33
|
Inoue K and Takano H: Urinary trypsin
inhibitor as a therapeutic option for endotoxin-related
inflammatory disorders. Expert Opin Investig Drugs. 19:513–520.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Maciejewski R, Burdan F, Burski K, Madej
B, Ziemiakowicz R, Dabrowski A and Wallner G: Selected biochemical
parameters and ultrastructural picture of pancreas due to
Ulinastatin treatment of experimental acute pancreatitis. Exp
Toxicol Pathol. 56:305–311. 2005. View Article : Google Scholar : PubMed/NCBI
|