Introduction
Ischemic heart disease (IHD) is a serious health
problem in the world with growing morbidity and mortality (1). Reconstruction of blood supply is not
only the most effective treatment for IHD, but also the
arch-criminal for myocardial ischemia reperfusion injury (MIRI) and
reducing the therapeutic benefit (2). We could not abandon the profit of
percutaneous coronary intervention, coronary artery bypass grafting
or thrombolytic agents but rescue reperfusion injury. The procedure
of MIRI refers to a series of complicated pathological processes,
including inflammatory response (3), calcium overload (4), complement activation (5), cell autophagy (6) and apoptosis (7). The inflammatory response with
Toll-like receptor (TLR) signaling is considered to be a pivotal
link to myocardial ischemia/reperfusion induced tissue injury
(8,9). Toll-like receptor 4 (TLR4)
transducted signal with an extracellular leucine rich repeat (LRR)
domain and a Toll/IL-1 receptor (TIR) domain plays a critical role
in the induction of the inflammatory response during MIRI (10). TLR4 was the first discovered
mammalian TLR and the only one of the TLRs family member that can
activate the myeloid differentiation factor 88 (MyD88)-dependent
pathways and TIR domain containing adapter inducing interferon β
(TRIF) dependent-pathway (11).
TLR4/MyD88 and TLR4/TRIF activated the transcriptional activity of
nuclear factor-κB (NF-κB) and interferon regulating factor 3 (IRF3)
respectively, inducing a series of inflammatory factors.
A variety of studies have demonstrated that
radioprotective 105 kDa protein (RP105) is a specific inhibitor of
the TLR4-triggered inflammatory response in dendritic cells,
macrophages and monocytes (12-14). RP105 is the specific homologue of
TLR4 of TLRs family containing an LRR domain, but TIR domain, which
could not transmit signaling alone (15). Surface expression and signal
transmission of TLR4 depends upon co-expression of myeloid
differential protein-2 (MD-2), while surface expression and TLR4
signaling inhibition of RP105 relies on co-localization of MD-1
homogenized to MD1 (15). The
physiological combination of RP105/MD-1 complex and TLR4/MD-2
complex contributes to the specific inhibition of LPS-induced TLR4
inflammation signaling by RP105 (16).
RP105 suppresses the MIRI-induced TLR4 signal in
cardiomyocytes in vivo. Our previous study proved that
overexpression of RP105 in cardiomyocytes resulted in de-activation
of the TLR4/P38MAPK/AP-1 signaling pathways, and was followed by
repressing myocardial cell apoptosis to protect MIRI (17). In addition, our unpublished data
show that RP105 transmission into cardiac myocytes markedly
repressed the pro-inflammatory action and remitted myocardial
damage during MIRI via TLR4/MyD88 signaling pathway. Nevertheless,
the mechanism of RP105 cardioprotection is complicated and still
incomplete. In this study, we utilized adenoviral transfection of
RP105 into cardiomyocytes to explore and consummate underlying
anti-inflammation mechanism further in an animal model of MIRI. Our
data illustrates that RP105 efficiently alleviated ischemia
reperfusion induced myocardial damage by negative regulation of
TLR4/TRIF signal pathway.
Materials and methods
Animal care and adenovirus vector
construction
All experiments were approved by the Insitutional
Animal Care Committee of the Faculty of Medicine, China Three
Gorges University, Yichang, China. Sprague-Dawley rats weighing
220-250 g were obtained from the Animal Center, China Three Gorges
University, Yichang, China. All the animals were housed in specific
pathogen-free (SPF) barrier environment during the procedure of
feeding and surgery.
Targeting gene RP105 was obtained by PCR and linked
to the shuttle vector Gv135 (CMv-MCS-EGFP) to form the linker
production. The recombinant vector CMv-RP105-MCS-EGFP was
transformed to E. coli DH5a cells to obtain a large number
of positive clones and confirmed by enzyme digestion and DNA
sequencing. AdMax virus packaging system was used for the
cotransfection of HEK293 cells by RP105 recombinant shuttle vector
and auxiliary packaging plasmid. By means of Cre/loxP recombinant
enzyme system, we acquired recombinant adenovirus Ad-RP105-EGFP and
its negative control Ad-EGFP. After the packaging, amplification
and purification of recombinant adenovirus vector, we obtained the
virus titer of 1×109 PFU/ml according to the
manufacturer's protocol.
Rats were randomly divided into four equal groups
(n=10): i) sham, normal non-ischemic group; ii) IR, myocardial
ischemia reperfusion group infected with saline; iii) Ad-RP105,
myocardial ischemia reperfusion infected with Ad-EGFP-RP105; and
iv) Ad-EGFP, myocardial ischemia reperfusion infected with
Ad-EGFP.
Surgical procedure of adenovirus
transfection and MIRI model establishment
All the surgical procedures of adenovirus transfer
and MIRI model establishment proceeded as previously described
(17,18) in the SPF animal laboratory,
avoiding the post-operation infections and other disturbing
elements of rats before their sacrifice. The animals were
anesthetized by intraperitoneal injection sodium pentobarbital (40
mg/kg) and fastened to the animal operating table. Rats were
ventilated via tracheal intubation of venous indwelling needle (24
G) with room air from the small animal ventilator at the rate of 80
breath/min and the inspiratory/expiratory ratio of 2:1. The heart
was exposed through a left thoracotomy between the fourth and fifth
ribs and transfected with 100 μl normal saline,
Ad-RP105-EGFP and Ad-EGFP at the heart apex of three separate sites
in respective group. Three days later, the second anesthetization,
ventilation and thoracotomy in the same manner was performed. After
the heart re-exposed, the left anterior descending coronary artery
(LAD) was ligated by 6-0 silk suture with medical latex tubing
(inner diameter, 1.5 mm, socket) to permit reversible occlusion of
LAD. Myocardial ischemia was monitored by lead II ECG with ST
segment elevation and ischemic region with pallor. Animals
underwent 30 min of myocardial ischemia, followed by 120 min of
reperfusion. Sham-operated rats were subjected the same procedures
without the blockage of the LAD or repatency. After surgery of
ischemic and reperfusion, all rats were sacrificed to collect blood
samples and myocardial tissue for the next step.
Immunofluorescent assay
We assessed the expression of RP105 successful
transfection in cardiac muscle tissue via immunofluorescent assay.
Myocardial tissues were fixed in 4.0% paraformaldehyde, embedded in
paraffin, and sectioned into 4 μm slices. After sections
were dewaxed and antigen fixing, slices were sealed by 1% bovine
serum albumin (BSA) for 30 min at room temperature, incubated with
anti-RP105 antibody (dilution 1:80; Santa Cruz Biotechnology, Inc.,
Santa Cruz, CA, USA) overnight at 4°C, hatched with Fluor Cy3
(red)-conjugated rabbit anti-bovine IgG (dilution 1:100; Boster
Biotechnology, Wuhan, China) for 60 min at 20-37°C in a humidified
box and stained with 4′,6′-diamidino-2-phenylindole (DAPI) under
the conditions of protection from light. Immunofluorescent assay
was executed with fluorescence microscopy (BX51; Olympus America,
Melville, NY, USA) in the dark.
Biochemical analysis
The blood serum lactate dehydrogenase (LDH) and
creatine kinase MB (CK-MB) were measured using a commercially
available analytical kit (Beijing Kemeidongya Biotechnology Ltd.,
Wuhan, China).
