Introduction
Glaucoma is the most common cause of irreversible
blindness worldwide and severely affects the visual function and
quality of life of the patients (1,2).
Filtration surgery is the most common treatment for reducing
intraocular pressure, but excessive scar tissue formation in the
filtration area reduces the postoperative 5-year survival rate of
the filtering bleb to <50% (3,4).
Several anti-scarring regimens, such as mitomycin C (MMC) or
5-fluorouracil (5-FU), are currently used to improve the results of
glaucoma surgery, but are of limited use clinically due to severe
complications, such as filtering bleb leakage, corneal epithelial
dysfunction, late-onset endophthalmitis and macular degeneration
(5–8). Therefore, safer and more effective
anti-scarring agents are urgently required.
Human conjunctival fibroblasts (HConFs) are located
in subconjunctival connective tissue, which plays an important role
in scar tissue reactions of the filtering bleb. Filtration surgery
may stimulate conjunctival fibroblast proliferation, migration,
differentiation from fibroblasts to myofibroblasts, and promote the
secretion of extracellular matrix (ECM); these are important
processes of wound healing, but may also lead to scarring (9–11).
The role of ion channels (chloride channels in particular) in the
development of fibrosis have attracted increasing attention in
recent years (12–15). Previous findings have demonstrated
that chloride channels are widely distributed in mammalian tissues
and organs and various types of cells, and play important roles in
regulating cell cycle progression, cell proliferation, migration
and apoptosis by maintaining the cell volume balance (16–24). The phosphatidylinositide 3-kinase
(PI3K)/protein kinase B (AKT) signaling pathway is a survival
pathway that has recently become known as a key regulator of cell
proliferation, migration and apoptosis (25,26). The PI3K/AKT pathway also plays a
key role in regulating ECM synthesis (27). We previously cloned the ClC-2
gene, which was sensitive to changing cell volumes, and revealed
its important roles in the processes of cell differentiation,
proliferation and division (17,18). The chloride channel blocker
5-nitro-2-(3-phenylpropylamino) benzoic acid (NPPB) is known to
selectively block volume-sensitive, hyperpolarization-activated and
medium-conductance Cl channels. NPPB is not highly selective, but
is used by numerous investigators to examine the pharmacological
properties of cl ion currents in various cells. Cheng et al
(19) obserevd that NPPB
increased apoptosis in human bronchial epithelial cells. We
previously found that the chloride channel blocker NPPB inhibited
the transition of quiescent (G0) fibroblasts into the cell cycle
(17). However, it remains
unclear whether or how NPPB affects the proliferation, migration
and ECM synthesis of conjunctival fibroblasts.
In the present study, HConFs were cultured to
investigate the affects of the chloride channel blocker NPPB on the
proliferation, migration, apoptosis and ECM synthesis of HConFs. It
was further investigated whether NPPB exerts the above-mentioned
effects on HConFs via the PI3K/AKT signaling pathway to provide
novel insights into prevention of glaucoma filtration surgery scar
formation.
Materials and methods
Drugs
NPPB and lubiprostone (both from Sigma-Aldrich;
Merck KGaA, St. Louis, MO, USA) were prepared as 0.4 and 0.1 M
stock solutions in dimethyl sulfoxide (DMSO) (Beyotime Institute of
Biotechnology, Jiangsu, China), respectively, stored at a
temperature <−20°C, and protected from light until use. NPPB and
lubiprostone were diluted to 10–200 μM and 10–300 nM,
respectively, in Dulbecco's modified Eagle's medium (DMEM)
(Sigma-Aldrich; Merck KGaA) containing 10% fetal bovine serum (FBS)
(Invitrogen/Gibco; Thermo Fisher Scientific, Grand Island, NY, USA)
on the day of the experiment. The highest concentration of DMSO in
the test solutions was 0.1%. To exclude the possibility that
proliferation was inhibited by DMSO, cells were exposed to a final
DMSO concentration of 0.1% in DMEM containing 10% FBS.
Cell lines and cell culture
HConFs from ScienCell Research Laboratories (San
Diego, CA, USA) were isolated from the human conjunctiva. HConFs
are characterized by a spindle-shaped morphology and positivity for
immunofluorescence staining with anti-fibronectin antibodies.
HConFs were maintained and cultured at 37°C in a humidified
incubator with 5% CO2 in fibroblast medium (ScienCell
Research Laboratories) containing fibroblast growth supplement
(undisclosed formulation), 2% FBS, 100 U/ml penicillin and 100
μg/ml streptomycin.
Cell proliferation assay [Cell Counting
Kit-8 (CCK-8) assay]
HConF suspensions were transferred to a 96-well
plates (200 µl/well) with a density of 0.5×104
cells/well for 24 h. The cells were then treated with media
containing different reagents for 48 h. Subsequently, each well was
incubated with 10 µl CCK-8 solution (BestBio, Jiangsu,
China) for a further 3 h at 37°C. The absorbance, expressed as
optical density (OD), was recorded at 450 nm using an automated
microplate reader (model 3001-1387; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA).
Cell apoptosis assay
Annexin V binding to HConF was performed using an
Annexin V-FITC Apoptosis kit (BestBio) to measure apoptosis. HConFs
(5×105/well) were plated into 6-well plates and then
treated with medium containing different reagents for 48 h.
Briefly, the cells, after being rinsed twice with
phosphate-buffered saline (PBS), were resuspended in 400 µl
of 1X binding buffer (10 mM HEPES, 140 mM NaCl, 2.5 mM
CaCl2; pH 7.4), to which 5 µl of Annexin V-FITC
was added and mixed well. After a 15-min incubation period at 2–8°C
in the dark, 10 µl of propidium iodide (PI) was added to the
cells and mixed well. The cells were incubated for a further 5 min
at 2–8°C in the dark, after which time flow cytometry (Cytomics FV
500; Beckman Coulter, Brea, CA, USA) was performed within 15
min.
Cell cycle analysis
The cell cycle status was assessed by flow cytometry
using a Cell Cycle and Apoptosis Analysis kit (Beyotime Institute
of Biotechnology). Briefly, the cells were grown in 100-mm plates
with 10% FBS for 24 h. After treating the cells with media
containing different reagents for 48 h, they were collected and
fixed in 75% ethanol for 24 h at 4°C. After rinsing the cells with
PBS, they were stained with PI buffer (containing 500 µl
staining buffer, 25 µl of 20X PI and 10 µl of 50X
RNase). The cell cycle distribution was assessed by flow cytometry
(Cytomics FV 500; Beckman Coulter).
