Ferroptosis is a novel form of cell death discovered
in recent years, and is characterised by an excessive accumulation
of cellular levels of lipid peroxide, caused by elevated levels of
reactive oxygen species (ROS) owing to a severe imbalance in the
intracellular redox state. This process is closely linked to
intracellular iron homeostasis, where an accumulation of the
strongly oxidising ferrous ion, which becomes a labile iron pool,
can generate ROS via the Fenton or Haber-Weiss reaction, thereby
initiating the ferroptosis process. Ferroptosis is classified as a
form of regulatory necrosis, a class of genetically regulated cell
death. Its similarity to necrosis is the disruption of plasma
membrane integrity and the release of cytoplasmic contents, which
usually leads to a potent inflammatory response (1,2).
However, unlike necrosis, the unregulated decadence process
resulting from extreme adverse conditions, different types of
regulated necrosis have different downstream execution mechanisms
and stimulatory molecular pathways (1,2).
Ferroptosis is regulated by a variety of factors, such as the
glutathione peroxidase 4 (GPX4) antioxidant system, dihydroorotate
dehydrogenase (DHODH), the ferroptosis suppressor protein 1
(FSP1)-mediated ferroptosis protection mechanism and the
lipoxygenase trigger mechanism, which are independent of each other
and together influence the occurrence of ferroptosis (3). An increasing number of studies have
revealed that ferroptosis is a key mechanism involved in the
development and progression of cancer, as well as in the
development of drug resistance. Therefore, it is crucial to
elucidate the regulatory mechanisms that underlie ferroptosis.
CUGBP ELAV-like family (CELF) proteins are a family
of RNA-binding proteins (RBPs) that, similar to the majority of
RBPs, play broad and diverse roles in RNA regulation. CELF2 is the
second member of this family and has been found to play a critical
role in cancer development. CELF2 functions as a tumour suppressor
in a variety of tumours, and its downregulation is associated with
a poor patient prognosis (4,5).
The tumour-suppressive effects of CELF2 are dependent on the
post-transcriptional regulation of various genes, such as heme
oxygenase-1 (HO-1) and cyclooxygenase 1 (COX-1)
(6,7), and its inhibition of various
cellular signalling pathways.
The present review discusses in detail the possible
mechanisms through which CELF2 regulates the mitogen-activated
protein kinase (MAPK) signalling pathway, PI3K/AKT signalling
pathway, endoplasmic reticulum (ER)-associated protein degradation
(ERAD) pathway, autophagy and the Wnt/β-catenin pathway.
Subsequently, the role of these pathways in influencing ferroptosis
was further summarized. It was hypothsized that the
tumour-suppressive effects of CELF2 may be partially dependent on
ferroptosis mechanisms.
MAPK is an intracellular signalling pathway that has
been extensively studied. This cascade is activated by a sequence
of three to five hierarchical layers of protein kinases known as
the MAPK kinase kinase kinase (MAPKKKK) class, MAPK kinase kinase
(MAPKKK) class, MAPK kinase (MAPKK) class, MAPK and MAPK-activated
protein kinase (MAPKAPK) (8). The
first three layers are considered the basic core units that
recognise and conduct various signals inside and outside the cell
via phosphorylation. MAPK activation is a critical step in the MAPK
signalling pathway that mainly involves the extracellular
signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK) and
p38 protein families. These proteins can activate a variety of
MAPKAPKs, thereby regulating the expression of downstream genes or
proteins and allowing cells to respond to intra- and extracellular
signals including proliferation, differentiation, apoptosis,
senescence and carcinogenesis (9,10).
Targeting the MAPK pathway, which is the most commonly mutated
signalling pathway in human cancers, has long been considered a
promising strategy for cancer therapy. An increasing number of
recent studies have demonstrated that influencing the ferroptosis
process is a key mechanism by which the MAPK signalling pathway
promotes tumour development (11-14). The role of CELF2 in the MAPK
signalling pathway and its possible role in ferroptosis is
illustrated in Fig. 1.
ERK1/2, two key members of the ERK/MAPK pathway, are
normally found in the cytoplasm and, when activated, can enter the
nucleus and regulate the activity of various transcription factors
and gene expression, playing a crucial role in cell differentiation
and proliferation (15).
Additionally, ERK1/2 activation can regulate ferroptosis in cancer
cells via multiple pathways.
FBW7 is a target of the ERK/MAPK signalling pathway,
and ERK1 can directly bind and phosphorylate the Thr205 site of
FBW7, which promotes the ubiquitinated degradation of FBW7 in a
Pin1-dependent manner, thereby inhibiting FBW7 expression (16). FBW7 is an E3 ubiquitin ligase that
ubiquitinates target substrates, usually via K11 or K48 linkages,
and consequently degrades target proteins that include a number of
crucial human cancer proteins (17,18). Thus, FBW7 functions as a tumour
suppressor; its expression is downregulated in several tumours and
is associated with patient prognosis (16,18). As previously reported, FBW7
induces cancer cell death by promoting ferroptosis. For example,
FBW7 has been shown to reduce the binding of nuclear receptor
subfamily 4 group A member 1 to the promoter of stearoyl-CoA
desaturase 1 (SCD1), an enzyme that converts saturated fatty
acids to monounsaturated fatty acids, and to inhibit its
transcription via an unknown mechanism, thereby promoting
ferroptosis in pancreatic cancer cells (19). Furthermore, FBW7 has been found to
be able to recognize and ubiquitinate c-Myc in a manner dependent
on Thr58 and Ser62 phosphorylation, thereby regulating the level of
c-Myc in cancer cells and likely influencing the process of
ferroptosis in cancer cells (20,21).
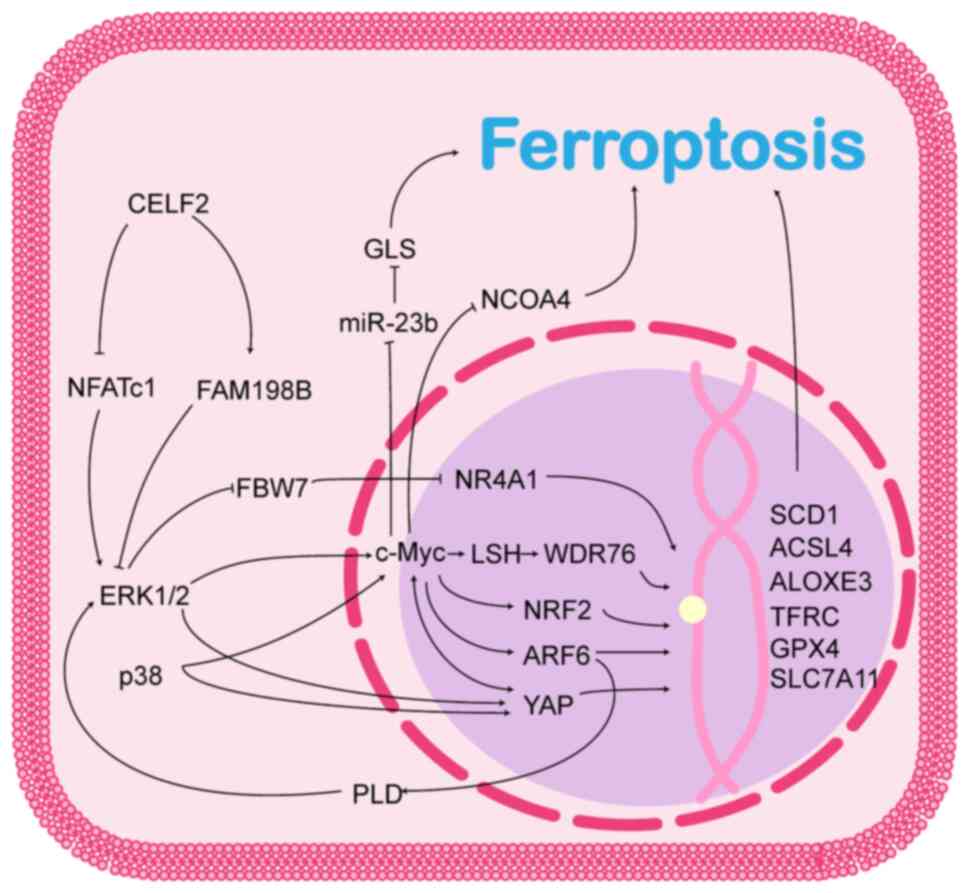
c-Myc is located downstream of the ERK/MAPK
signalling pathway. In addition to indirectly regulating c-Myc
levels through FBW7, ERK1/2 directly phosphorylates Ser62 of c-Myc
and activates its transcription under conditions of oxidative
stress (22). Moreover, p38a,
another MAPK parallel to ERK, promotes c-Myc expression by directly
phosphorylating Ser64 and Ser67 and inhibiting its
proteasome-dependent degradation (23), or transcriptionally activating
c-Myc by directly binding and phosphorylating β-catenin (24). c-Myc is considered an oncogenic
transcription factor that binds to the promoters of various
oncogenes to promote their transcription. It has been reported that
c-Myc is able to transcriptionally activate a variety of
ferroptosis suppressor genes, conferring cancer cells the ability
to resist ferroptosis. For example, c-Myc is enriched at the solute
carrier family 7 member 11 (SLC7A11) and γ-glutamylcysteine
synthetase promoters, and transcriptionally activates these genes
to help cancer cells resist oxidative stress through a pathway that
promotes glutathione synthesis, which also renders cancer cells
resistant to chemo- and radiotherapy (22,25-27). c-Myc activates lymphoid specific
helicase gene transcription, which promotes WD40-repeat protein 76
enrichment at the SCD1/fatty acid desaturase 2 promoter and
inhibits ferroptosis through a pathway that affects lipid
metabolism (28). Furthermore,
c-Myc can bind to the E-box on the nuclear factor E2-related factor
2 (NRF2) promoter to activate NRF2 transcription, thereby
maintaining intracellular redox homeostasis (29). The non-transcription factor
activities of c-Myc have also been identified; for example, c-Myc
has been found to be able to directly inhibit the expression of
miR-23b, which regulates cellular ferroptosis by targeting
glutaminase expression at the 3′-untranslated region (3′-UTR)
(30,31). c-Myc was also demonstrated to
directly bind and inhibit nuclear receptor coactivator 4
(NCOA4) mRNA expression, suppressing ferroptosis by
inhibiting ferritinophagy (32).