Assessment of myocardial infarct
size
Myocardial infarct area was measured by the Evans
blue/triphenyltetrazolium chloride (TTC) double-staining as
previously described (17,18).
At the end of 2 h reperfusion, 1 ml 2.0% Evans blue dye
(Sigma-Aldrich, St. Louis, MO, USA) was injected via jugular vein
with LAD re-ligated. The heart was isolated from in vivo,
frozen at −80°C for 30 sec, sliced into 5 sections (2 mm) and
stained in 1.5% TTC (Sigma Chemicals) at 37°C for 15 min. There are
three colors in the sections, including blue representing the
viable non-ischemic area, red expressing the myocardium area at
risk (AAR) and white presenting infarct area (IA). The assessment
of myocardial infarct size is presented by the percentage of IA/AAR
that was calculated in Image-Pro Plus 5.0 software.
Hematoxylin and eosin (H&E)
staining
Hearts were fixed with 4% buffered formaldehyde for
24 h at room temperature, embedding in paraffin wax, and cutting
into slices (5 μm). Subsequently, sections were stained with
H&E and observed under a light microscope.
Enzyme-linked immunosorbent assay
(ELISA)
The levels of interferon-β (IFN-β) and tumor
necrosis factor-α (TNF-α) in myocardial tissue were detected by
ELISA following the manufacturer's instructions, using ELISA kits
(Abcam, Cambridge, MA, USA).
Western blot analysis
We processed western blot analysis to examine the
protein levels of TRL4/TRIF signaling pathway downstream in
myocardial tissue. Protein extraction was exerted by the RIPA Lysis
Buffer and concentration detection was determined using a
bicinchoninic acid protein assay kit (both from Beyotime, Jiangsu,
China). Equal amounts of protein were separated by 12% sodium
dodecyl sulfate-poly-acrylamide gel electrophoresis (SDS-PAGE) and
transferred to a nitrocellulose membrane by electroblotting. The
membrane was blocked by 5% defatted milk with Tris-buffered saline
Tween-20 (TBST) for 2 h at room temperature in case of nonspecific
binding. Then, the rinsed membrane was incubated with the
respective specific primary antibody overnight at 4°C, followed by
a peroxidase conjugated secondary antibodies. We used anti-TRIF
(dilution 1:600), anti-TBK1 (dilution 1:1,000) (both from Santa
Cruz Biotechnology, Inc.), anti-IRF3 (dilution 1:1,000) and
anti-p-IRF3 (dilution 1:600) (both from Cell Signaling Technology,
Danvers, MA, USA). horseradish peroxidase glyceraldehyde
3-phosphate dehydrogenase (GAPDH) was counted as a housekeeping
gene to normalize the intensity of different samples. Band was
measured with an enhanced chemiluminescence (ECL) reagent (Thermo
Fisher Scientific, Waltham, MA, USA) and a analyzed with BandScan
5.0 software.
Quantitative reverse
transcription-polymerase chain reaction (qRT-PCR)
Total RNA was extracted from myocardial tissue with
TRIzol Reagent (Invitrogen, Carlsbad, CA, USA) and
reverse-transcribed into complementary DNA (cDNA) with commercial
cDNA synthesis kit (Thermo Fisher Scientific). qRT-PCR was
conducted with the ABI Prism 7500 system using
SYBR-Green/flourescein qPCR Master Mix kit (Thermo Fisher
Scientific). Amplification conditions were: 50°C (2 min), 95°C (10
min), followed by 40 cycles of 95°C (30 sec), 60°C (30 sec).
Primers used to amplify TRL4/TRIF signaling pathway relevant gene
fragments are listed in Table I.
Target gene mRNA expression was normalized by their ratios to the
reference gene β-actin.
 | Table IPrimers used for qRT-RCR. |
Table I
Primers used for qRT-RCR.
| Gene | Sense | Antisense |
|---|
| β-actin |
CACGATGGAGGGGCCGGACTCATC |
TAAAGACCTCTATGCCAACACAGT |
| TRIF |
TGAGGAGGGTTTCTGGTAGC |
GGATGCCCAGAAGAACTTGT |
| TBK-1 |
AGATGTGGTGGGCGGAATGA |
CCGTGGCTGCGTGGTAGAAT |
| IRF3 |
ATGGCTGACTTTGGCATCTT |
GCTAATCGCAACACTTCTTTCC |
Electrophoretic mobility shift assay
(EMSA)
EMSA for the DNA-binding activity of IRF3 in
cardiomyocytes was performed. IRF3 binding consensus
oligonucleotides containing the ISREs in the rat IFN-β promoter
(5′-GAAAACTGAAAGGGAGAACTGAAAGTGGG-3′) were annealed and end-labeled
with biotin. Nucleoprotein extracted from myocardial cell of
respective group was incubated for 20 min on ice with binding
buffer before probe accretion. The mixture with addition of the
probe was further incubated for 15 min at 16°C, using LightShift
Chemiluminescent EMSA kit (viagene Biotech, Ningbo, China).
Unlabeled probe (5′-GAACCTGACCAGGGAGCCTGACCAGTGGG-3′) was used for
the competition assays. Protein with labeled or unlabeled probe
mixture was separated by electrophoresis on a 5.5% polyacrylamide
gel. Subsequently, gel was dried and exposed to X-ray film after
electrophoresis.
Statistical analysis
Statistical analyses were performed using SPSS
software (version 19.0). Data are expressed as mean ± SD.
comparisons between two groups were performed using the Student's
t-test, while multiple comparisons were conducted by one-way
analysis of variance with a Bonferroni post hoc test. The value of
P<0.05 was considered to be statistically significant.