Scratch-wound assay
HConFs (5×105 cells/well) were plated
into 6-well plates in culture medium. At 24 h after seeding, the
culture medium was replaced with fresh medium supplemented with 4
µg/ml MMC (Zhejiang Hisun Pharmaceutical Co., Ltd., Taizhou,
China). After a 2-h incubation, a line was scratched into confluent
cell monolayers using a sterile 200-µl pipette tip, and the
cells were washed 3 times with PBS. The scratch wound was allowed
to heal for 12 h in the presence of different reagents. Micrographs
were captured for each sample at 0 and 12 h, and the migration
capacity of HConFs was evaluated by measuring the width of the
scratch wound at both time-points, using ImageJ software, version
1.5 (produced by Java2HTML).
Transwell migration assay
The chemomigration assay was performed in Transwell
plates (pore size, 8 µm; Corning Costar, Inc., Corning, NY,
USA). The chambers were inserted into a 24-well plate. Cells
(1×105) were suspended in 200 µl DMEM and added
to the upper chamber. Our preliminary experiments demonstrated that
an FBS concentration of 10% is optimal for observing cell migration
(data not shown). Therefore, DMEM with 10% FBS was added into the
lower chamber of each well, and the cells were incubated for 12 h.
The medium and non-migrated cells in the upper chamber were removed
gently with a cotton swab, whereas the migrated cells in the lower
chamber were fixed with paraformaldehyde (4%) and stained with
crystal violet. Images were captured at a magnification of ×200.
Cells in 6 different fields were counted.
Western blot analysis
Cells were washed with pre-cooled PBS and lysed in
radioimmunoprecipitation assay buffer containing the protease
inhibitor phenylmethylsulfonyl fluoride and the phosphatase
inhibitor Na3VO4 (Nanjing KeyGen Biotech Co.,
Ltd., Nanjing, China) on ice. Subsequently, the cells were gently
scraped with a rubber policeman and centrifuged at 1,500 × g for 12
min at 4°C. Protein concentrations were measured using the
bicinchoninic acid (BCA) method with the enhanced BCA protein assay
kit (cat. no. P0009; Beyotime institute of Biotechnology).
A total of 20 µg of protein per sample was
separated by 6–15% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis and transferred onto polyvinylidene fluoride
membranes. The membranes were then blocked with 5% non-fat dry milk
in Tris-buffered saline with 0.1% Tween-20 (TBST) for 1 h at 37°C
and incubated overnight at 4°C in TBST with mouse monoclonal
anti-collagen I (1:2,500 dilution, cat. no. ab88147) and rabbit
polyclonal anti-fibronectin (1:1,500 dilution, cat. no. ab2375)
(both from Abcam, Cambridge, UK), mouse monoclonal anti-PI3 kinase
p85 (1:500 dilution, cat. no. sc-1637), mouse polyclonal anti-p-PI
3-kinase p85α (1:500 dilution, cat. no. sc-12929), mouse monoclonal
anti-Akt1 (1:500 dilution, cat. no. sc-5298), mouse monoclonal
anti-p-Akt1 (1:500 dilution, cat. no. sc-293125) (all from Santa
Cruz Biotechnology, Inc., Santa Cruz, CA, USA), and/or mouse
monoclonal anti-β-actin (1:1,000 dilution, cat. no. AF0003;
Beyotime Institute of Biotechnology) antibodies. Following two
washes with TBST, the membrane was incubated with horseradish
peroxidase-conjugated goat anti-rabbit IgG secondary antibody
(1:5,000 dilution, cat. no. A0208) or goat anti-mouse IgG secondary
antibody (1:5,000 dilution, cat. no. A0129) (both from Beyotime
Institute of Biotechnology) for another 1 h at room temperature and
washed 3 times with TBST. Detection was performed with enhanced
chemiluminescence western blotting reagents (Beyotime Institute of
Biotechnology) and the membranes were visualized by exposure to
Kodak X-ray film. The results were scanned and analyzed using
ImageJ software (National Institutes of Health, Bethesda, MD,
USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from HConFs using the RNAiso
Plus reagent (Takara Bio, Inc., Shiga, Japan), according to the
manufacturer's instructions. Single-stranded cDNA templates were
prepared from 500 ng total RNA using the RT-for-PCR kit (Takara
Bio, Inc.), according to the manufacturer's instructions. Specific
cDNAs were subsequently amplified by PCR using the following
primers (Shanghai R&S Biotechnology Co., Ltd., Shanghai,
China): Collagen I forward, 5′-TCCTCTTTAGCACCCTTTCG-3′ and reverse,
5′-GGACCAGCAACACCATCTG-3′; fibronectin forward,
5′-CCAGCAGAGGCATAAGGTTC-3′ and reverse, 5′-CACTCATCTCCAACGGCATA-3′;
GAPDH forward, 5′-CAGGAGGCATTGCTGATGAT-3′ and reverse,
5′-CAGGAGGCATTGCTGATGAT-3′. PCR amplification from cDNA was
performed using a LightCycler® 480 system (Roche
Diagnostics, Basel, Switzerland) in a final reaction volume of 20
µl containing 2X SYBR-Green mix (10 µl; Toyobo Co.,
Ltd., Osaka, Japan), 1 µl primer mix, 1 µl template
DNA and 8 µl diethylpyrocarbonate water. The following
cycling conditions were used: Initial denaturation at 95°C for 30
sec; 40 cycles of denaturation at 95°C for 15 sec, annealing at
59°C for 20 sec, elongation at 72°C for 20 sec; and a final
extension at 72°C for 10 min. The data were normalized to GAPDH
mRNA expression, using the ΔΔCq method (22).
Statistical analysis
Data are expressed as the mean ± standard error
(number of observations) and were analyzed using the Student's
t-test and one-way analysis of variance. Statistical significance
was defined as P<0.05. All experiments were repeated 4
times.