ADP-ribosylation factor 6 (ARF6), a small GTPase
belonging to the Ras superfamily, is mainly localised in the plasma
membrane and endosomal compartments, and plays a crucial role in
plasma membrane endocytosis, cytokinesis, endosomal recycling,
cytokinesis and actin cytoskeletal reorganisation (33). ARF6 is located downstream of the
ERK/MAPK signalling pathway and ERK1/2 activates ARF6 transcription
via c-Myc (34). Notably, ARF6
continuously activates the ERK/MAPK pathway by interacting with
phospholipase D (35). This forms
a positive feedback mechanism that maintains high levels of c-Myc
in cancer cells (34). Recent
studies have revealed that although ARF6 does not directly regulate
lipid peroxidation, it can alter the sensitivity of cancer cells to
oxidative stress, rendering them less sensitive to ferroptosis
induced by RSL3 and erastin, and thereby participating in the
development of drug resistance in cancer cells (36,37). ARF6 has been reported to inhibit
the expression of acyl-CoA synthetase long-chain family member 4
(ACSL4) (36) and GPX4 (37) at the transcriptional level,
allowing pancreatic and gastric cancer cells to develop tolerance
to tabine analogues.
YAP is a key effector of the Hippo pathway and is
frequently dysregulated in human cancers. Aberrantly activated YAP
has emerged as a key driver of tumorigenesis, chemoresistance and
tumour metastasis (38). YAP is a
co-transcription factor that interacts with DNA-binding
transcription factors to regulate diverse cellular behaviours
(39). Recent studies have
revealed that YAP is located downstream of the MAPK pathway and
that ERK1/2/5 (40-42), p38 (41), mechanistic target of rapamycin
(mTOR)C2 (43) and c-Myc
(44) can activate YAP in a
non-Hippo pathway-dependent manner. Notably, the reciprocal
upregulation of c-Myc and YAP has been observed in hepatocellular
carcinoma. In hepatocellular carcinoma cells, c-Myc activates YAP
transcription by interacting with hepatitis B X-interacting protein
(44), while YAP promotes c-Myc
transcription by binding to c-Abl (45). Taken together, YAP and c-Myc
promote the development of hepatocellular carcinoma. However, its
role in ferroptosis remains unclear. YAP enhances the binding of
transcriptional enhanced associate domain (TEAD)4 to transferrin
receptor protein (TFRC), ACSL4, and arachidonate
lipoxygenase (ALOX)E3 (46-48), thereby enhancing the sensitivity
of cancer cells to ferroptosis, whereas the YAP-TEAD complex
inhibits the expression of threonine tyrosine kinase and TFRC via
s-phase kinase-associated protein 2 (49), thereby protecting cancer cells
from ferroptosis. Moreover, the YAP-activating transcription factor
(ATF)4 complex can bind to the SLC7A11 promoter and promote
its transcription, thereby promoting the resistance of
hepatocellular carcinoma cells to sorafenib (50). These studies suggest that the role
of YAP in ferroptosis is complex and may be related to the cancer
cell type and genetic background, and that the role of YAP in
ferroptosis requires further exploration.
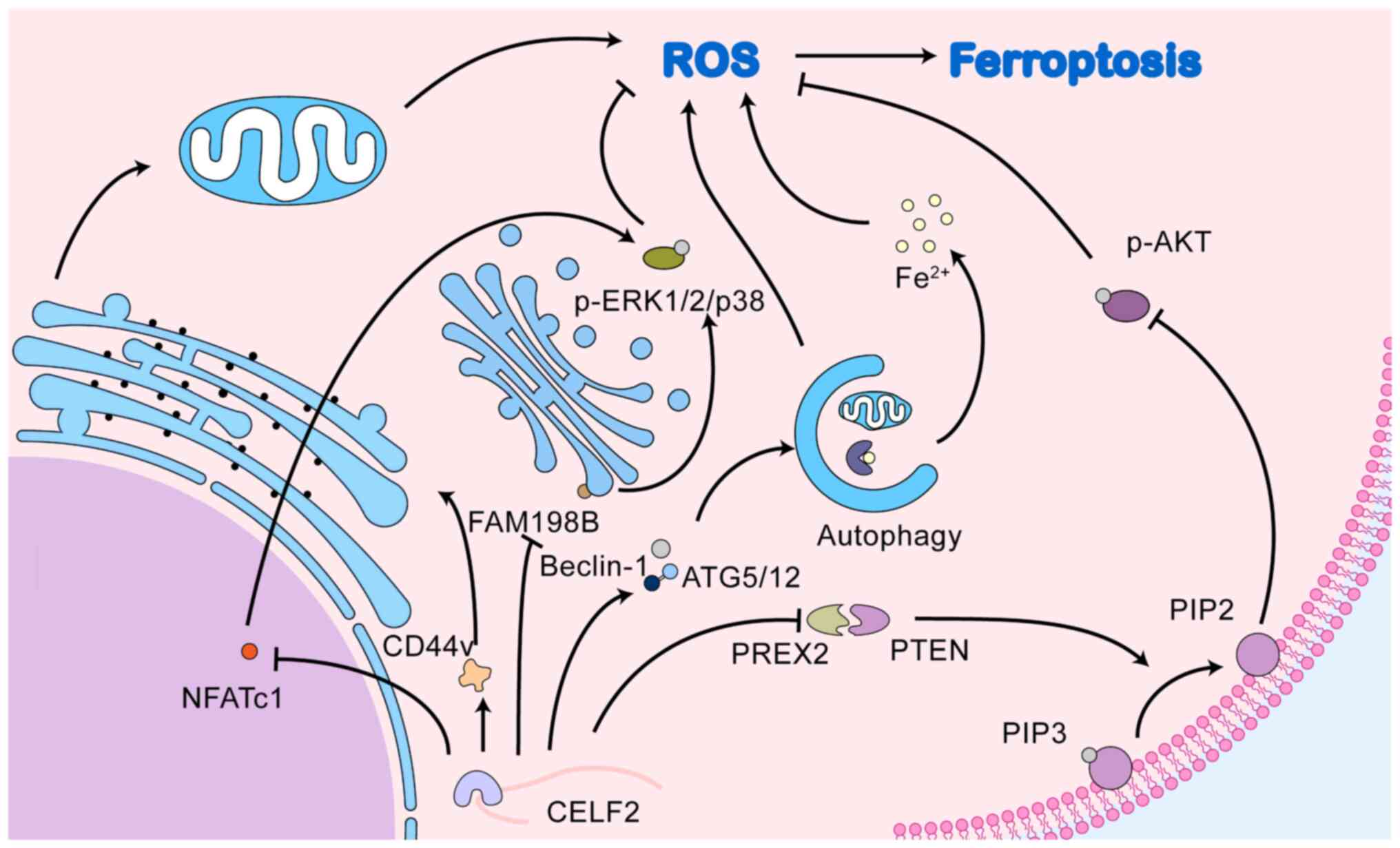
Based on the data from available studies, CELF2 may
affect the MAPK pathway through both the human family with sequence
similarity 198, member B (FAM198B) and nuclear factor of activated
T-cell c1 (NFATc1) pathways. FAM198B is an N-linked
glycoprotein with unknown functions that is localised to the Golgi
membrane (51). CELF2 stabilises
FAM198B mRNAs by binding to its AREs (AU/U-rich elements) within
the 3′-UTR and then upregulates the expression of FAM198B (52). FAM198B is downregulated in lung
and ovarian adenocarcinomas and is associated with a poor patient
prognosis (51,52). FAM198B also functions in the
tumour microenvironment. For example, Zheng et al (53) observed that the expression level
of FAM198B in macrophages of colon cancer tissues correlated with
patient prognosis. FAM198B regulates M2 polarisation in macrophages
and promotes colon cancer progression by targeting SMAD2 (53). Existing research demonstrates that
FAM198B exerts its tumour-suppressive effects mainly by inhibiting
the ERK/MAPK pathway (51,52);
however, the exact mechanisms involved remain elusive. The only
factor that can be determined is that the inhibitory effect of
FAM198B on the ERK/MAPK pathway depends on its three major
glycosylation sites, namely, Asn98, Asn289 and Asn322, and that
defects in the glycosylation sites would result in FAM198B being
unable to inhibit the ERK/MAPK signalling pathway (51).