Results
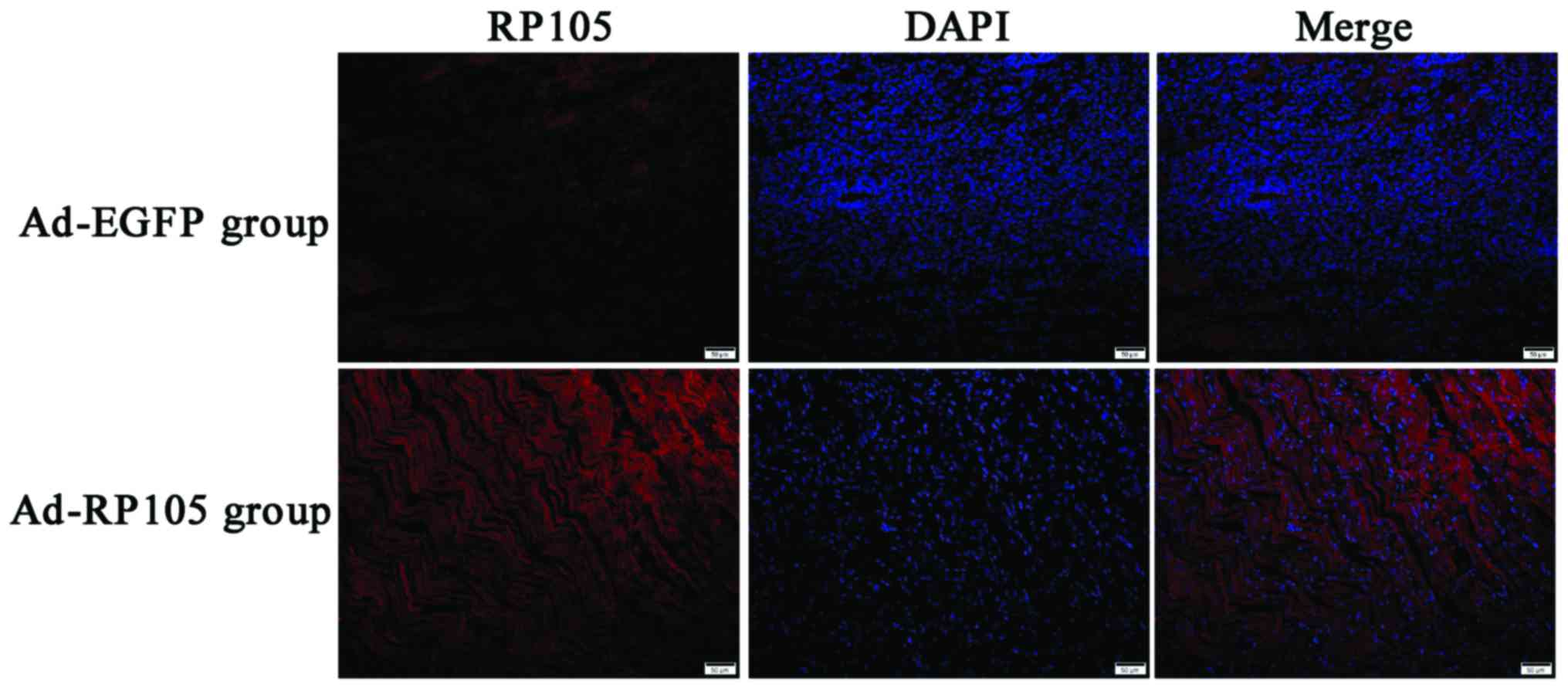
Successful delivery of RP105 into rat
cardiac muscle tissue
We injected the adenoviral vector Ad-EGFP-RP105 into
three separate sites at the apex of the rat heart, exploring the
anti-inflammatory mechanism of RP105 cardioprotection in MIRI
model. RP105 transduction was obviously visible in Ad-EGFP-RP105
group, whereas only very low levels of endogenous RP105 were
detected in Ad-EGFP group (Fig.
1). The DAPI-labeled nuclei density was higher in the latter
group, which was considered to be the nuclei of inflammatory
cells.
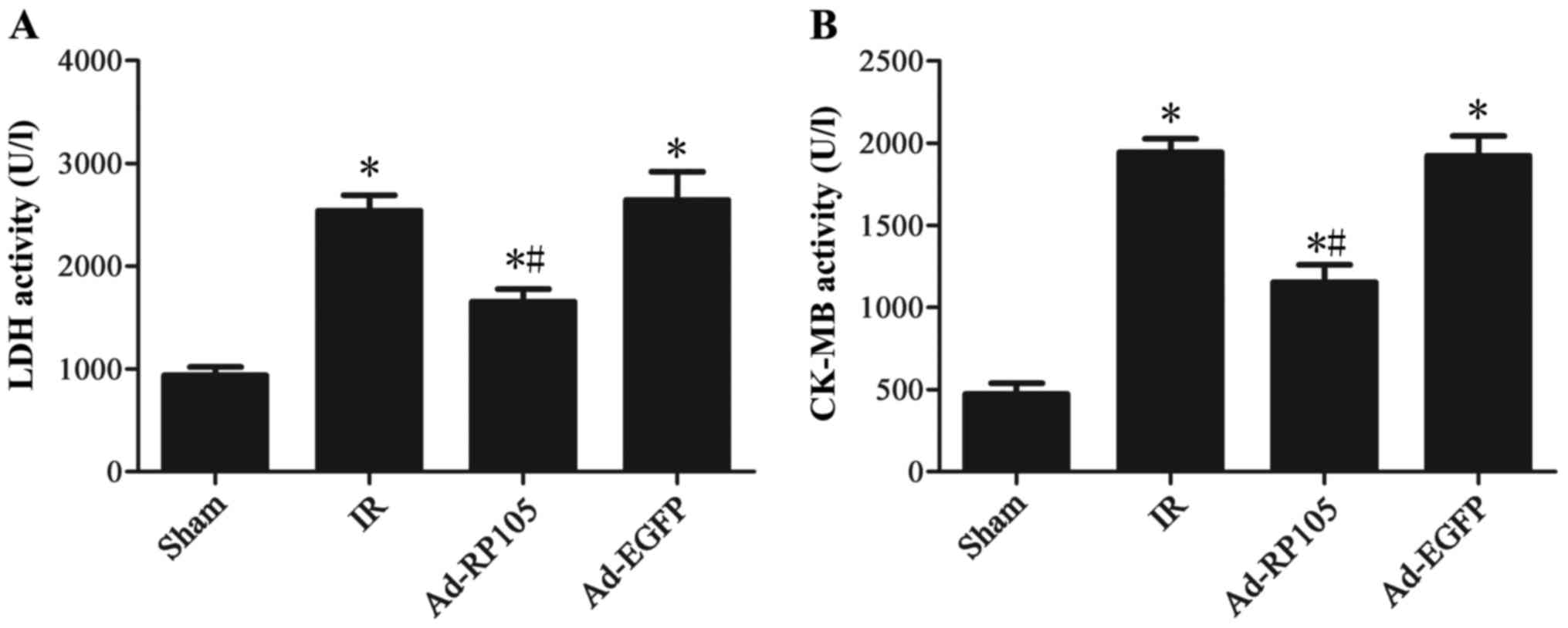
Upregulation of RP105 reduces serum
myocardial enzyme activities during MIRI
Fig. 2 illustrates
that 30 min ischemia followed by 2 h reperfusion resulted in a
significant increase in the activities of serum myocardial enzyme
CK-MB and LDH compared to the sham group (sham group vs. IR group,
P<0.05). After RP105 transduction into myocardium, the CK-MB and
LDH level decreased significantly compared to IR group or Ad-EGFP
group (Ad-RP105 group vs. IR group, P<0.05; Ad-RP105 group vs.
Ad-EGFP group, P<0.05).