Results
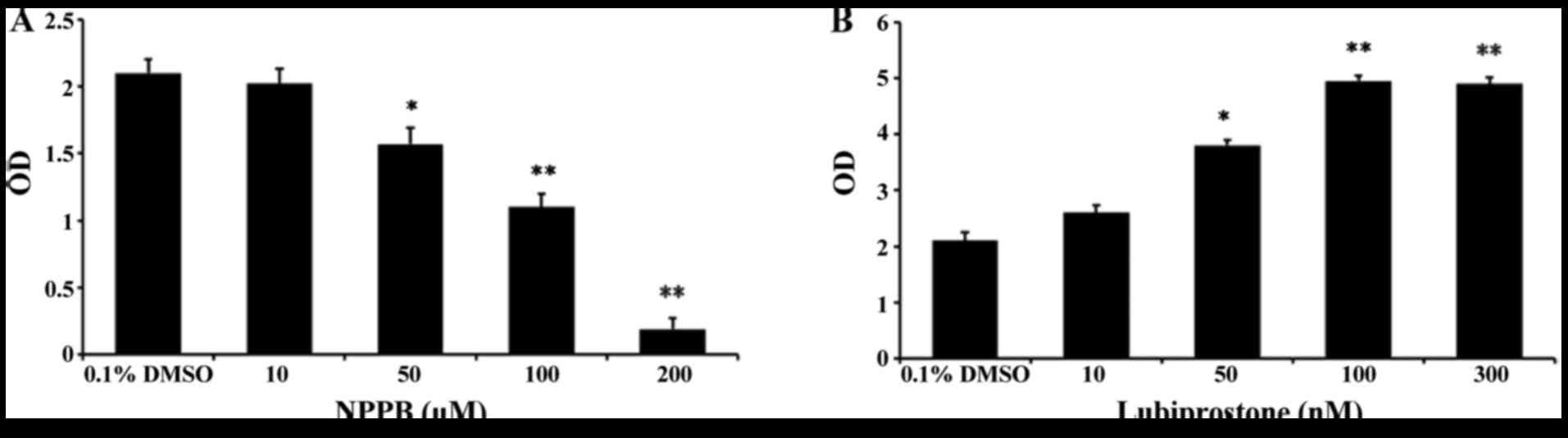
Effect of NPPB on cell proliferation
To assess the effects of the chloride channel
blocker NPPB and the chloride channel activator lubiprostone on
HConF cell proliferation, HConFs were first stimulated with
different concentrations of NPPB (10, 50, 100 and 200 µM)
for 48 h using 0.1% DMSO as control. NPPB inhibited HConF
proliferation in a dose-dependent manner, and 100 µM NPPB
inhibited cell proliferation by 47.62±1.99% (P<0.01 vs. control;
Fig. 1A). Conversely, HConF
stimulation with lubiprostone (10, 50, 100 and 300 nM) promoted
cell proliferation in a dose-dependent manner, the effect of which
peaked at 100 nM (Fig. 1B).
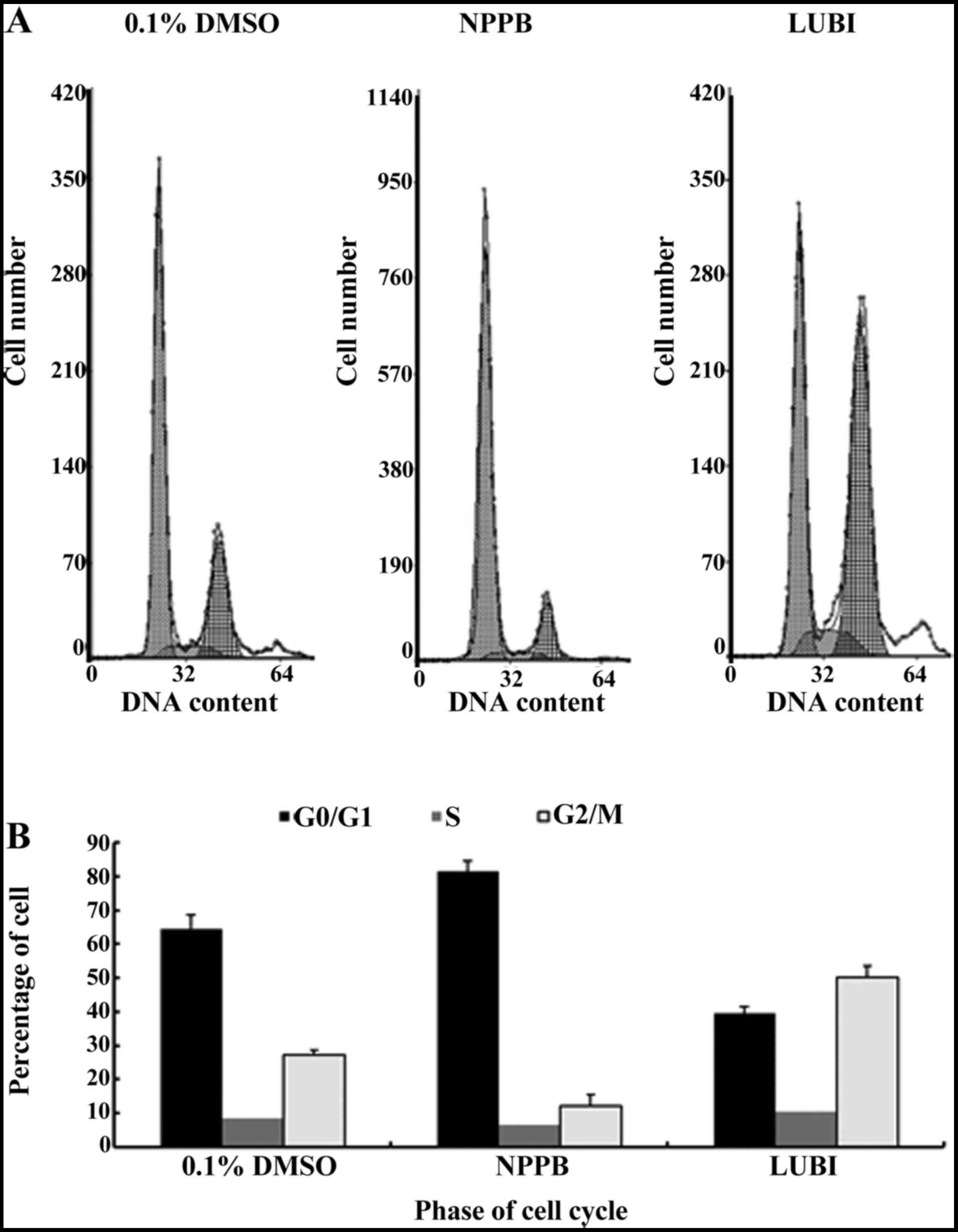
Effect of NPPB on the cell cycle
Next, HConFs were divided into 3 groups, which were
exposed to 100 µM NPPB (NPPB group), 100 nM lubiprostone
(LUBI group) or 0.1% DMSO as control (0.1% DMSO group). The effects
of these treatments on cell cycle progression were tested by flow
cytometry. The proportion of cells in G0/G1 in the 0.1% DMSO, NPPB
and LUBI groups was 64.43±4.21, 81.44±3.15 and 39.40±2.14%,
respectively. The proportion of cells in S phase in the 0.1% DMSO,
NPPB and LUBI groups was 8.37±2.02, 6.56±2.21 and 10.30±3.09%,
respectively. The proportion of cells in G2/M phase in the 0.1%
DMSO, NPPB and LUBI groups was 27.20±1.32, 12.00±3.56 and
50.30±3.23%, respectively. Therefore, NPPB inhibited cell cycle
progression by arresting the cells in the G0/G1 phase, but
lubiprostone promoted cell cycle progression by advancing the cells
from G0/G1 to the S and G2/M phase (Fig. 2).