NFATc1 is a major transcription factor involved in
osteoblast differentiation. Recent studies have demonstrated that
it is upregulated in a variety of cancer types and mediates the
malignant behaviour of cancer cells. The cancer-promoting function
of NFATc1 is largely dependent on c-Myc expression. In addition to
directly binding to the TGFβ inhibitory element of the c-Myc
promoter (54), NFATc1
upregulates c-Myc expression by activating the ERK1/2/p38 MAPK
signalling pathway (55), thereby
promoting the progression of ovarian (55), lung (56) and pancreatic (54) cancers. The knockdown of NFATc1
reduces ERK1/2 phosphorylation, whereas the pharmacological
inhibition of ERK1/2 similarly impairs NFATc1 expression (57). NFATc1 and the ERK1/2 MAPK
signalling pathway appear to have a mutually reinforcing
relationship. The mechanisms through which NFATc1 regulates the
MAPK signalling pathway remain unclear. One possible explanation is
that NFATc1 maintains the activation of the MAPK signalling pathway
by interacting with STAT3 and promoting the transcription of
proteins upstream of the MAPK signalling pathway (58). CELF2 exerts tumour-suppressive
effects by regulating NFATc1 expression. A previous animal study
discovered that tumour size and weight were substantially reduced
in mice overexpressing CELF2, and that the overexpression of CELF2
was associated with reduced NFATc1 levels in tumour tissue
(59). Furthermore, NFATc1
overexpression significantly reversed the CELF2-mediated reduction
in the viability and invasive capacity of MCF-7 cells (59). Taken together, these results
suggest that CELF2 affects tumour progression via the NFATc1/MAPK
pathway.
PI3K is a lipid kinase that phosphorylates the 3-OH
moiety of phosphatidylinositol in the plasma and cell membranes.
There are several PI3K classes, among which, the most extensively
studied is class I PI3K, whose activation is involved in a variety
of biological behaviours in cancer cells. The main focus of the
present review was PI3K signalling pathway. Class I PI3K converts
phosphatidylinositol-3,4-bisphosphate (PIP2) on the plasma membrane
to phosphatidylinositol 3,4,5-triphosphate (PIP3), which binds AKT
and pyruvate dehydrogenase lipoamide kinase isozyme 1 to PH domains
and facilitates their interaction to phosphorylate and activate AKT
(60,61). Activated AKT phosphorylates
several downstream effectors, ultimately leading to cell growth,
survival, and proliferation. The PI3K/AKT signalling pathway has
been shown to promote tumour progression and drug resistance
development by inhibiting ferroptosis (62,63). The role of CELF2 in the PI3K/AKT
signalling pathway and its possible role in ferroptosis is
illustrated in Fig. 2.
NRF2 is a critical transcription factor that
regulates antioxidant responses and plays a key role in preventing
ferroptosis. In response to oxidative stress, the nuclear
translocation of NRF2 is caused by the suppressed inhibitory effect
of kelch-like ECH-associated protein 1 (Keap1) on NRF2. In the
nucleus, NRF2 interacts with antioxidant response elements located
in the mRNA promoter region, which encodes a subset of antioxidant
genes, such as metallothionein-1G, HO-1, NAD(P)H:quinone
oxidoreductase 1, ferritin heavy chain (FTH)1 and
SLC7A11, ultimately activating and targeting gene
transcription, thereby enhancing resistance to ferroptosis and
promoting the development of drug resistance in cancer cells. The
activation of the PI3K/AKT pathway induces the development of
sorafenib resistance in cancer cells by upregulating NRF2 (64-66). Indeed, the PI3K/AKT pathway can
phosphorylate and inhibit glycogen synthase kinase-3β (GSK3β),
which attenuates the inhibitory effect of GSK3β on Fyn, which can
phosphorylate the Tyr568 site of NRF2, leading to the nuclear
export, ubiquitination and degradation of NRF2 (67-69).
mTOR is a protein kinase that regulates cell growth,
survival, metabolism and immunity, and is a major effector of the
PI3K/AKT signalling pathway. Although mTOR can phosphorylate and
activate p62, promoting the binding of p62 to Keap1 and
upregulating NRF2 expression (70), its inhibitory effect on
ferroptosis is not largely dependent on NRF2 (71). mTOR inhibits ferroptosis by
directly upregulating GPX4 and SLC7A11 expression (72,73). In addition, mTOR upregulates
sterol regulatory element binding protein-1 (SREBP-1) at
both the transcriptional and post-translational levels (74,75), and the overexpression of SREBP-1
promotes resistance to ferroptosis through a pathway that affects
lipid metabolism in cancer cells (71).
HIF-1α is another gene downstream of the
PI3K/AKT/mTOR signalling pathway. The PI3K/AKT/mTOR pathway
promotes the translation of HIF-1α by regulating eukaryotic
translation initiation factor 4E (eIF4E)-binding protein 1
(eIF-4EBP1), eIF-4E and TRPV1-ribosomal protein 70 S6 kinase
(p70S6K) via phosphorylation (76,77). However, the regulation of HIF-1α
by the PI3K/AKT signalling pathway is cell-specific and is
influenced by environmental oxygenation. The inhibition of the
PI3K/AKT pathway significantly inhibits HIF-1α accumulation under
normoxic conditions, but not under hypoxic conditions (78,79). Under hypoxic conditions, the
PI3K/AKT pathway plays a minimal role in regulating HIF-1α.
Additionally, the overexpression of HIF-1α can promote AKT
phosphorylation, which is dependent on the activation of autocrine
growth factor genes, such as IGF-II and TGF-α by
HIF-1α (80). The positive
feedback effects of the PI3K/AKT pathway and HIF-1α combine to
promote cancer progression (80).
HIF-1α has also been found to play an inhibitory role in
ferroptosis. For example, under hypoxic conditions, HIF-1α, which
is highly expressed in gliomas and gastric cancer, promotes SLC7A11
expression by increasing the stability of SLC7A11 mRNA via the poly
(methacrylic acid-niclosamide) polymer/ELAV-like RNA binding
protein 1 pathway, thereby promoting resistance to ferroptosis and
sulfasalazine (81,82). Moreover, HIF-1α can also promote
the production of NADPH from glucose into the pentose phosphate
pathway to maintain intracellular redox homeostasis (83).
CELF2 influences tumour progression by affecting the
PI3K/AKT signalling pathway (84,85). Indeed, the ability of CELF2 to
bind to PREX2 and reduce the interaction of PREX2 with PTEN
increases the activity of PTEN phosphatase, which reverses the
conversion of PIP2 to PIP3, thereby inhibiting activation of the
PI3K/AKT signalling pathway (86,87). However, whether CELF2 influences
ferroptosis through the PI3K/AKT signalling pathway requires
further investigation.
Recent studies have revealed that autophagy plays a
crucial role in ferroptosis; for example, ferritinophagy, the
degradation of ferritin by autophagy, is a main cause of cellular
ferroptosis (91-93). During ferritinophagy, FTH binds
specifically to NCOA4 to form a complex and forms cohesions in
response to interactions between NCOA4 protein molecules (94,95), which are then degraded by
lysosomes via the macroautophagic (92,96) or macroautophagic (97-99) pathways, releasing Fe2+
and causing ferroptosis. During this process, NCOA4 functions as an
autophagic cargo receptor and is essential for ferritinophagy.
Therefore, NCOA4 is a key target of ferritinophagy. In addition,
NCOA4 has been reported to mediate mitochondrial autophagy in the
context of iron homeostasis imbalance. For example, deferiprone, an
iron chelator, causes cellular iron depletion that increases
mitochondrial ferritin (FTMT) expression through the
HIF-1α/transcription factor specificity protein 1 axis and
localises its precursor form to the outer mitochondrial membrane,
whereas NCOA4 interacts with the precursor form of FTMT and
triggers mitochondrial autophagy to inhibit hepatocellular
carcinoma cell growth (100,101).
The mitochondria play a critical role in
ferroptosis. Ferroptosis is accompanied by mitochondrial
dysfunction and the accumulation of mitochondrial ROS (mtROS).
Mitochondria release mtROS into the cytoplasm to exacerbate their
accumulation in the cytoplasm via several mechanisms (102), among which, mitochondrial
autophagy may be a key mechanism (103-105). The excessive accumulation of
mtROS and its resultant mitochondrial dysfunction, manifested as
mitochondrial depolarisation, has been well documented as a cause
of mitochondrial autophagy (105-107). Indeed, the excessive
accumulation of mtROS and mitochondrial dysfunction can induce the
activation of mitochondrial PTEN-induced kinase 1 (PINK1) (108) and its localisation to the outer
mitochondrial membrane (109,110), leading to PINK1-mediated
ubiquitin-dependent mitochondrial autophagy. Moreover,
mitochondrial DNA damage caused by the excessive accumulation of
mtROS leads to an increase in intracytoplasmic mitochondrial DNA,
which can trigger mitochondrial autophagy via the GCAS (cyclic
GMP-AMP synthase)-STING1 (stimulator of interferon response cGAMP
interactor 1) signalling pathway (111). Damaged mitochondria are
enzymatically cleaved in endolysosomes, which is a cellular defence
mechanism that removes dysfunctional mitochondria, thereby
preventing excessive ROS from damaging the cell. This also obscures
the role of mitochondrial autophagy in tumour development. The
analyses of public databases (112,113) have demonstrated that PINK1
expression is decreased in a variety of tumours and plays
contradictory roles in various tumours and even in different cells.