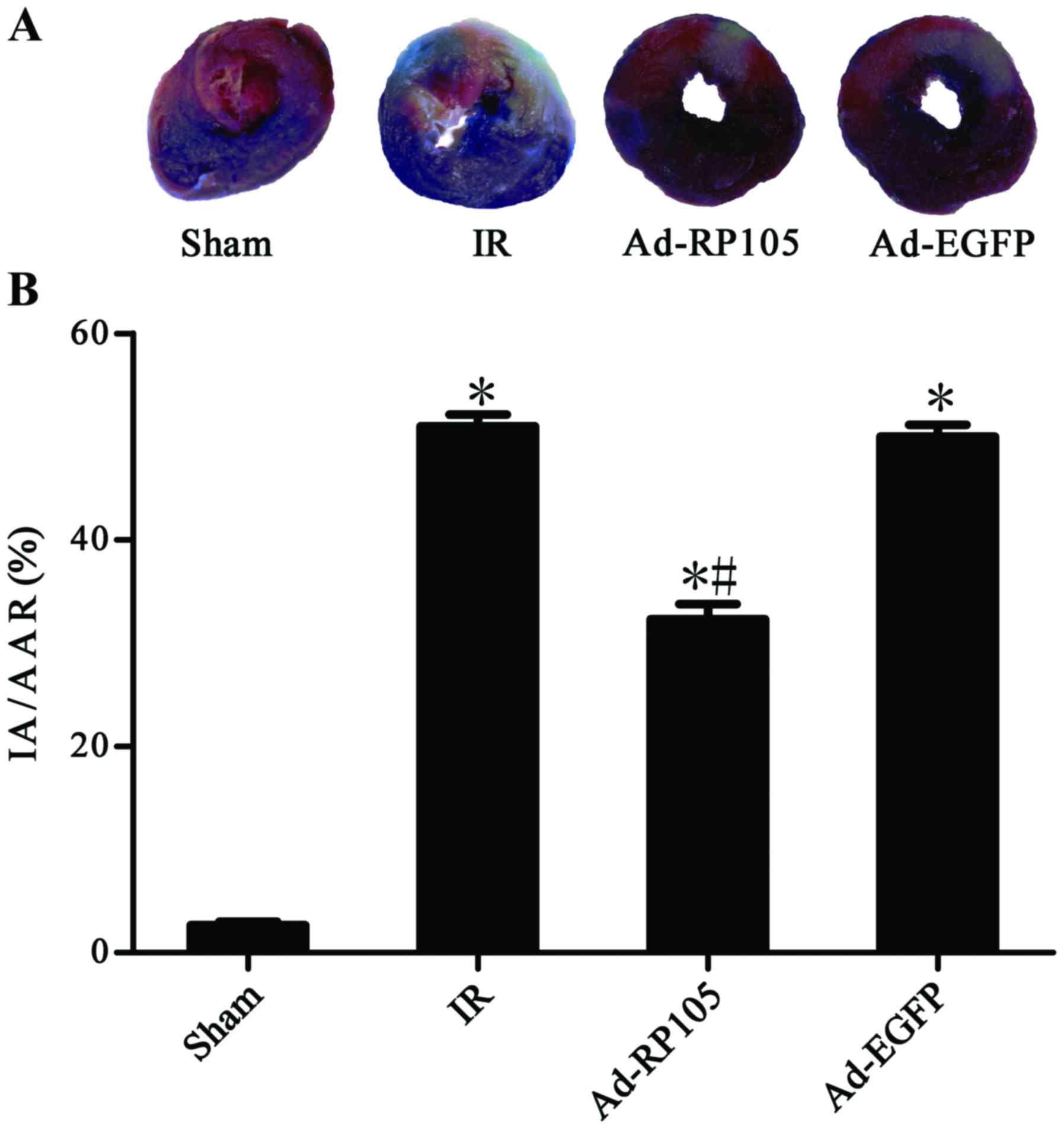
Upregulation of RP105 decreases
myocardial infarction during MIRI
Evans blue/TTC double-staining showed obvious white
infarct area after MIRI (Fig. 3A)
and myocardial infarct size was 51.20±1.21% in IR group and
50.43±1.21% in Ad-EGFP group (Fig.
3B) (IR group vs. Ad-EGFP group, P>0.05). Interestingly, the
myocardium transduced with Ad-EGFP-RP105 exerted cardioprotective
effect and had less infarct tissue 32.71±1.51% (Ad-RP105 group vs.
IR group, P<0.05; Ad-RP105 group vs. Ad-EGFP group, P<0.05).
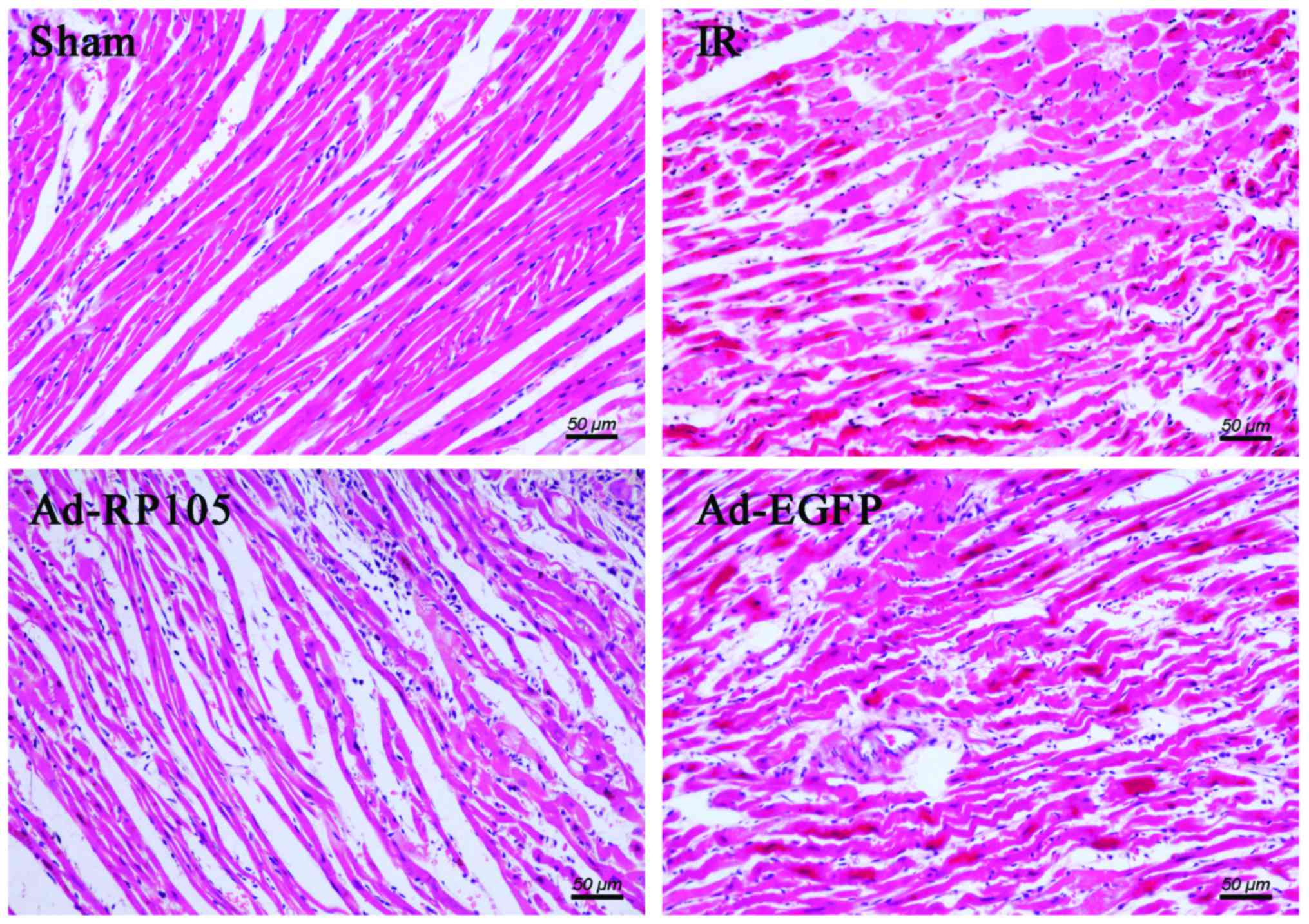
In light microscopy, we found that infarcted myocardium performed
myocardial fiber deformation and disorganization, myocardial cell
swelling and rupturing, and inflammatory cell infiltration was
distinct in IR group and Ad-EGFP group (Fig. 4). Whereas, myocardial tissues from
sham group were arranged regularly in clear striations without
apomorphosis or necrosis. However, delivery of RP105 partially
rescued myocardium with approximately normal structure, slight
edema and a tiny amount of inflammatory cell infiltration.