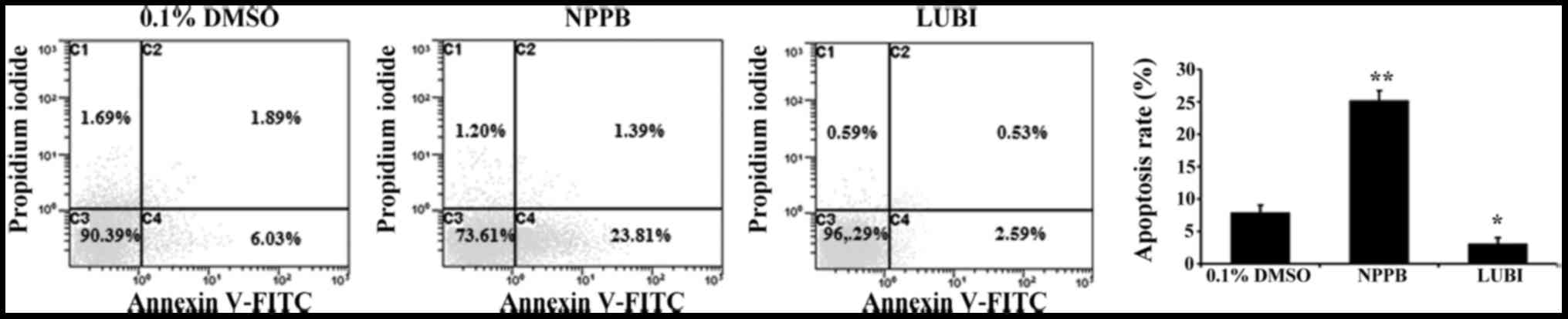
Effect of NPPB on cell apoptosis
To investigate the effect of NPPB on HConF
apoptosis, HConFs were stimulated with 100 µM NPPB or 100 nM
lubiprostone for 48 h, using 0.1% DMSO as control. Treatment with
100 µM NPPB for 48 h significantly increased the number of
apoptotic HConFs compared with control treatment (25.2±1.5 vs.
7.92±1.1%; P<0.01). Treatment of HConFs with 100 nM lubiprostone
resulted in a lower rate of apoptosis (3.12±0.88%; P<0.05 vs.
control; Fig. 3).
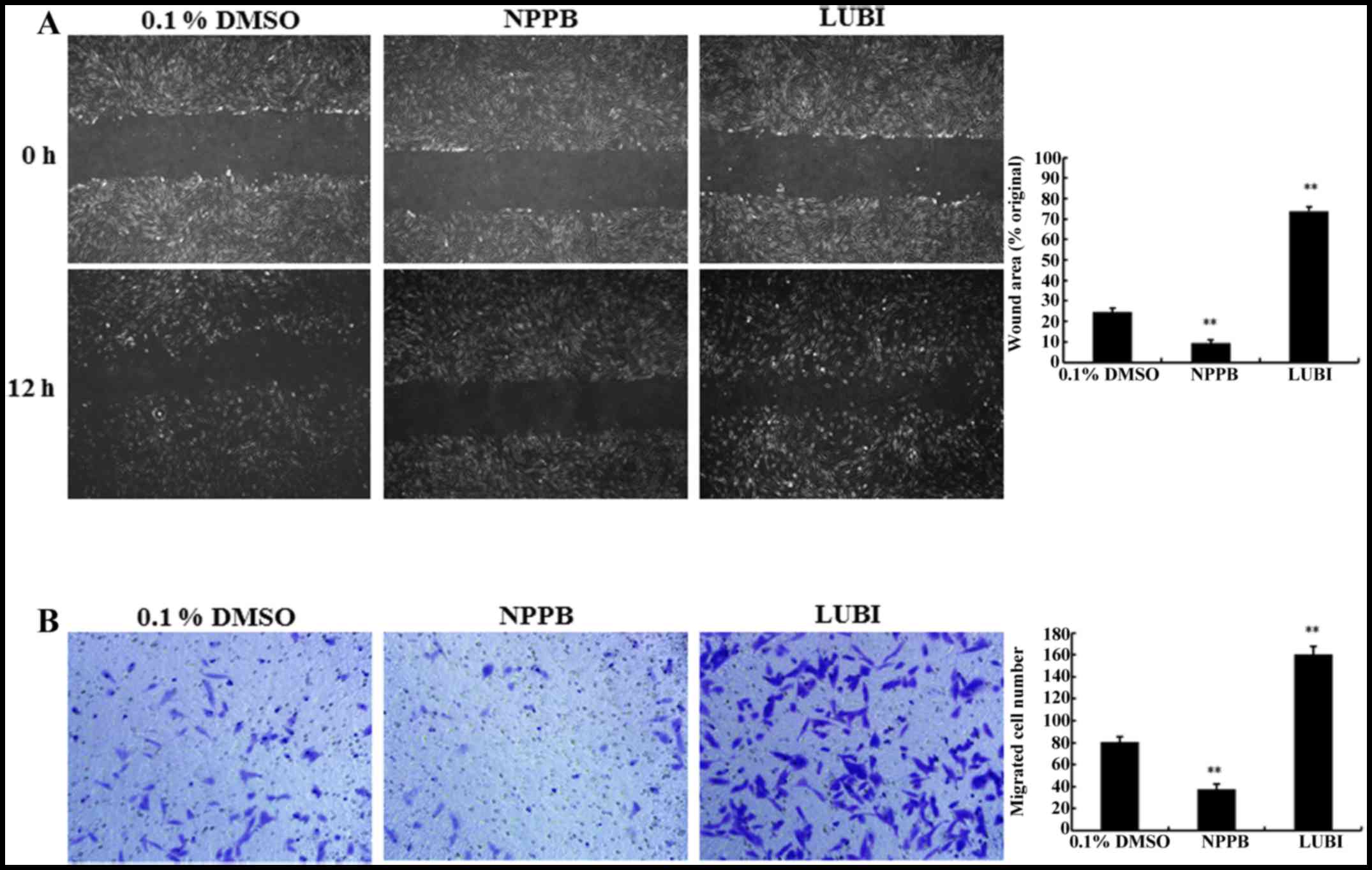
Effect of NPPB on cell migration
The migration capacity of HConFs was measured via
in vitro scratch and Transwell migration assays. The
scratch-wound assay revealed that treatment with NPPB significantly
inhibited wound healing compared with that observed in the control
group (9.36±1.44 vs. 24.54±1.82%, respectively; P<0.01).