For example, in hepatocellular carcinoma, some studies have
demonstrated a tumour-suppressive effect of PINK1-mediated
mitochondrial autophagy (114,115), whereas other studies have
revealed opposing results (113,116). These conflicting results
indicate that mitochondrial autophagy may be influenced by certain
factors that determine the ultimate effect of mitochondrial
autophagy on the cell, whether facilitated or inhibited.
Endolysosomes are key sites for removing damaged
organelles and abnormal proteins from cells. Endolysosomes contain
significant amounts of iron and play a crucial role in maintaining
cellular iron homeostasis (117,118). In acidic endolysosomes, iron is
mainly present in the ferrous form and excessive iron content
renders the endolysosomal membrane more susceptible to oxidative
damage by ROS (117,119). It has been reported that in
erastin- or RSL3-induced ferroptosis, the ROS content of the
endolysosome increases rapidly and leads to endolysosomal membrane
permeabilization, resulting in the release of ROS into the
cytoplasm, producing a cytoplasmic ROS burst and triggering
ferroptosis (120,121). Moreover, excess levels of
Fe2+ can also lead to changes in the endolysosomal
function and structure. For example, FAC, an iron agent, can
significantly increase the number and surface area of endolysosomes
and can lead to their dephosphorylation, which drives
Fe2+ within endolysosomes into the cytoplasm via
divalent metal transporter 1 and the related non-selective two-pore
cation channels, thereby exacerbating cytoplasmic iron overload and
promoting ferroptosis (122,123). In summary, these findings
illustrate that endolysosomes play a role in promoting ferroptosis
in the context of iron and ROS overload. During ferroptosis,
ferritinophagy and mitochondrial autophagy provide large amounts of
Fe2+ and ROS to endolysosomes, which causes
endolysosomal dysfunction and exacerbates the ferroptosis-promoting
function of the endolysosome, which partially explains the
conflicting roles of mitochondrial autophagy.
Therefore, the following conclusions can be inferred
about autophagy and ferroptosis: During ferroptosis, autophagy is
activated to eliminate excess Fe2+ and ROS from the
cell; however, dysfunctional endolysosomes do not assist the cell
to deal with the excess ROS and even leak iron and ROS from the
lysosome, causing a burst of intracytoplasmic ROS and allowing
ferroptosis to occur in cancer cells.
CELF2, an RNA-binding protein, can directly bind to
the mRNA of autophagy-related factors, allowing CELF2 to be
directly associated with cellular autophagy. In colorectal cancer,
the overexpression of CELF2 induced by radiotherapy is able to bind
to Beclin1 and autophagy related gene (ATG)5/12
mRNAs, increasing their half-life and promoting the onset of
autophagic cell death (124).
In addition to directly regulating autophagy-related
factors, CELF2 may indirectly regulate cellular autophagy via the
MAPK and PI3K/AKT pathways. The p38/MAPK signalling pathway plays a
dual role in autophagy. p38 can inhibit the activity of unc-51 like
autophagy activating kinase 1 (ULK1) (125,126), ATG5 (127), ATG8 (128) and mATG9 (129), thereby reducing autophagic flux;
by contrast, p38 can also promote autophagy in some cases. For
example, p38 has been found to promote autophagy through the heat
shock protein 27/CREB pathway, which activates the transcription of
ATG7 (130); the
proteasome inhibitor MG231 is also able to induce LC3II production
via the p38/GSK3β pathway, thereby activating autophagy (131). p38 also inhibits TP53
ubiquitinated degradation via phosphorylation modifications, the
latter activating the downstream DNA damage-regulated autophagy
modulator 1 gene, mediating autophagy induced by ROS accumulation
(132). In the JNK/MAPK
signalling pathway, activated JNK can enter the nucleus and promote
the transcription of various autophagy regulators, such as LC3,
Beclin1, Sestrin2 and ATG5/7 (133-137). Simultaneously, JNK can also
promote autophagy through non-transcriptional mechanisms in some
cases, such as through phosphorylation of BCL2. In addition, ERK1/2
and JNK, but not p38, can localise to the mitochondria, increasing
the stability of PINK1 and promoting mitochondrial autophagy
(138,139). In recent years, researchers have
found that the MAPK and PI3K/AKT pathways activate p62 via the
NRF2-Keap1 axis, which functions as an autophagic cargo receptor
that binds to LC3 and promotes autophagosome formation (140).
mTORC1, located downstream of the MAPK and PI3K/AKT
pathways, is a key junction in the regulation of autophagy. mTORC1
is a critical inhibitor of autophagy, and available studies have
shown that mTORC1 affects autophagy through several mechanisms.
First, ULK1 is the primary downstream target of mTORC1 that can
affect ULK1 activity through phosphorylation modifications and
post-translational pathways. Activated mTORC1 interacts with ULK1
and joins the ULK1-ATG13-FAK-family interacting protein of 200 kDa
complex, whereas mTORC1 directly phosphorylates the Ser757 site in
ULK1 (141,142). Ser757 is a key regulatory site
of ULK1. The phosphorylation of ULK1 Ser757 not only prevents the
activation of ULK1 by AMPK, which inhibits the phosphatase kinase
activity of ULK1 (143), but
also disrupts the interaction of ULK1 with ATG13, which helps
localise ULK1 to the detached membrane, thereby inhibiting the
initiation of autophagy (125).
Moreover, mTORC1 promotes the ubiquitination of ULK1 by tumour
necrosis factor receptor-associated factor 6 by phosphorylating the
Ser52 site of the activating molecule in Beclin1-regulated
autophagy protein 1, thereby reducing the stability of ULK1
(144). Second, activated mTORC1
phosphorylates the Ser113 and Ser120 (nuclear receptor binding
factor 2 (NRBF2) sites. When mTORC1 is inhibited, the
dephosphorylated form of NRBF2 binds ATG14-BECN1, facilitating the
assembly of the Ptdlns3K complex and stimulating the production of
Ptdlns3P on the isolated membrane, which can link to ULK1 and
activate autophagy (145).
Third, mTORC1 binds and phosphorylates the Ser498 site of the UV
radiation resistance-associated gene (UVRAG). Phosphorylated UVRAG
is able to inhibit the activity of Vps34 via RUN domain Beclin
1-interacting and cysteine-rich containing protein, whereas its
function to activate homotypic fusion and vacuole protein sorting
is diminished, resulting in a decrease in ras-like small GTPase
superfamily member 7 activity, thereby inhibiting the initiation of
autophagy and the maturation of autophagosomes and endosomes
(146). Finally, mTORC1 can also
reduce autophagic flux by phosphorylating autophagy regulators,
such as death-associated protein 1 (147) and p70S6K (148).
The Wnt/β-catenin signalling pathway has beeb found
to be a negative regulator of autophagy. This pathway inhibits
autophagosome maturation mainly by suppressing p62/SQSTM1
expression (149,150). Fan et al (151) found that miR-363-3p led to the
activation of the Wnt/β-catenin signalling pathway by targeting
CELF2 and induced epithelial-mesenchymal transition (EMT) in glioma
cells. That study demonstrated for the first time that CELF2 may be
located upstream of the Wnt/β-catenin signalling pathway; however,
it did not explain the specific regulatory mechanisms. The
mechanisms through which CELF2 affects autophagy through the
Wnt/β-catenin signalling pathway require further investigation. The
association between CELF2 and autophagy and its possible role in
ferroptosis is illustrated in Fig.
3.
The ER is a crucial site for protein synthesis and
processing, and its function is vulnerable to external factors. A
variety of conditions, such as nutritional deprivation, hypoxia,
viral infection, oxidative stress and calcium depletion can cause
an imbalance in cellular compartment homeostasis and lead to ER
stress, which is characterised by the accumulation of misfolded
proteins within the ER lumen.