Upregulation of RP105 mitigates
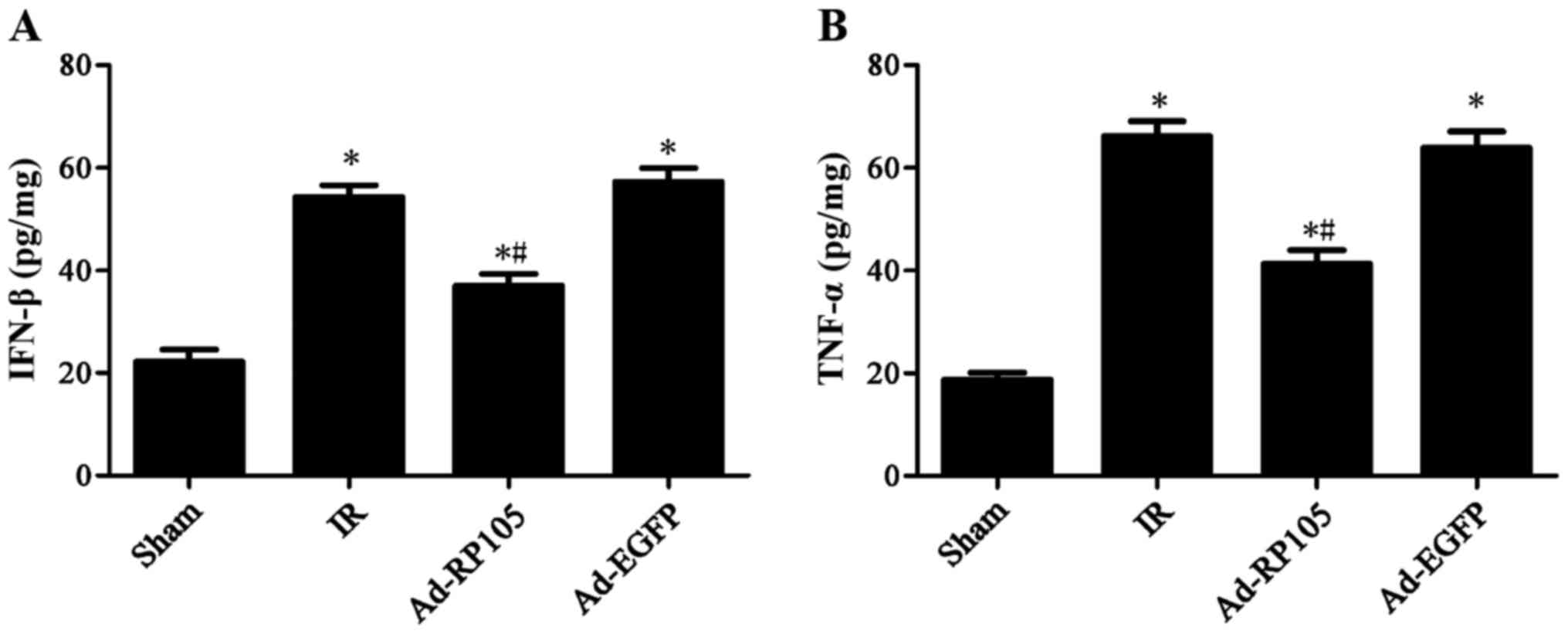
inflammatory factors IFN-β and TNF-α during MIRI
Rats subjected to MIRI exhibited a remarkable
increase in inflammatory cytokine IFN-β by 54.37±1.30 and TNF-α by
66.16±2.91 in IR group, as compared to the sham group (Fig. 5) (sham group vs. IR group, P-value
both <0.05). Notably, upregulation of RP105 in cardiac muscle
cells mitigated IFN-β to 37.01±1.31 and TNF-α to 41.33±2.73
(Ad-RP105 group vs. IR group, both P<0.05). Adenoviral
transfection of Ad-EGFP showed no significant difference on the two
cytokines comparing to IR group (IR group vs. Ad-EGFP group,
P>0.05).
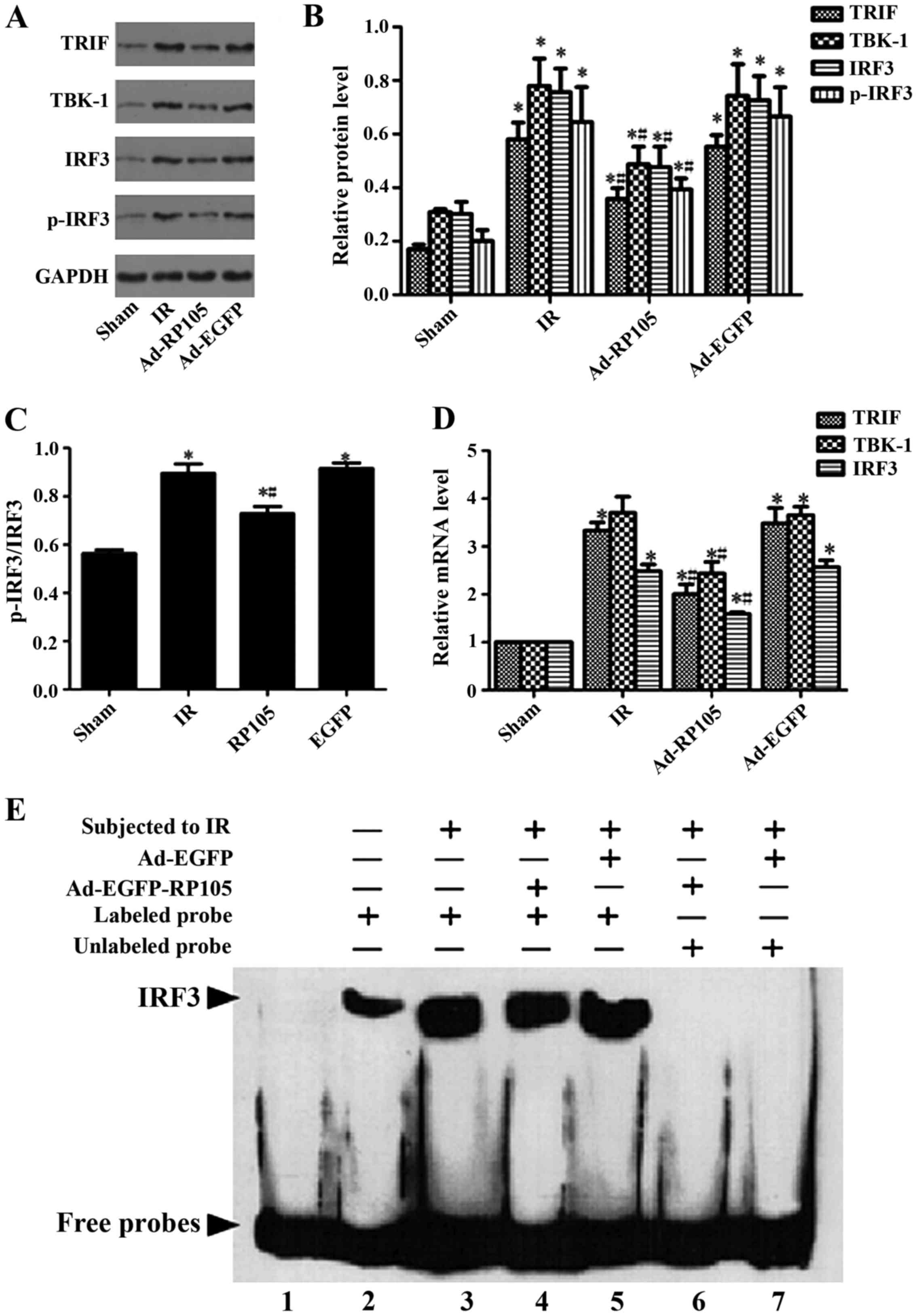
Upregulation of RP105 suppresses
inflammation during MIRI via inhibiting TLR4/TRIF signaling
pathway
We verified the relative protein and corresponding
mRNA in TLR4/TRIF/ IRF3 signaling pathway, as well as IRF3
transcription activity, to explore the specific cardioprotection
mechanism of RP105 during MIRI. Comparing with sham group, the
expression of relative protein TRIF, TBK-1, IRF3, p-IRF3 and the
ratio of p-IRF3 and IRF3 were obviously upregulated after 30 min
ischemia and 2 h reperfusion (Fig.