Treatment of HConFs with 100 nM lubiprostone significantly
increased wound healing compared to that observed in the control
group (73.83±2.26, P<0.01 vs. control; Fig. 4A).
To confirm the results of the scratch-wound assay, a
Transwell-migration assay was performed. As shown in Fig. 4B, significantly fewer HConFs
migrated through the Transwell membrane in the NPPB group compared
with the control group (7.41±0.83 vs. 20.55±1.02 cells,
respectively; P<0.01). A significantly greater number of cells
migrated through the Transwell membrane in the LUBI group compared
with the control group (41.58±0.89; P<0.01 vs. control). These
result indicated that NPPB inhibited and that lubiprostone promoted
HConF migration (Fig. 4B).
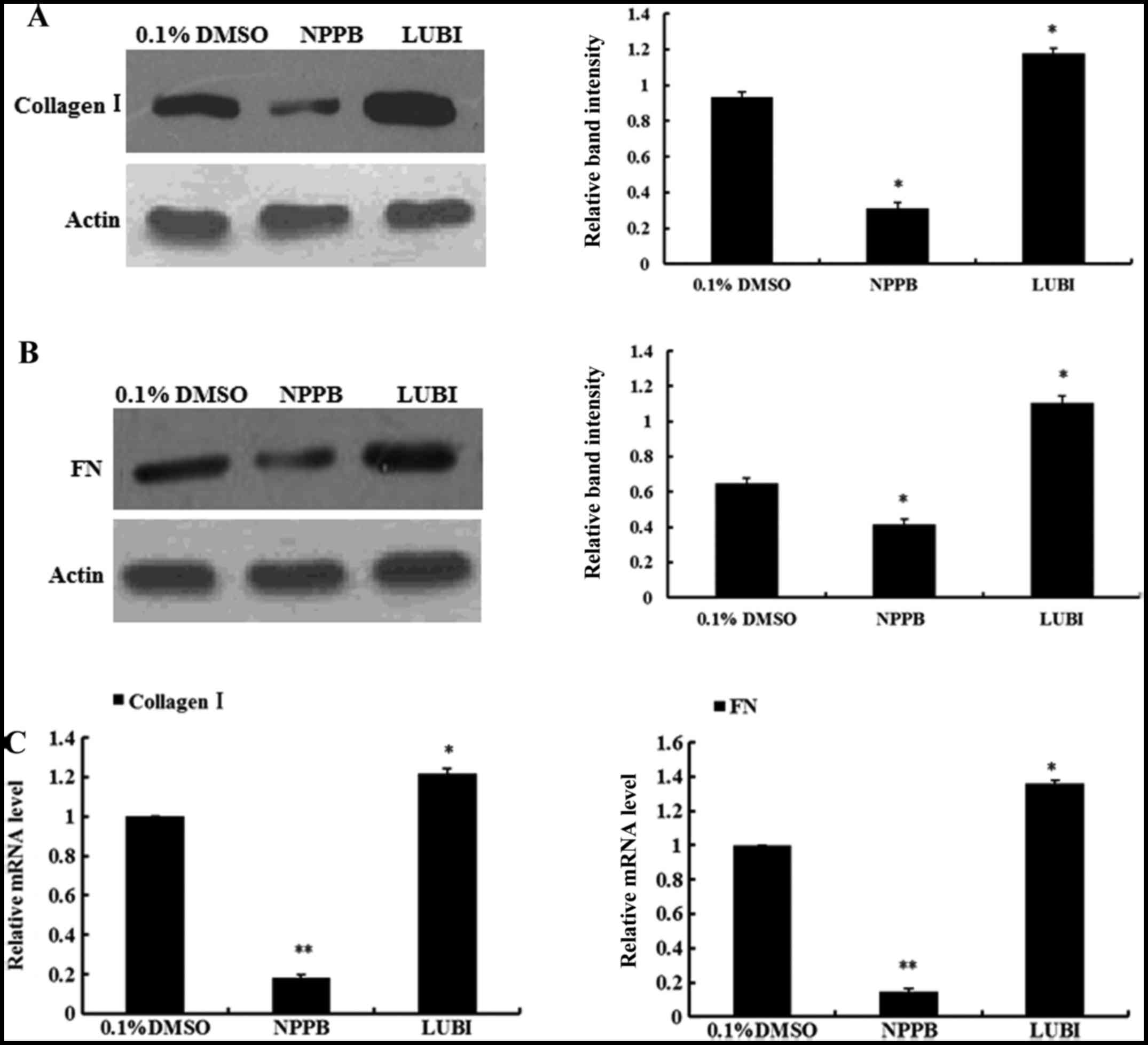
Effect of NPPB on collagen I and
fibronectin expression
To evaluate the effects of NPPB on ECM production,
the mRNA and protein expression of collagen I and fibronectin in
the three groups were measured. The relative quantification results
revealed that HConFs treated with NPPB exhibited significantly
inhibited collagen I and fibronectin expression, at both the mRNA
and protein levels. By contrast, the mRNA and protein expression of
collagen I and fibronectin significantly increased in the
lubiprostone group (Fig. 5).