ER stress is also involved in ferroptosis. Increased
acidity and viscosity within the ER have been reported in
erastin-induced ferroptosis, and the combination of erastin and
dithiothreitol, an ER stress inducer, caused significant increases
in acidification and viscosity in the ER over a short period,
suggesting the involvement of ER stress in the ferroptosis process
(152,153). However, the contribution of ER
stress to ferroptosis is so complex that it cannot be explained
merely by changes in ER content. ER stress exerts varying, or even
contrasting, effects on ferroptosis under different conditions. For
example, in renal tubular epithelial cells (154) and hepatocytes (155), ER stress induced by cadmium
exposure leads to ferritinophagy, whereas in lung cancer, ER stress
caused by Ca2+ bursts can lead to the reprogramming of
Ca2+ distribution and mitochondrial dysfunction,
facilitating the ferroptosis process through ROS production
(156). Moreover, another study
reported that the pharmacological inhibition or siRNA knockdown of
zrt-like, and Irt-like protein family member 7 induced ER stress by
affecting zinc metabolism, which promoted the transcription of
homocysteine-responsive endoplasmic reticulum-resident
ubiquitin-like domain member 1 and protected MDA-MB-231, RCC1 and
HT1080 cells from ferroptosis damage through an unknown mechanism
(157). The unfolded protein
response (UPR), a signalling mechanism induced by ER stress aimed
at resolving misfolded ER proteins and restoring ER homeostasis, is
one of the most critical downstream pathways of ER stress and plays
an unknown role in ferroptosis. Protein kinase R-like endoplasmic
reticulum kinase) and ATF6α, two major UPR effectors, have been
found to play opposing roles in ferroptosis (158-161). In summary, these studies
collectively suggest that ER stress is involved in ferroptosis and
that its function is influenced by a variety of factors.
Similar to the UPR, ERAD is a key quality control
mechanism in cells capable of degrading natural or misfolded
proteins within the ER and maintaining ER homeostasis (162). The ERAD process is broadly
divided into three stages. First, ERAD substrates are recognised by
chaperone proteins or chaperone-like lectins and are retained
within the ER. Depending on their nature or sorting mechanisms
within the ER, ERAD substrates are ubiquitinated by various E3
ubiquitin-linked enzymes and transported to the cytoplasm by
p97/VCP proteins in an ATP-dependent manner. Finally, the substrate
proteins are degraded by the proteasome (163,164). This involves complex molecular
mechanisms that will not be described herein, as they exceed the
scope of the present review. During ER stress, both the UPR and
ERAD are activated and both mechanisms play a crucial role in
restoring ER homeostasis. For example, the activation of the UPR
promotes protein folding, while also increasing the expression of
ERAD-related proteins and promoting the role of ERAD in degrading
misfolded proteins; by contrast, defects in ERAD can also lead to
the accumulation of misfolded proteins within the ER, resulting in
sustained ER stress, subsequently leading to cell death (165). Therefore, these two mechanisms
have complementary, synergistic and irreplaceable functions. The ER
and mitochondria are highly functionally associated; therefore, the
status of the ERAD pathway also appears to be associated with
mitochondrial function. Eeyarestatin I, an ERAD inhibitor,
reportedly reorganises the overall mitochondrial activity in HepG2
cells, resulting in mitochondrial dysfunction by elevating the
intramitochondrial Ca2+ and ROS levels (166). In addition, the inhibition of
the ERAD pathway leads to the accumulation of various substrate
proteins on the ER membrane, such as sigma non-opioid intracellular
receptor 1 (SigmaR1) (167) and
diacylglycerol o-acyltransferase 2 (168), and the excessive accumulation of
these proteins can affect mitochondrial function through various
pathways and may therefore be involved in the ferroptosis process
(169).
In addition to resisting ER stress, ERAD maintains a
basal state of activation under normal conditions and is involved
in the degradation of normal proteins within the ER. A previous
study identified SigmaR1 as an ERAD substrate in brown adipocytes
(167). That study demonstrated
that the knockdown of the Sel1L-Hrd1 complex, the most conserved
form of ERAD from yeast to humans, resulted in SigmaR1 accumulation
on ER membranes, which allowed the mitochondria to fuse in response
to cold stimulation and reduced the mitochondrial utilisation of
lipid droplets. This phenomenon was independent of ER stress
(167). Of note, SigmaR1 has
also been found in various cancer cells (170-172) and has been shown to function as
an inhibitor of ferroptosis in hepatocellular carcinoma cells
(173,174). Furthermore, cytochrome P450, an
upstream regulator of ferroptosis (175), can be degraded as a substrate
for ERAD (176). Therefore, the
ERAD pathway may function as an upstream regulator of
ferroptosis.
CD44 is a hyaluronan-binding cell surface signal
transduction receptor that plays a crucial role in the genesis,
invasion and metastasis of a number of tumours, and is widely
considered a marker of cancer stem cells. CD44 contains two
variable regions encoded by variable exons; therefore, there are
multiple isoforms of CD44, including standard CD44 (CD44s) and
variant CD44 (CD44v). The dysregulation of alternative splicing
frequently occurs in cancer, resulting in a shift from CD44s to
CD44v, which can profoundly affect tumour biology (177). Lai et al (178) observed that CELF2, an important
factor for mRNA alternative splicing, was involved in the
alternative splicing of CD44 and led to a transition from CD44s to
CD44v. Lai et al (178)
also found that the role of CD44v in promoting pancreatic cancer
development was dependent on the ERAD pathway, and that an
inhibitor of the ERAD pathway was effective in reversing the
effects of CD44 on cancer cells. Although the exact mechanisms of
ERAD regulation by CD44 have not yet been elucidated, it is
suggested that CELF2 functions as an upstream regulator of the ERAD
pathway. The association between CELF2 and ERAD and their possible
role in ferroptosis are illustrated in Fig. 4.
In the classical Wnt/β-catenin signalling pathway,
the Wnt protein binds to the Frizzled and low-density lipoprotein
receptor-related protein 5/6 (LRP5/6) co-receptors, thereby
activating Dishevelled protein (DVL). This prevents adenomatous
polyposis coli, axis inhibition protein (AXIN) and GSK3β from
forming a destructive complex that prevents the phosphorylation and
subsequent degradation of β-catenin. The accumulated β-catenin then
translocates to the nucleus and activates downstream genes by
binding to different co-transcription factors to form
transcriptional complexes. The Wnt/β-catenin signalling pathway
plays an inhibitory role in ferroptosis. β-catenin translocated to
the nucleus promotes the activation of ferroptosis regulatory
genes, such as GPX4 (179), COX2
(182), SCD1 (181), peroxisome proliferator-activated
receptor-γ coactivator 1-α (181), matrix metalloproteinases
(182) and c-Myc (182,183), and promotes tolerance to
platinum-based chemotherapeutic agents through ferroptosis
resistance in gastric (184) and
ovarian (179) cancers and brain
metastases from lung adenocarcinoma (185).
The MAPK cascade, and PI3K/AKT and Wnt/β-catenin
signalling pathways crosstalk in a variety of tumours, and all
three pathways play synergistic or alternative roles in promoting
tumour progression. The pro-tumorigenic effects of the MAPK and
PI3K/AKT pathways are partly dependent on the activation of the
Wnt/β-catenin signalling pathway. Thus, the MAPK and PI3K/AKT
pathways may play regulatory roles upstream of the Wnt/β-catenin
signalling pathway. As previously reported, p38, JNK and ERK1/2 are
all able to phosphorylate the Ser1490 and Thr1572 sites of LRP6,
which allows LRP6 to provide more AXIN1 and GSK3β binding sites,
while isolating these two proteins from the β-catenin destruction
complex and thereby reducing the degradation of β-catenin (186,187). Therefore, GSK3β is a critical
regulatory target. AKT directly phosphorylates the Ser9 site of
GSK3β to negatively regulate GSK3β activity, inhibit β-catenin
degradation and promote Wnt/β-catenin pathway activation (188). In addition, β-catenin is a
direct target of the MAPK and PI3K/AKT pathways. (p21-Activated
kinase 1, located downstream of the PI3K/AKT and ERK/MAPK
signalling pathways, activates the Wnt/β-catenin pathway by
directly phosphorylating β-catenin and promoting its nuclear
localization (189); ERK2 also
promotes the nuclear translocation of β-catenin by inhibiting the
linkage of α-catenin and β-catenin through the phosphorylation of
casein kinase 2α (190).
Taken together, these results suggest that CELF2
may affect the Wnt/β-catenin signalling pathway via MAPK, PI3K/AKT
and autophagy. The association between CELF2 and the Wnt/β-catenin
pathway and its possible role in ferroptosis are illustrated in
Fig. 5.
Ferroptosis is a novel form of cell death
characterised by the excessive accumulation of intracellular lipid
peroxide, which is dependent on an increase in intracellular
iron-dependent ROS. Ferroptosis involves a variety of factors, such
as the GPX4 antioxidant system, the ALOX and
Ca2+-independent phospholipase A2β pathways,
DHODH and FSP1, and occurs under the combined regulatory effect of
these factors. Moreover, the pro-tumour effects of signalling
pathways, such as MAPK, PI3K/AKT and Wnt/β-catenin, are partly
dependent on the resistance of cancer cells to ferroptosis.
Ferroptosis plays a crucial role in the development of cancer cells
and may serve as a mechanism for tumour therapy.