6A–C) (sham group vs. IR group, P<0.05). Transduction of
RP105 into myocardium markedly decreased above protein expression
(Ad-RP105 vs. IR group, P<0.05). qRT-PCR confirmed the above
analogical results in mRNA level. The mRNA levels of TRIF, TBK-1,
IRF3 and p-IRF3 were notably higher in IR group than in the sham
group (Fig. 6D, sham group vs. IR
group, P<0.05). Similarly, upregulation of RP105 reduced the
mRNA expression after delivering RP105 (Ad-RP105 vs. IR group,
P<0.05). Ad-EGFP did not affect IR-induced increase in protein
or mRNA level of TLR4/TRIF pathway (Ad-EGFP vs. IR group, P-value
both >0.05). We implemented EMSA assay to assess the
transcriptional activity of IRF3 in cardiomyocytes. Myocardial
ischemia reperfusion markedly increased the DNA-binding and
transcriptional activity of IRF3 (Fig. 6E) (sham group vs. IR group,
P<0.05), yet transfection of RP105 could attenuate it to some
degree (Ad-RP105 vs. IR group, P<0.05).
Discussion
Although myocardial ischemia and reperfusion
occurred in an aseptic environment, cell debris and endogenous
molecules like fibronectin-EDA and heat shock protein released from
myocardium, the so-called danger-associated molecular patterns
(DAMPs), would activate abacterial myocardial innate immune
response and injure cardiac muscle tissue via TLRs signaling
excitation (19). TLR4 plays a
vital role in the regulation of immune response and the activation
of inflammatory response during MIRI, as a result of
pro-inflammatory cytokine induction and chemokine factor
infiltration (11). In the
present study, we demonstrated that RP105 exerted a dramatic
inhibition of TLR4 inflammatory signaling pathway, developing
potent protective effect against myocardial ischemia and
reperfusion induced inflammation. RP105 overexpression with
adenovirus vectors reduced serum myocardial enzyme (CK-MB and LDH)
activities, decreased myocardial infarct size and mitigated
inflammatory factors IFN-β and TNF-α during MIRI, and TLR4/TRIF
signal pathway relative proteins and mRNAs (TRIF, TBK-1, IRF3 and
p-IRF3) were repressed with a low transcriptional activity of IRF3.
These findings first expounded that RP105 suppressed the ischemia
reperfusion induced inflammatory status in the heart via inhibiting
TLR4/ TRIF signaling pathway.
RP105 performes disparate regulatory roles in
diverse diseases owing to different cell types. RP105 was initially
discovered in the proliferation and radioactivation of murine B
cells (20). The proliferation
and activation, dependent upon TLR4 signaling, were distinctly
reduced during the LPS- induced humoral immune response in the B
cells derived from RP105 knockout mice (21). In other words, RP105 carried out a
promotional function in B cell activated inflammatory reponse.
Contrarily, the role of RP105 in myeloid cells (monocyte and
macrophagocyte) was the opposite. RP105-/- monocytes
increased the mean fluorescence intensity (MFI) of monocyte
activated marker CD11b and promoted the production of
pro-inflammation factors TNF-α and IL-6 stimulated by LPS, compared
to WT mice (22). Pre-incubated
with inhibitory anti-MD-1 antibody used to block the function of
the RP105/ MD-1 complex in macrophages, there was increased
secretion of pro-inflammatory cytokines TNF-α and IL-12p40 derived
from TLR4/MyD88 pathway and dreased anti-inflammatory cytokine
IL-10 (23). RP105 developed a
physiological mediator in myeloid cell inflammatory response
induced by TLR4 signal negatively. There is a hypothesis to explain
this phenomenon. Dichotomous roles of RP105 on TLR4 signaling in B
cells and myeloid cells are on account of disparate expression of
TLR4 in these different cell types, leading to the diverse
interactions with cell surface molecular partners (13). TLR4/MD-2 has a higher affinity for
homodimerization with itself than for heterodimerization with
RP105-MD-1 (13). High expression
of TLR4/MD-2 [e.g., on macrophages (24)] induced lower affinity
heterodimeric interactions to inhibit TLR4 multimerization and
signaling, while low expression of TLR4/MD-2 [e.g., on B cells
(21)] promoted heterodimeric
interactions to facilitate further TLR4 recruitment and signaling.
The fact that RP105 displays promotion role in B cell activation
and inhibition role in myeloid cells activation, may decipher the
RP105 controversial regulation in diverse conditions. The roles of
RP105 in the heart are very few and the mechanism remains unclear
and imperfect. The study of Louwe et al proved that
deficiency of the RP105 caused a boost in the inflammatory status
and a remarkable cardiac dilatation after induction of myocardial
infarction by amplifying TLR4 signaling (25). Our data confirmed that RP105
contributed to cardioprotection and negatively regulated the
TLR4/TRIF- induced inflammatory signal pathway.
TRIF was discovered as a TIR domain-containing
adaptor protein of TLR3 and TLR4 for signal transmission, inducing
type I IFNs strongly (26).
Stimulation of TRIF-dependent signaling pathway resulted in the
revitalization of the the transcriptional regulator IRF3, the
late-phase activation of NF-κB, and the activation of
mitogen-activated protein kinase (MAPK) (27,28). As TLR4 excitation, TRIF-related
adaptor molecule (TRAM) mediated the interaction between TIR domain
and TRIF to facilitate the conformation changes of TRIF and
recruitment of the downstream TNF receptor-associated factor 3
(TRAF3) and TRAF6 (29,30). TRAF6 promoted the recruitment and
ubiquitylation of receptor interacting protein 1 (RIP1) that
compound with transforming growth factor-β-activated kinase 1
(TAK1) and MAPKs, leading to the nuclear translocation of NF-κB and
AP-1, respectively. TRAF3 is the trigger of kinase TBK1 activation,
as a result of IRF3 phosphorylation and translocation into the
nucleus. The TLR4/TRIF/IRF3 signaling pathway induces type I
interferon and IFN-inducible genes (31,32). IFN-β in turn is recognized by the
surface receptor and triggers the activation and transduction of
JAK/STAT1 signaling, inducing the expression of inducible genes,
such as iNOS (33), IL-6
(34) TNF-α (35) and CXCL10 (33). Pro-inflammatory cytokines
transcribed by TLR4/TRIF/IRF3 signal pathway damage various
organisms. Several studies reported that inhibition of TRIF
signaling cascade exerted a protective role during ischemia
reperfusion injury in the liver, intestines and retina (36-41). Kang et al (36) and Kang and Lee (37) suggested that melatonin ameliorated
the liver ischemia reperfusion damage via suppressing the type I
IFN signaling pathway downstream of TLR4/TRIF/IRF3. Further studies
indicated that ischemia reperfusion induced liver damage could be
improved by inhibiting TLR4-triggered MyD88 and TRIF- dependent
pathways (38,39). In the analysis of intestinal and
retinal ischemia reperfusion injury, TRIF signaling contributed
more significantly to disease development than MyD88 signaling, and
TRIF-/- had an obviously reduced pro-inflammatory status
after IR (40,41). This study demonstrated that
TLR4/TRIF/IRF3 signaling played a crucial role in ischemia
reperfusion-induced myocardial damage and inhibition of it could
develop cardioprotection through RP105 recombinant adenovirus
transfection.