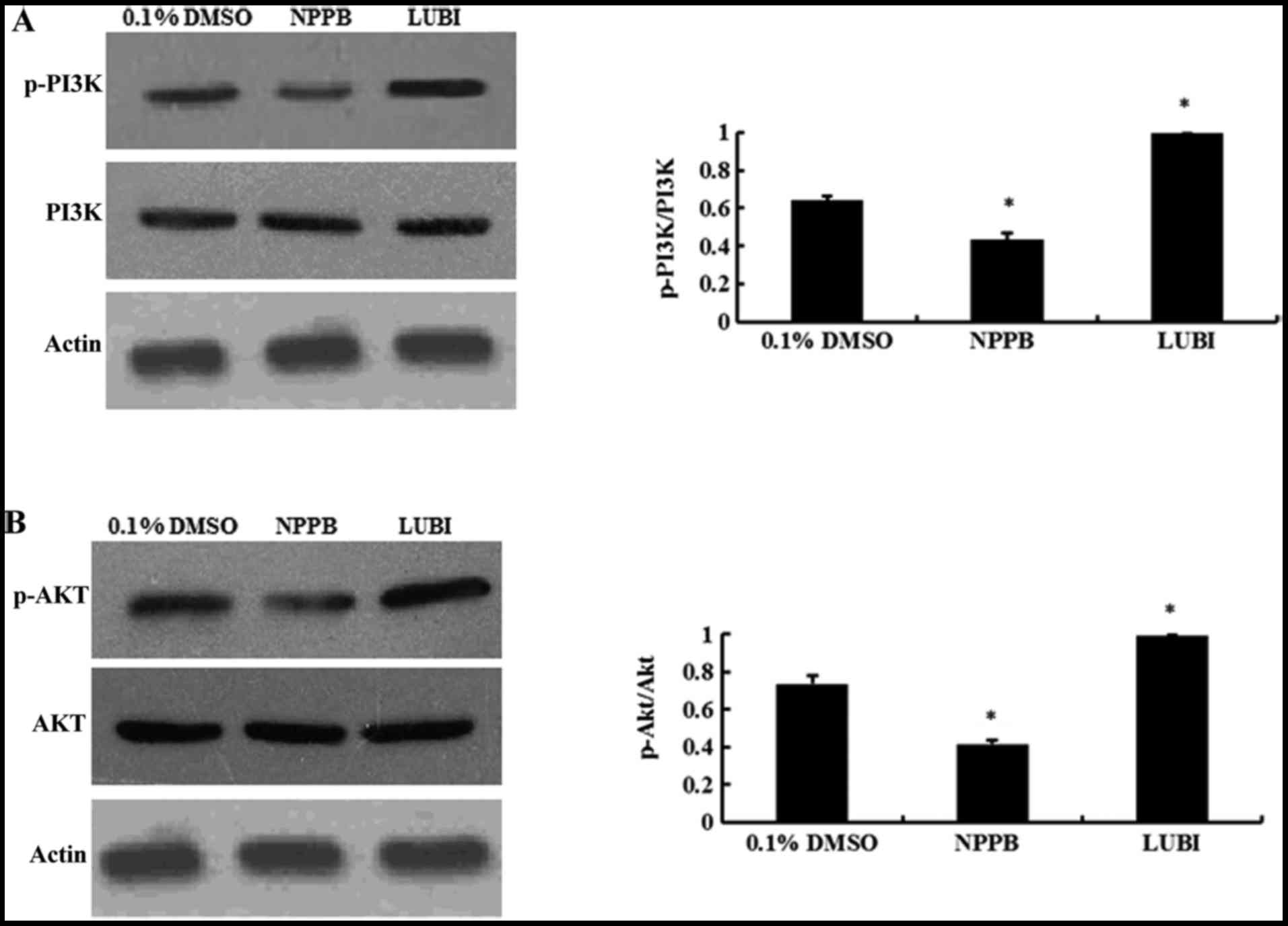
NPPB repressed the PI3K/Akt pathways
Western blot analysis revealed that NPPB treatment
significantly decreased the levels of p-PI3K and p-AKT (both
P<0.05 vs. control). Lubiprostone significantly increased the
expression of p-PI3K and p-Akt (P<0.05 vs. control; Fig. 6).
Discussion
Excessive postoperative conjunctival scarring at
filtering bleb sites is the main reason underlying trabeculectomy
failure. In general, this process consists of a series of events,
including conjunctival fibroblast migration and proliferation,
deposition, and contraction of dense collagen fibers after the
proliferation of fibroblasts, which leads to aqueous outflow
blockage by creating adhesions between the conjunctiva and
episclera, as well as between the scleral flap and underlying
tissues (28,29). Accumulating evidence suggests that
chloride channels are critical for cell proliferation and apoptosis
(30–32). NPPB was found to inhibit cell
proliferation in a concentration- and time-dependent manner in
nasopharyngeal carcinoma cells (30). NPPB also reduced cell
proliferation in a concentration-dependent manner in mouse
mesenchymal stem cells (31).
Consistent with previous data, we observed that NPPB inhibited
HConF proliferation in a dose-dependent manner. Data from our
previous study demonstrated that the chloride channel blocker NPPB
inhibits the transition of quiescent (G0) fibroblasts synchronized
by serum deprivation/replenishment reentry into the proliferating
phase by impairing passage through G1/S (17). Tao et al (31) reported that inhibiting
Cl− currents using NPPB retained mouse mesenchymal stem
cells in the G0/G1 phase and decreased the distribution of cells in
S phase. NPPB inhibited the volume-activated chloride current and
proliferation of nasopharyngeal carcinoma cells in a dose-dependent
manner, inhibited cell cycle progression and arrested cells at the
G0/G1 phase boundary (32). To
further investigate how NPPB participates in HConF proliferation,
flow cytometry experiments were conducted and demonstrated that
NPPB inhibited cell cycle progression and arrested cells at the
G0/G1 phase boundary. Moreover, NPPB increased the abundance of the
apoptotic HConF population, which was prevented by the chloride
channel activator lubiprostone. In agreement with our findings,
Souktani et al (33) found
that NPPB increased apoptosis in rabbit myocardial cells. Cheng
et al (19) reported that,
both stimulation with NPPB and knocking down the expression of the
CLC-3 chloride channel increased apoptosis; by contrast,
overexpression of CLC-3 prevented TGF-β1-induced apoptosis in human
bronchial epithelial cells. Contradictory data were also reported,
indicating that NPPB inhibited apoptosis, instead of accelerating
it (34,35). In CNE-2Z cells, NPPB prevented
apoptosis induced by paclitaxel (36). The selectivity of NPPB for
chloride channels in different cell types may account for the
conflicting effects of NPPB on apoptosis.
The PI3K/AKT pathway is a survival pathway that
regulates cell proliferation, apoptosis, differentiation and
migration (37). The PI3K/AKT
pathway may play key roles in the physiology and pathophysiology of
several types of cells (38–41). The key enzyme of this pathway,
PI3K, converts phosphatidylinositol 4,5-biphosphate to
phosphatidylinositol 3,4,5-triphosphate, which binds both AKT and
3-phosphoinositide-dependent protein kinase 1 (PDK1), enabling PDK1
to phosphorylate AKT (41,42).