CELF2 contains three RNA recognition motifs, two at
the N-terminus and one at the C-terminus, and a segment of a
divergent structural domain that may mediate interactions with RNA
(192). This determines the
RNA-binding properties of CELF2 (192). Indeed, CELF2 expression is
reduced in a variety of cancers and is significantly associated
with tumour stage and a poor prognosis of patient patients with
various types of cancer, including in non-small cell lung (87), colorectal (5,193), glioblastoma (151), nasopharyngeal (194), gastric (195), breast (4), ovarian (52) and pancreatic cancers (178), and CELF2 may be a key locus for
the action of various dysregulated miRNAs or lncRNAs (84,86,151,193-195). The overexpression of CELF2 in
these tumour cell lines has been reported to inhibit their
biological behaviours, including proliferation, invasion,
migration, EMT, and resistance to radio- and chemotherapy (7,85,124,193-199), although the exact mechanisms
involved remain unclear. The activation of MAPK or PI3K/AKT
signalling pathways owing to the downregulation of CELF2 has been
found to be sufficient for inducing proliferation, invasion and
migration of cancer cells in a variety of tumours (84,86,87), but CELF2 may act through multiple
mechanisms throughout tumour development. For example, in gliomas,
the migration and invasion of cancer cells caused by CELF2
downregulation may be associated with the activation of EMT and
Wnt/β-catenin signalling pathways (151). In pancreatic cancer, the
CELF2-mediated CD44 alternative splicing affects apoptosis and cell
stemness by regulating the ERAD signalling pathway (178). Furthermore, the downregulation
of CELF2 confers chemoresistance to cancer cells through HO-1- and
COX-2-mediated cytoprotective effects (6,7).
Thus, CELF2 appears to function as a key target in tumour
development. The anticancer potential of CELF2 was initially
demonstrated in several in vitro studies (7,52,200). Curcumin, a natural polyphenolic
compound derived from turmeric, enhances the sensitivity of
pancreatic and ovarian cancer cell lines to gemcitabine and
cisplatin, respectively, by upregulating CELF2 (7,52,200). Although previous studies have
identified the effects of CELF2 on the genes that regulate
ferroptosis (6,7), the specific role it plays in
ferroptosis remains unclear.
The present review summarised the downstream
targets of CELF2 in detail and speculated on their role in
ferroptosis in a continuous context, which also poses as a
limitation of the present review. The present review identified
several avenues for further research to improve the understanding
of ferroptosis. Fig. 6 broadly
illustrates that CELF2 affects ferroptosis through a variety of
mechanisms. Overall, CELF2 can exert its oncogenic effects through
multiple pathways that may be partly dependent on ferroptosis.
Not applicable.
JiahaoL and LX conceived the study. ZZ was involved
in the search methods for relevant literature, as well as the
structure of the review. JiahaoL provided the software used to
prepare the figures (Adobe Illustrator CC 2018), and was also
involved in determining the novelty and innovation of the direction
of the topic, and in the writing and preparation of the original
draft. WZ, RZ, WX and JiahaoL were involved in reading and
evaluating the retrieved literature to determine whether it could
be included in the review. YW was involved in the evaluation of the
retrieved literature for inclusion in the review. LX, WX and
JiaruiL were involved in the writing, reviewing and editing of the
manuscript. ZZ and WX were involved in visualization. JiaruiL
supervised the study. JiaruiL was involved in project
administration. JiaruiL was involved in funding acquisition. All
authors have read and agreed to the published version of the
manuscript. Data authentication is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
The present study was funded by the Jilin Provincial Science and
Technology Foundation (grant no. 20200201487JC).
|
1
|
Sun Y, Chen P, Zhai B, Zhang M, Xiang Y,
Fang J, Xu S, Gao Y, Chen X, Sui X and Li G: The emerging role of
ferroptosis in inflammation. Biomed Pharmacother. 127:1101082020.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Peng JJ, Song WT, Yao F, Zhang X, Peng J,
Luo XJ and Xia XB: Involvement of regulated necrosis in blinding
diseases: Focus on necroptosis and ferroptosis. Exp Eye Res.
191:1079222020. View Article : Google Scholar
|
|
3
|
Ma T, Du J, Zhang Y, Wang Y, Wang B and
Zhang T: GPX4-independent ferroptosis-a new strategy in disease's
therapy. Cell Death Discov. 8:4342022. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang L, Liu Z, Liu L, Guo C, Jiao D, Li L,
Zhao J, Han X and Sun Y: CELF2 is a candidate prognostic and
immunotherapy biomarker in triple-negative breast cancer and lung
squamous cell carcinoma: A pan-cancer analysis. J Cell Mol Med.
25:7559–7574. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ramalingam S, Ramamoorthy P, Subramaniam D
and Anant S: Reduced expression of RNA binding protein CELF2, a
putative tumor suppressor gene in colon cancer.
Immunogastroenterology. 1:27–33. 2012. View Article : Google Scholar
|
|
6
|
Sureban SM, Murmu N, Rodriguez P, May R,
Maheshwari R, Dieckgraefe BK, Houchen CW and Anant S: Functional
antagonism between RNA binding proteins HuR and CUGBP2 determines
the fate of COX-2 mRNA translation. Gastroenterology.
132:1055–1065. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jakstaite A, Maziukiene A, Silkuniene G,
Kmieliute K, Dauksa A, Paskauskas S, Gulbinas A and Dambrauskas Z:
Upregulation of cugbp2 increases response of pancreatic cancer
cells to chemotherapy. Langenbecks Arch Surg. 401:99–111. 2016.
View Article : Google Scholar
|
|
8
|
Guo YJ, Pan WW, Liu SB, Shen ZF, Xu Y and
Hu LL: ERK/MAPK signalling pathway and tumorigenesis. Exp Ther Med.
19:1997–2007. 2020.PubMed/NCBI
|
|
9
|
Lee S, Rauch J and Kolch W: Targeting MAPK
Signaling in Cancer: Mechanisms of Drug Resistance and Sensitivity.
Int J Mol Sci. 21:11022020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sui X, Kong N, Ye L, Han W, Zhou J, Zhang
Q, He C and Pan H: p38 and JNK MAPK pathways control the balance of
apoptosis and autophagy in response to chemotherapeutic agents.
Cancer Lett. 344:174–179. 2014. View Article : Google Scholar
|
|
11
|
Chang WT, Bow YD, Fu PJ, Li CY, Wu CY,
Chang YH, Teng YN, Li RN, Lu MC, Liu YC and Chiu CC: A Marine
terpenoid, heteronemin, induces both the apoptosis and ferroptosis
of hepatocellular carcinoma cells and involves the ROS and MAPK
pathways. Oxid Med Cell Longev. 2021:76890452021. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhou D, Wu Q, Qiu H, Li M and Ji Y:
Simvastatin inhibits endometrial cancer malignant behaviors by
suppressing R AS/ M itogen-Activated protei n k i nase ( M A PK)
Pathway-Mediated reactive oxygen species (ROS) and ferroptosis.
Evid Based Complement Alternat Med. 2022:61774772022. View Article : Google Scholar
|
|
13
|
He T, Lin X, Yang C, Chen Z, Wang L, Li Q,
Ma J, Zhan F, Wang Y, Yan J and Quan Z: Theaflavin-3,3′-Digallate
Plays a ROS-Mediated dual role in ferroptosis and apoptosis via the
MAPK pathway in human osteosarcoma cell lines and xenografts. Oxid
Med Cell Longev. 2022:89663682022. View Article : Google Scholar
|
|
14
|
Bhatt V, Lan T, Wang W, Kong J, Lopes EC,
Wang J, Khayati K, Raju A, Rangel M, Lopez E, et al: Inhibition of
autophagy and MEK promotes ferroptosis in Lkb1-deficient
Kras-driven lung tumors. Cell Death Dis. 14:612023. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Santarpia L, Lippman SM and El-Naggar AK:
Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy.
Expert Opin Ther Targets. 16:103–119. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ji S, Qin Y, Shi S, Liu X, Hu H, Zhou H,
Gao J, Zhang B, Xu W, Liu J, et al: ERK kinase phosphorylates and
destabilizes the tumor suppressor FBW7 in pancreatic cancer. Cell
Res. 25:561–573. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Davis RJ, Welcker M and Clurman BE: Tumor
suppression by the Fbw7 ubiquitin ligase: mechanisms and
opportunities. Cancer Cell. 26:455–464. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li Y, Hu K, Xiao X, Wu W, Yan H, Chen H,
Chen Z and Yin D: FBW7 suppresses cell proliferation and G2/M cell
cycle transition via promoting γ-catenin K63-linked ubiquitylation.
Biochem Biophys Res Commun. 497:473–479. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ye Z, Zhuo Q, Hu Q, Xu X, Mengqi Liu,
Zhang Z, Xu W, Liu W, Fan G, Qin Y, et al: FBW7-NRA41-SCD1 axis
synchronously regulates apoptosis and ferroptosis in pancreatic
cancer cells. Redox Biol. 38:1018072021. View Article : Google Scholar
|
|
20
|
Yada M, Hatakeyama S, Kamura T, Nishiyama
M, Tsunematsu R, Imaki H, Ishida N, Okumura F, Nakayama K and
Nakayama KI: Phosphorylation-dependent degradation of c-Myc is
mediated by the F-box protein Fbw7. EMBO J. 23:2116–2125. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen J, Ding C, Chen Y, Hu W, Lu Y, Wu W,
Zhang Y, Yang B, Wu H, Peng C, et al: ACSL4 promotes hepatocellular
carcinoma progression via c-Myc stability mediated by
ERK/FBW7/c-Myc axis. Oncogenesis. 9:422020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Benassi B, Fanciulli M, Fiorentino F,
Porrello A, Chiorino G, Loda M, Zupi G and Biroccio A: c-Myc
phosphorylation is required for cellular response to oxidative
stress. Mol Cell. 21:509–519. 2006. View Article : Google Scholar
|
|
23
|
Lepore Signorile M, Grossi V, Fasano C,
Forte G, Disciglio V, Sanese P, De Marco K, La Rocca F, Armentano
R, Valentini AM, et al: c-MYC protein stability is sustained by
MAPKs in colorectal cancer. Cancers (Basel). 14:48402022.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lepore Signorile M, Grossi V, Di Franco S,
Forte G, Disciglio V, Fasano C, Sanese P, De Marco K, Susca FC,
Mangiapane LR, et al: Pharmacological targeting of the novel
β-catenin chromatin-associated kinase p38α in colorectal cancer
stem cell tumorspheres and organoids. Cell Death Dis. 12:3162021.