RP105 delivery into myocardium via recombinant
adenovirus vector to alleviate MIRI is a kind of gene therapy in
our present study. The basic concept of gene therapy is to transfer
functional genetic material with viral vectors or non-viral vectors
into human organs, tissues, or cells to supply therapeutic effects
in order to prevent or treat disease (42). Genetics has become a promising
therapeutic approach to treat and heal cardiovascular diseases and
able to recover the heart from dysfunctional to a functional state
(43). There is a successful
case. Intracoronary administration of an adenovirus vector encoding
the human HGF gene (Ad-HGF) in patients with coronary heart
disease, led to high expression of HGF and its receptor (c-met),
augmented serum concentration of HGF, vEGF, MCP-1 and IL-10 and
reduced serum concentration of IL-8, and increased the percentage
of CD34+ and CD117+ cells in peripheral blood
(44). However, gene therapy
efficacy is mainly restricted by insufficient transfection
efficiency of the target gene, negative immunoreaction to the
treatment and weak of therapeutic effect over time (45). Thus, the key point of gene therapy
is an appropriate targeting gene and an efficient and safe gene
delivery vector. We have confirmed the cardioprotection of RP105 in
MIRI, but its clinical application remains uncertain and needs
further study.
In conclusion, the present study demonstrated that
adenovirus transmission of RP105 into cardiocytes decreased serum
myocardial enzyme activities and pro-inflammatory cytokines via
inhibiting TLR4/TRIF signaling pathway to alleviate heart from
ischemia reperfusion injury. Our findings provide a theoretical
foundation of RP105 gene in MIRI.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (grant nos. 81170133, 81200088 and
81470387), the Hubei Province's Science Technology Support program
(grant no. 2015BKA340) and the Hubei Province's OutstandingMedical
Academic Leader program (grant no. 201304), China.
Notes
[1] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Go AS, Mozaffarian D, Roger VL, Benjamin
EJ, Berry JD, Blaha MJ, Dai S, Ford ES, Fox CS, Franco S, et al
American Heart Association Statistics Committee and Stroke
Statistics Subcommittee: Executive summary: Heart disease and
stroke statistics - 2014 update: a report from the American Heart
Association. Circulation. 129:399–410. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fang Y and Hu J: Toll-like receptor and
its roles in myocardial ischemic/reperfusion injury. Med Sci Monit.
17:RA100–RA109. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yang M, Chen J, Zhao J and Meng M:
Etanercept attenuates myocardial ischemia/reperfusion injury by
decreasing inflammation and oxidative stress. PLoS One.
9:e1080242014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ma HJ, Li Q, Ma HJ, Guan Y, Shi M, Yang J,
Li DP and Zhang Y: Chronic intermittent hypobaric hypoxia
ameliorates ischemia/reperfusion-induced calcium overload in heart
via Na/Ca2 exchanger in developing rats. Cell Physiol Biochem.
34:313–324. 2014. View Article : Google Scholar
|
|
5
|
Ilczuk T, Wasiutynski A, Wilczek E and
Gornicka B: The study of the protein complement in myocardial
infarction. Immunol Lett. 162:262–268. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xu J, Qin X, Cai X, Yang L, Xing Y, Li J,
Zhang L, Tang Y, Liu J, Zhang X, et al: Mitochondrial JNK
activation triggers autophagy and apoptosis and aggravates
myocardial injury following ischemia/reperfusion. Biochim Biophys
Acta. 1852:262–270. 2015. View Article : Google Scholar
|
|
7
|
Kim MY, Lim SH and Lee J: Intake of hot
water-extracted apple protects against myocardial injury by
inhibiting apoptosis in an ischemia/reperfusion rat model. Nutr
Res. 34:951–960. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Frangogiannis NG, Smith CW and Entman ML:
The inflammatory response in myocardial infarction. Cardiovasc Res.
53:31–47. 2002. View Article : Google Scholar
|
|
9
|
Chao W: Toll-like receptor signaling: A
critical modulator of cell survival and ischemic injury in the
heart. Am J Physiol Heart Circ Physiol. 296:H1–H12. 2009.
View Article : Google Scholar :
|
|
10
|
Ding HS, Yang J, Gong FL, Yang J, Ding JW,
Li S and Jiang YR: High mobility group [corrected] box 1 mediates
neutrophil recruitment in myocardial ischemia-reperfusion injury
through toll like receptor 4-related pathway. Gene. 509:149–153.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Akira S, Uematsu S and Takeuchi O:
Pathogen recognition and innate immunity. Cell. 124:783–801. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Divanovic S, Trompette A, Atabani SF,
Madan R, Golenbock DT, visintin A, Finberg RW, Tarakhovsky A, Vogel
SN, Belkaid Y, et al: Inhibition of TLR-4/MD-2 signaling by
RP105/MD-1. J Endotoxin Res. 11:363–368. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Divanovic S, Trompette A, Atabani SF,
Madan R, Golenbock DT, visintin A, Finberg RW, Tarakhovsky A, Vogel
SN, Belkaid Y, et al: Negative regulation of Toll-like receptor 4
signaling by the Toll-like receptor homolog RP105. Nat Immunol.
6:571–578. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wezel A, van der velden D, Maassen JM,
Lagraauw HM, de vries MR, Karper JC, Kuiper J, Bot I and Quax PH:
RP105 deficiency attenuates early atherosclerosis via decreased
monocyte influx in a CCR2 dependent manner. Atherosclerosis.
238:132–139. 2015. View Article : Google Scholar
|
|
15
|
Ohto U, Miyake K and Shimizu T: Crystal
structures of mouse and human RP105/MD-1 complexes reveal unique
dimer organization of the toll-like receptor family. J Mol Biol.
413:815–825. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kimoto M, Nagasawa K and Miyake K: Role of
TLR4/MD-2 and RP105/MD-1 in innate recognition of
lipopolysaccharide. Scand J Infect Dis. 35:568–572. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang J1, Guo X, Yang J, Ding JW, Li S,
Yang R, Fan ZX and Yang CJ: RP105 protects against apoptosis in
ischemia/reperfusion-induced myocardial damage in rats by
suppressing TLR4-mediated signaling pathways. Cell Physiol Biochem.