The primary direct downstream target protein of PI3K is AKT. The
activation of AKT causes a cascade of responses with downstream
targets that regulate cellular functions. For example, AKT
regulates cell migration via Rac1 and RhoA, increases cell survival
via Bcl-2, and increases cell proliferation via the activation of
the mammalian target of rapamycin (37,41). Zhou et al (26) reported that exendin-4 mediated
proliferation, migration and apoptosis via the PI3K/AKT pathway in
bone marrow mesenchymal stem cells. It has been reported that
activation of PI3K/AKT signaling may induce the expression of ECM
molecules in a number of cell types (43,44). Previous results demonstrated that
inhibiting CIC-2 expression by RNA interference or a chloride
channel blocker may attenuate cell proliferation and migration via
the PI3K/AKT signaling pathway (45,46). Li et al (47) observed that suppression of
PI3K/AKT signaling may decrease adhesion and migration of bone
marrow-derived mesenchymal stem cells. In the present study, NPPB
promoted cell apoptosis and inhibited proliferation, migration,
cell cycle progression and synthesis of ECM, which paralleled
suppressed expression of p-PI3K and p-AKT. Therefore, NPPB exerts
the abovementioned effects on HConFs by inhibiting PI3K-dependent
signaling.
References
|
1
|
Quigley HA and Broman AT: The number of
people with glaucoma worldwide in 2010 and 2020. Br J Ophthalmol.
90:262–267. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Foster PJ and Johnson GJ: Glaucoma in
China: how big is the problem. Br J Ophthalmol. 85:1277–1282. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Khaw PT, Chang L, Wong TT, Mead A, Daniels
JT and Cordeiro MF: Modulation of wound healing after glaucoma
surgery. Curr Opin Ophthalmol. 12:143–148. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lee DA: Antifibrosis agents and glaucoma
surgery. Invest Ophthalmol Vis Sci. 35:3789–3791. 1994.PubMed/NCBI
|
|
5
|
Mielke C, Dawda VK and Anand N:
Intraoperative 5-fluorouracil application during primary
trabeculectomy in Nigeria: a comparative study. Eye (Lond).
17:829–834. 2003. View Article : Google Scholar
|
|
6
|
Wong TT, Khaw PT, Aung T, Foster PJ, Htoon
HM, Oen FT, Gazzard G, Husain R, Devereux JG, Minassian D, et al:
The singapore 5-fluorouracil trabeculectomy study: effects on
intraocular pressure control and disease progression at 3 years.
Ophthalmology. 116:175–184. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shin DH, Ren J, Juzych MS, Hughes BA, Kim
C, Song MS, Yang KJ and Glover KB: Primary glaucoma triple
procedure in patients with primary open-angle glaucoma: the effect
of mitomycin C in patients with and without prognostic factors for
filtration failure. Am J Ophthalmol. 125:346–352. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Perkins TW, Gangnon R, Ladd W, Kaufman PL
and Heatley GA: Trabeculectomy with mitomycin C: intermediate-term
results. J Glaucoma. 7:230–236. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chang L, Crowston JG, Cordeiro MF, Akbar
AN and Khaw PT: The role of the immune system in conjunctival wound
healing after glaucoma surgery. Surv Ophthalmol. 45:49–68. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Atreides SP, Skuta GL and Reynolds AC:
Wound healing modulation in glaucoma filtering surgery. Int
Ophthalmol Clin. 44:61–106. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Desjardins DC, Parrish RK II, Folberg R,
Nevarez J, Heuer DK and Gressel MG: Wound healing after filtering
surgery in owl monkeys. Arch Ophthalmol. 104:1835–1839. 1986.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Song Y, Zhan L, Yu M, Huang C, Meng X, Ma
T, Zhang L and Li J: TRPV4 channel inhibits TGF-β1-induced
proliferation of hepatic stellate cells. PLoS One. 9:e1011792014.
View Article : Google Scholar
|
|
13
|
Roach KM, Duffy SM, Coward W,
Feghali-Bostwick C, Wulff H and Bradding P: The K+
channel KCa3.1 as a novel target for idiopathic
pulmonary fibrosis. PLoS One. 8:e852442013. View Article : Google Scholar
|
|
14
|
Roach KM, Feghali-Bostwick C, Wulff H,
Amrani Y and Bradding P: Human lung myofibroblast TGFβ1-dependent
Smad2/3 signalling is Ca(2+)-dependent and regulated by
KCa3.1 K(+) channels. Fibrogenesis Tissue Repair.
8:52015. View Article : Google Scholar
|
|
15
|
Sun H, Harris WT, Kortyka S, Kotha K,
Ostmann AJ, Rezayat A, Sridharan A, Sanders Y, Naren AP and Clancy
JP: Tgf-beta down-regulation of distinct chloride channels in
cystic fibrosis-affected epithelia. PLoS One. 9:e1068422014.
View Article : Google Scholar
|
|
16
|
Nilius B and Droogmans G: Amazing chloride
channels: an overview. Acta Physiol Scand. 177:119–147. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zheng YJ, Furukawa T, Tajimi K and Inagaki
N: Cl− channel blockers inhibit transition of quiescent
(G0) fibroblasts into the cell cycle. J Cell Physiol.
194:376–383. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zheng YJ, Furukawa T, Ogura T, Tajimi K
and Inagaki N: M phase-specific expression and
phosphorylation-dependent ubiquitination of the ClC-2 channel. J
Biol Chem. 277:32268–32273. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cheng G, Shao Z, Chaudhari B and Agrawal
DK: Involvement of chloride channels in TGF-beta1-induced apoptosis
of human bronchial epithelial cells. Am J Physiol Lung Cell Mol
Physiol. 293:L1339–L1347. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhu L, Yang H, Zuo W, Yang L, Zhang H, Ye
W, Mao J, Chen L and Wang L: Differential expression and roles of
volume-activated chloride channels in control of growth of normal
and cancerous nasopharyngeal epithelial cells. Biochem Pharmacol.
83:324–334. 2012. View Article : Google Scholar
|
|
21
|
Okada Y, Shimizu T, Maeno E, Tanabe S,
Wang X and Takahashi N: Volume-sensitive chloride channels involved
in apoptotic volume decrease and cell death. J Membr Biol.
209:21–29. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mao J, Chen L, Xu B and Wang L, Li H, Guo
J, Li W, Nie S, Jacob TJ and Wang L: Suppression of ClC-3 channel
expression reduces migration of nasopharyngeal carcinoma cells.