View Article : Google Scholar
|
|
25
|
Jiang X, Guo S, Xu M, Ma B, Liu R, Xu Y
and Zhang Y: TFAP2C-Mediated lncRNA PCAT1 inhibits ferroptosis in
docetaxel-resistant prostate cancer through c-Myc/miR-25-3p/SLC7A11
signaling. Front Oncol. 12:8620152022. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Benassi B, Zupi G and Biroccio A:
Gamma-glutamylcysteine synthetase mediates the c-Myc-dependent
response to antineoplastic agents in melanoma cells. Mol Pharmacol.
72:1015–1023. 2007. View Article : Google Scholar
|
|
27
|
Kim BY, Kwak SY, Yang JS and Han YH:
Phosphorylation and stabilization of c-Myc by NEMO renders cells
resistant to ionizing radiation through up-regulation of γ-GCS.
Oncol Rep. 26:1587–1593. 2011.PubMed/NCBI
|
|
28
|
Jiang Y, Mao C, Yang R, Yan B, Shi Y, Liu
X, Lai W, Liu Y, Wang X, Xiao D, et al: EGLN1/c-Myc induced
lymphoid-specific helicase inhibits ferroptosis through lipid
metabolic gene expression changes. Theranostics. 7:3293–3305. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liang C, Shi S, Liu M, Qin Y, Meng Q, Hua
J, Ji S, Zhang Y, Yang J, Xu J, et al: PIN1 maintains redox balance
via the c-Myc/NRF2 axis to counteract kras-induced mitochondrial
respiratory injury in pancreatic cancer cells. Cancer Res.
79:133–145. 2019. View Article : Google Scholar
|
|
30
|
Lu H, Yin H, Qu L, Ma X, Fu R and Fan D:
Ginsenoside Rk1 regulates glutamine metabolism in hepatocellular
carcinoma through inhibition of the ERK/c-Myc pathway. Food Funct.
13:3793–3811. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gao P, Tchernyshyov I, Chang TC, Lee YS,
Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT and
Dang CV: c-Myc suppression of miR-23a/b enhances mitochondrial
glutaminase expression and glutamine metabolism. Nature.
458:762–765. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jin Y, Qiu J, Lu X and Li G: C-MYC
inhibited ferroptosis and promoted immune evasion in ovarian cancer
cells through NCOA4 mediated ferritin autophagy. Cells.
11:41272022. View Article : Google Scholar :
|
|
33
|
Hongu T and Kanaho Y: Activation machinery
of the small GTPase Arf6. Adv Biol Regul. 54:59–66. 2014.
View Article : Google Scholar
|
|
34
|
Liang C, Qin Y, Zhang B, Ji S, Shi S, Xu
W, Liu J, Xiang J, Liang D, Hu Q, et al: ARF6, induced by mutant
Kras, promotes proliferation and Warburg effect in pancreatic
cancer. Cancer Lett. 388:303–311. 2017. View Article : Google Scholar
|
|
35
|
Knizhnik AV, Kovaleva OV, Komelkov AV,
Trukhanova LS, Rybko VA, Zborovskaya IB and Tchevkina EM: Arf6
promotes cell proliferation via the PLD-mTORC1 and p38MAPK
pathways. J Cell Biochem. 113:360–371. 2012. View Article : Google Scholar
|
|
36
|
Ye Z, Hu Q, Zhuo Q, Zhu Y, Fan G, Liu M,
Sun Q, Zhang Z, Liu W, Xu W, et al: Abrogation of ARF6 promotes
RSL3-induced ferroptosis and mitigates gemcitabine resistance in
pancreatic cancer cells. Am J Cancer Res. 10:1182–1193.
2020.PubMed/NCBI
|
|
37
|
Geng D and Wu H: Abrogation of ARF6 in
promoting erastin-induced ferroptosis and mitigating capecitabine
resistance in gastric cancer cells. J Gastrointest Oncol.
13:958–967. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yan F, Qian M, He Q, Zhu H and Yang B: The
posttranslational modifications of Hippo-YAP pathway in cancer.
Biochim Biophys Acta Gen Subj. 1864:1293972020. View Article : Google Scholar
|
|
39
|
Jang JW, Kim MK and Bae SC: Reciprocal
regulation of YAP/TAZ by the Hippo pathway and the Small GTPase
pathway. Small GTPases. 11:280–288. 2020. View Article : Google Scholar :
|
|
40
|
Meng XY, Zhang HZ, Ren YY, Wang KJ, Chen
JF, Su R, Jiang JH, Wang P and Ma Q: Pinin promotes tumor
progression via activating CREB through PI3K/AKT and ERK/MAPK
pathway in prostate cancer. Am J Cancer Res. 11:1286–1303.
2021.PubMed/NCBI
|
|
41
|
Lee CW, Nam JS, Park YK, Choi HK, Lee JH,
Kim NH, Cho J, Song DK, Suh HW, Lee J, et al: Lysophosphatidic acid
stimulates CREB through mitogen- and stress-activated protein
kinase-1. Biochem Biophys Res Commun. 305:455–461. 2003. View Article : Google Scholar
|
|
42
|
Ippolito F, Consalvi V, Noce V,
Battistelli C, Cicchini C, Tripodi M, Amicone L and Marchetti A:
Extracellular signal-Regulated Kinase 5 (ERK5) is required for the
Yes-associated protein (YAP) co-transcriptional activity. Cell
Death Dis. 14:322023. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Holmes B, Benavides-Serrato A, Saunders
JT, Kumar S, Nishimura RN and Gera J: mTORC2-mediated direct
phosphorylation regulates YAP activity promoting glioblastoma
growth and invasive characteristics. Neoplasia. 23:951–965. 2021.
View Article : Google Scholar :
|
|
44
|
Wang Y, Fang R, Cui M, Zhang W, Bai X,
Wang H, Liu B, Zhang X and Ye L: The oncoprotein HBXIP up-regulates
YAP through activation of transcription factor c-Myb to promote
growth of liver cancer. Cancer Lett. 385:234–242. 2017. View Article : Google Scholar
|
|
45
|
Xiao W, Wang J, Ou C, Zhang Y, Ma L, Weng
W, Pan Q and Sun F: Mutual interaction between YAP and c-Myc is
critical for carcinogenesis in liver cancer. Biochem Biophys Res
Commun. 439:167–172. 2013. View Article : Google Scholar
|
|
46
|
Qin Y, Pei Z, Feng Z, Lin P, Wang S, Li Y,
Huo F, Wang Q, Wang Z, Chen ZN, et al: Oncogenic activation of YAP
signaling sensitizes ferroptosis of hepatocellular carcinoma via
ALOXE3-mediated lipid peroxidation accumulation. Front Cell Dev
Biol. 9:7515932021. View Article : Google Scholar
|
|
47
|
Wu J, Minikes AM, Gao M, Bian H, Li Y,
Stockwell BR, Chen ZN and Jiang X: Intercellular interaction
dictates cancer cell ferroptosis via NF2-YAP signalling. Nature.
572:402–406. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Fang K, Du S, Shen D, Xiong Z, Jiang K,
Liang D, Wang J, Xu H, Hu L, Zhai X, et al: SUFU suppresses
ferroptosis sensitivity in breast cancer cells via Hippo/YAP
pathway. iScience. 25:1046182022. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Yang WH, Lin CC, Wu J, Chao PY, Chen K,
Chen PH and Chi JT: The Hippo pathway effector YAP promotes
ferroptosis via the E3 ligase SKP2. Mol Cancer Res. 19:1005–1014.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Gao R, Kalathur RKR, Coto-Llerena M, Ercan
C, Buechel D, Shuang S, Piscuoglio S, Dill MT, Camargo FD,
Christofori G and Tang F: YAP/TAZ and ATF4 drive resistance to
Sorafenib in hepatocellular carcinoma by preventing ferroptosis.
EMBO Mol Med. 13:e143512021. View Article : Google Scholar :
|
|
51
|
Hsu CY, Chang GC, Chen YJ, Hsu YC, Hsiao
YJ, Su KY, Chen HY, Lin CY, Chen JS, Chen YJ, et al: FAM198B is
associated with prolonged survival and inhibits metastasis in lung
adenocarcinoma via blockage of ERK-mediated MMP-1 expression. Clin
Cancer Res. 24:916–926. 2018. View Article : Google Scholar
|
|
52
|
Guo Q, Wu Y, Guo X, Cao L, Xu F, Zhao H,
Zhu J, Wen H, Ju X and Wu X: The RNA-binding protein CELF2 inhibits
ovarian cancer progression by stabilizing FAM198B. Mol Ther Nucleic
Acids. 23:169–184. 2021. View Article : Google Scholar
|
|
53
|
Zheng X, Chen J, Nan T, Zheng L, Lan J,
Jin X, Cai Y, Liu H and Chen W: FAM198B promotes colorectal cancer
progression by regulating the polarization of tumor-associated
macrophages via the SMAD2 signaling pathway. Bioengineered.