36:2137–2148. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yang J, Chen L, Yang J, Ding J, Li S, Wu
H, Zhang J, Fan Z, Dong W and Li X: MicroRNA-22 targeting CBP
protects against myocardial ischemia-reperfusion injury through
anti-apoptosis in rats. Mol Biol Rep. 41:555–561. 2014. View Article : Google Scholar
|
|
19
|
vilahur G and Badimon L:
Ischemia/reperfusion activates myocardial innate immune response:
The key role of the toll-like receptor. Front Physiol. 5:4962014.
View Article : Google Scholar
|
|
20
|
Miyake K, Yamashita Y, Hitoshi Y, Takatsu
K and Kimoto M: Murine B cell proliferation and protection from
apoptosis with an antibody against a 105-kD molecule:
Unresponsiveness of X-linked immunodeficient B cells. J Exp Med.
180:1217–1224. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ogata H, Su I, Miyake K, Nagai Y, Akashi
S, Mecklenbräuker I, Rajewsky K, Kimoto M and Tarakhovsky A: The
toll-like receptor protein RP105 regulates lipopolysaccharide
signaling in B cells. J Exp Med. 192:23–29. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bastiaansen AJ, Karper JC, Wezel A, de
Boer HC, Welten SM, de Jong RC, Peters EA, de vries MR, van
Oeveren-Rietdijk AM, van Zonneveld AJ, et al: TLR4 accessory
molecule RP105 (CD180) regulates monocyte-driven arteriogenesis in
a murine hind limb ischemia model. PLoS One. 9:e998822014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hoebe K, Du X, Georgel P, Janssen E,
Tabeta K, Kim SO, Goode J, Lin P, Mann N, Mudd S, et al:
Identification of Lps2 as a key transducer of MyD88-independent TIR
signalling. Nature. 424:743–748. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Akashi S1, Shimazu R, Ogata H, Nagai Y,
Takeda K, Kimoto M and Miyake K: Cutting edge: cell surface
expression and lipopolysaccharide signaling via the toll-like
receptor 4-MD-2 complex on mouse peritoneal macrophages. J Immunol.
164:3471–3475. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Louwe MC, Karper JC, de vries MR, Nossent
AY, Bastiaansen AJ, van der Hoorn JW, Willems van Dijk K, Rensen
PC, Steendijk P, Smit JW, et al: RP105 deficiency aggravates
cardiac dysfunction after myocardial infarction in mice. Int J
Cardiol. 176:788–793. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu S, Cai X, Wu J, Cong Q, Chen X, Li T,
Du F, Ren J, Wu YT, Grishin NV, et al: Phosphorylation of innate
immune adaptor proteins MAvS, STING, and TRIF induces IRF3
activation. Science. 347:aaa26302015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Covert MW, Leung TH, Gaston JE and
Baltimore D: Achieving stability of lipopolysaccharide-induced
NF-kappaB activation. Science. 309:1854–1857. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fitzgerald KA, McWhirter SM, Faia KL, Rowe
DC, Latz E, Golenbock DT, Coyle AJ, Liao SM and Maniatis T:
IKKepsilon and TBK1 are essential components of the IRF3 signaling
pathway. Nat Immunol. 4:491–496. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kagan JC, Su T, Horng T, Chow A, Akira S
and Medzhitov R: TRAM couples endocytosis of Toll-like receptor 4
to the induction of interferon-beta. Nat Immunol. 9:361–368. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
O'Neill LA and Bowie AG: The family of
five: TIR-domain-containing adaptors in Toll-like receptor
signalling. Nat Rev Immunol. 7:353–364. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bowie AG and Haga IR: The role of
Toll-like receptors in the host response to viruses. Mol Immunol.
42:859–867. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Perry AK, Chen G, Zheng D, Tang H and
Cheng G: The host type I interferon response to viral and bacterial
infections. Cell Res. 15:407–422. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Toshchakov V, Jones BW, Perera PY, Thomas
K, Cody MJ, Zhang S, Williams BR, Major J, Hamilton TA, Fenton MJ,
et al: TLR4, but not TLR2, mediates IFN-beta-induced
STAT1alpha/beta-dependent gene expression in macrophages. Nat
Immunol. 3:392–398. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
34
|
Samavati L, Rastogi R, Du W, Hüttemann M,
Fite A and Franchi L: STAT3 tyrosine phosphorylation is critical
for interleukin 1 beta and interleukin-6 production in response to
lipopolysaccharide and live bacteria. Mol Immunol. 46:1867–1877.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hui KP1, Lee SM, Cheung CY, Ng IH, Poon
LL, Guan Y, Ip NY, Lau AS and Peiris JS: Induction of
proinflammatory cytokines in primary human macrophages by influenza
A virus (H5N1) is selectively regulated by IFN regulatory factor 3
and p38 MAPK. J Immunol. 182:1088–1098. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kang JW, Koh EJ and Lee SM: Melatonin
protects liver against ischemia and reperfusion injury through
inhibition of toll-like receptor signaling pathway. J Pineal Res.
50:403–411. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kang JW and Lee SM: Melatonin inhibits
type 1 interferon signaling of toll-like receptor 4 via heme
oxygenase-1 induction in hepatic ischemia/reperfusion. J Pineal
Res. 53:67–76. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Huang HF, Zeng Z, Wang KH, Zhang HY, Wang
S, Zhou WX, Wang ZB, Xu WG and Duan J: Heme oxygenase-1 protects
rat liver against warm ischemia/reperfusion injury via
TLR2/TLR4-triggered signaling pathways. World J Gastroenterol.
21:2937–2948. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Mahmoud MF, Gamal S and El-Fayoumi HM:
Limonin attenuates hepatocellular injury following liver ischemia
and reperfusion in rats via toll-like receptor dependent pathway.
Eur J Pharmacol. 740:676–682. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Dvoriantchikova G, Santos AR, Danek D,
Dvoriantchikova X and Ivanov D: The TIR-domain-containing adapter
inducing interferon-β-dependent signaling cascade plays a crucial
role in ischemia-reperfusion-induced retinal injury, whereas the
contribution of the myeloid differentiation primary response
88-dependent signaling cascade is not as pivotal. Eur J Neurosci.
40:2502–2512. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang J, He GZ, Wang YK, Zhu QK, Chen W and
Guo T: TLR4-HMGB1-, MyD88- and TRIF-dependent signaling in mouse
intestinal ischemia/reperfusion injury. World J Gastroenterol.
21:8314–8325. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lyon AR, Sato M, Hajjar RJ, Samulski RJ
and Harding SE: Gene therapy: Targeting the myocardium. Heart.
94:89–99. 2008. View Article : Google Scholar
|
|
43
|
Mason D, Chen YZ, Krishnan HV and Sant S:
Cardiac gene therapy: Recent advances and future directions. J
Control Release. 215:101–111. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yang ZJ, Xu SL, Chen B, Zhang SL, Zhang
YL, Wei W, Ma DC, Wang LS, Zhu TB, Li CJ, et al: Hepatocyte growth
factor plays a critical role in the regulation of cytokine
production and induction of endothelial progenitor cell
mobilization: A pilot gene therapy study in patients with coronary
heart disease. Clin Exp Pharmacol Physiol. 36:790–796. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sasano T, Kikuchi K, McDonald AD, Lai S
and Donahue JK: Targeted high-efficiency, homogeneous myocardial
gene transfer. J Mol Cell Cardiol. 42:954–961. 2007. View Article : Google Scholar : PubMed/NCBI
|