Biochem Pharmacol. 75:1706–1716. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mao J, Yuan J, Wang L, Zhang H, Jin X, Zhu
J, Li H, Xu B and Chen L: Tamoxifen inhibits migration of estrogen
receptor-negative hepatocellular carcinoma cells by blocking the
swelling-activated chloride current. J Cell Physiol. 228:991–1001.
2013. View Article : Google Scholar
|
|
24
|
Guan YY, Wang GL and Zhou JG: The ClC-3
Cl− channel in cell volume regulation, proliferation and
apoptosis in vascular smooth muscle cells. Trends Pharmacol Sci.
27:290–296. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sang H, Li T, Li H and Liu J: Gab1
regulates proliferation and migration through the PI3K/Akt
signaling pathway in intrahepatic cholangiocarcinoma. Tumour Biol.
36:8367–8377. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhou H, Li D, Shi C, Xin T, Yang J, Zhou
Y, Hu S, Tian F, Wang J and Chen Y: Effects of exendin-4 on bone
marrow mesenchymal stem cell proliferation, migration and apoptosis
in vitro. Sci Rep. 5:128982015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zeng R, Xiong Y, Zhu F, Ma Z, Liao W, He
Y, He J, Li W, Yang J, Lu Q, et al: Fenofibrate attenuated
glucose-induced mesangial cells proliferation and extracellular
matrix synthesis via PI3K/AKT and ERK1/2. PLoS One. 8:e768362013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hitchings RA and Grierson I: Clinico
pathological correlation in eyes with failed fistulizing surgery.
Trans Ophthalmol Soc U K. 103:84–88. 1983.PubMed/NCBI
|
|
29
|
Addicks EM, Quigley HA, Green WR and Robin
AL: Histologic characteristics of filtering blebs in glaucomatous
eyes. Arch Ophthalmol. 101:795–798. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen L, Wang L, Zhu L, Nie S, Zhang J,
Zhong P, Cai B, Luo H and Jacob TJ: Cell cycle-dependent expression
of volume-activated chloride currents in nasopharyngeal carcinoma
cells. Am J Physiol Cell Physiol. 283:C1313–C1323. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tao R, Lau CP, Tse HF and Li GR:
Regulation of cell proliferation by intermediate-conductance
Ca2+-activated potassium and volume-sensitive chloride
channels in mouse mesenchymal stem cells. Am J Physiol Cell
Physiol. 295:C1409–C1416. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen LX, Zhu LY, Jacob TJ and Wang LW:
Roles of volume-activated Cl− currents and regulatory
volume decrease in the cell cycle and proliferation in
nasopharyngeal carcinoma cells. Cell Prolif. 40:253–267. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Souktani R, Ghaleh B, Tissier R,
d'Anglemont de Tassigny A, Aouam K, Bedossa P, Charlemagne D,
Samuel J, Henry P and Berdeaux A: Inhibitors of swelling-activated
chloride channels increase infarct size and apoptosis in rabbit
myocardium. Fundam Clin Pharmacol. 17:555–561. 2003. View Article : Google Scholar
|
|
34
|
Small DL, Tauskela J and Xia Z: Role for
chloride but not potassium channels in apoptosis in primary rat
cortical cultures. Neurosci Lett. 334:95–98. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Maeno E, Ishizaki Y, Kanaseki T, Hazama A
and Okada Y: Normotonic cell shrinkage because of disordered volume
regulation is an early prerequisite to apoptosis. Proc Natl Acad
Sci USA. 97:9487–9492. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang H, Li H, Yang L, Deng Z, Luo H, Ye
D, Bai Z, Zhu L, Ye W, Wang L, et al: The ClC-3 chloride channel
associated with microtubules is a target of paclitaxel in its
induced-apoptosis. Sci Rep. 3:26152013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chen J: The IL-23/IL-17 axis may be
important in obesity-associated cancer by way of the activation of
multiple signal pathways. Int J Obes. 34:1227–1229. 2010.
View Article : Google Scholar
|
|
38
|
Martelli AM, Evangelisti C, Chiarini F,
Grimaldi C, Cappellini A, Ognibene A and McCubrey JA: The emerging
role of the phosphatidylinositol 3-kinase/Akt/mammalian target of
rapamycin signaling network in normal myelopoiesis and
leukemogenesis. Biochim Biophys Acta. 1803:991–1002. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Weichhart T and Säemann MD: The
PI3K/Akt/mToR pathway in innate immune cells: emerging therapeutic
applications. Ann Rheum Dis. 67(Suppl 3): iii70–iii74. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhao T, Qi Y, Li Y and Xu K: PI3 kinase
regulation of neural regeneration and muscle hypertrophy after
spinal cord injury. Mol Biol Rep. 39:3541–3547. 2012. View Article : Google Scholar
|
|
41
|
Liu P, Cheng H, Roberts TM and Zhao JJ:
Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev
Drug Discov. 8:627–644. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Cantley LC: The phosphoinositide 3-kinase
pathway. Science. 296:1655–1657. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Liu Y, Li W, Liu H, Peng Y, Yang Q, Xiao
L, Liu Y and Liu F: Inhibition effect of small interfering RNA of
connective tissue growth factor on the expression of extracellular
matrix molecules in cultured human renal proximal tubular cells.
Ren Fail. 36:278–284. 2014. View Article : Google Scholar
|
|
44
|
Qin D, Zhang GM, Xu X and Wang LY: The
PI3K/Akt signaling pathway mediates the high glucose-induced
expression of extracellular matrix molecules in human retinal
pigment epithelial cells. J Diabetes Res 2015. 920280:2015.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Pan F, Guo R, Cheng W, Chai L, Wang W, Cao
C and Li S: High glucose inhibits ClC-2 chloride channels and
attenuates cell migration of rat keratinocytes. Drug Des Devel
Ther. 9:4779–4791. 2015.PubMed/NCBI
|
|
46
|
Heo KS, Ryoo SW, Kim L, Nam M, Baek ST,
Lee H, Lee AR, Park SK, Park Y, Myung CS, et al: Cl− channel is
essential for LDL-induced cell proliferation via the activation of
Erk1/2 and PI3K/Akt and the upregulation of Egr-1 in human aortic
smooth muscle cells. Mol Cells. 26:468–473. 2008.PubMed/NCBI
|
|
47
|
Xia Li L, Wang Y, Cao Z, Da X, Guo Z, Qian
G, Liu J, Fan X, Sun YL, et al: Suppression of the PI3K-Akt pathway
is involved in the decreased adhesion and migration of bone
marrow-derived mesenchymal stem cells from non-obese diabetic mice.
Cell Biol Int. 35:961–966. 2011. View Article : Google Scholar : PubMed/NCBI
|