13:12435–12445. 2022. View Article : Google Scholar :
|
|
54
|
Buchholz M, Schatz A, Wagner M, Michl P,
Linhart T, Adler G, Gress TM and Ellenrieder V: Overexpression of
c-myc in pancreatic cancer caused by ectopic activation of NFATc1
and the Ca2+/calcineurin signaling pathway. EMBO J. 25:3714–3724.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Xu W, Gu J, Ren Q, Shi Y, Xia Q and Wang
J, Wang S, Wang Y and Wang J: NFATC1 promotes cell growth and
tumorigenesis in ovarian cancer up-regulating c-Myc through
ERK1/2/p38 MAPK signal pathway. Tumour Biol. 37:4493–4500. 2016.
View Article : Google Scholar
|
|
56
|
Ren F, Zhu K, Wang Y, Zhou F, Pang S and
Chen L: Proliferation, apoptosis and invasion of human lung cancer
cells are associated with NFATc1. Exp Ther Med. 25:492023.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Russo R, Mallia S, Zito F and Lampiasi N:
Long-lasting activity of ERK kinase depends on NFATc1 induction and
is involved in cell migration-fusion in murine macrophages
RAW264.7. Int J Mol Sci. 21:89652020. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Baumgart S, Chen NM, Siveke JT, König A,
Zhang JS, Singh SK, Wolf E, Bartkuhn M, Esposito I, Heßmann E, et
al: Inflammation-induced NFATc1-STAT3 transcription complex
promotes pancreatic cancer initiation by KrasG12D. Cancer Discov.
4:688–701. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zhou L and Xie X: RNA-binding protein
CELF2 inhibits breast cancer cell invasion and angiogenesis by
downregulating NFATc1. Exp Ther Med. 22:8982021. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Faes S and Dormond O: PI3K and AKT:
Unfaithful partners in cancer. Int J Mol Sci. 16:21138–21152. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Hashemi M, Taheriazam A, Daneii P,
Hassanpour A, Kakavand A, Rezaei S, Hejazi ES, Aboutalebi M,
Gholamrezaie H, Saebfar H, et al: Targeting PI3K/Akt signaling in
prostate cancer therapy. J Cell Commun Signal. Nov 11–2022.
View Article : Google Scholar : Epub ahead of
print. PubMed/NCBI
|
|
62
|
Ma RH, Ni ZJ, Thakur K, Cespedes-Acuña CL,
Zhang JG and Wei ZJ: Transcriptome and proteomics conjoint analysis
reveal metastasis inhibitory effect of 6-shogaol as ferroptosis
activator through the PI3K/AKT pathway in human endometrial
carcinoma in vitro and in vivo. Food Chem Toxicol. 170:1134992022.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Lu Y, Mao J, Xu Y, Pan H, Wang Y and Li W:
Ropivacaine represses the ovarian cancer cell stemness and
facilitates cell ferroptosis through inactivating the PI3K/AKT
signaling pathway. Hum Exp Toxicol. 41:96032712211206522022.
View Article : Google Scholar
|
|
64
|
Wang L, Wang J and Chen L: TIMP1 represses
sorafenib-triggered ferroptosis in colorectal cancer cells by
activating the PI3K/Akt signaling pathway. Immunopharmacol
Immunotoxicol. 45:419–425. 2022. View Article : Google Scholar
|
|
65
|
Liu H, Zhao L, Wang M, Yang K, Jin Z, Zhao
C and Shi G: FNDC5 causes resistance to sorafenib by activating the
PI3K/Akt/Nrf2 pathway in hepatocellular carcinoma cells. Front
Oncol. 12:8520952022. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Huang W, Chen K, Lu Y, Zhang D, Cheng Y,
Li L, Huang W, He G, Liao H, Cai L, et al: ABCC5 facilitates the
acquired resistance of sorafenib through the inhibition of
SLC7A11-induced ferroptosis in hepatocellular carcinoma. Neoplasia.
23:1227–1239. 2021. View Article : Google Scholar :
|
|
67
|
Jain AK and Jaiswal AK: GSK-3beta acts
upstream of Fyn kinase in regulation of nuclear export and
degradation of NF-E2 related factor 2. J Biol Chem.
282:16502–16510. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Rizvi F, Shukla S and Kakkar P: Essential
role of PH domain and leucine-rich repeat protein phosphatase 2 in
Nrf2 suppression via modulation of Akt/GSK3 beta/Fyn kinase axis
during oxidative hepatocellular toxicity. Cell Death Dis.
5:e11532014. View Article : Google Scholar
|
|
69
|
Liao S, Wu JN, Liu RM, Wang SX, Luo J,
Yang Y, Qin Y, Li T, Zheng X, Song J, et al: A novel compound DBZ
ameliorates neuroinflammation in LPS-stimulated microglia and
ischemic stroke rats: Role of Akt(Ser473)/GSK3β(Ser9)-mediated Nrf2
activation. Redox Biol. 36:1016442020. View Article : Google Scholar
|
|
70
|
Ichimura Y, Waguri S, Sou YS, Kageyama S,
Hasegawa J, Ishimura R, Saito T, Yang Y, Kouno T, Fukutomi T, et
al: Phosphorylation of p62 activates the Keap1-Nrf2 pathway during
selective autophagy. Mol Cell. 51:618–631. 2013. View Article : Google Scholar
|
|
71
|
Yi J, Zhu J, Wu J, Thompson CB and Jiang
X: Oncogenic activation of PI3K-AKT-mTOR signaling suppresses
ferroptosis via SREBP-mediated lipogenesis. Proc Natl Acad Sci USA.
117:31189–31897. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Liu Y, Wang Y, Liu J, Kang R and Tang D:
Interplay between MTOR and GPX4 signaling modulates
autophagy-dependent ferroptotic cancer cell death. Cancer Gene
Ther. 28:55–63. 2021. View Article : Google Scholar
|
|
73
|
Zhang L, Liu W, Liu F, Wang Q, Song M, Yu
Q, Tang K, Teng T, Wu D, Wang X, et al: IMCA induces ferroptosis
mediated by SLC7A11 through the AMPK/mTOR pathway in colorectal
cancer. Oxid Med Cell Longev. 2020:16756132020. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Li S, Oh YT, Yue P, Khuri FR and Sun SY:
Inhibition of mTOR complex 2 induces GSK3/FBXW7-dependent
degradation of sterol regulatory element-binding protein 1 (SREBP1)
and suppresses lipogenesis in cancer cells. Oncogene. 35:642–650.
2016. View Article : Google Scholar
|
|
75
|
Yang Q, Mao Y, Wang J, Yu H, Zhang X, Pei
X, Duan Z, Xiao C and Ma M: Gestational bisphenol A exposure
impairs hepatic lipid metabolism by altering mTOR/CRTC2/SREBP1 in
male rat offspring. Hum Exp Toxicol. 41:96032712211298522022.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Masoud GN and Li W: HIF-1α pathway: Role,
regulation and intervention for cancer therapy. Acta Pharm Sin B.
5:378–389. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
van den Beucken T, Koritzinsky M and
Wouters BG: Translational control of gene expression during
hypoxia. Cancer Biol Ther. 5:749–755. 2006. View Article : Google Scholar
|
|
78
|
Alvarez-Tejado M, Alfranca A, Aragonés J,
Vara A, Landázuri MO and del Peso L: Lack of evidence for the
involvement of the phosphoinositide 3-kinase/Akt pathway in the
activation of hypoxia-inducible factors by low oxygen tension. J
Biol Chem. 277:13508–13517. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Arsham AM, Plas DR, Thompson CB and Simon
MC: Phosphatidylinositol 3-kinase/Akt signaling is neither required
for hypoxic stabilization of HIF-1 alpha nor sufficient for
HIF-1-dependent target gene transcription. J Biol Chem.
277:15162–15170. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Tanaka H, Yamamoto M, Hashimoto N,
Miyakoshi M, Tamakawa S, Yoshie M, Tokusashi Y, Yokoyama K,
Yaginuma Y and Ogawa K: Hypoxia-independent overexpression of
hypoxia-inducible factor 1alpha as an early change in mouse
hepatocarcinogenesis. Cancer Res. 66:11263–11270. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Sun S, Guo C, Gao T, Ma D, Su X, Pang Q
and Zhang R: Hypoxia enhances glioma resistance to
sulfasalazine-induced ferroptosis by upregulating SLC7A11 via
PI3K/AKT/HIF-1α axis. Oxid Med Cell Longev. 2022:78624302022.
View Article : Google Scholar
|
|
82
|
Lin Z, Song J, Gao Y, Huang S, Dou R,
Zhong P, Huang G, Han L, Zheng J, Zhang X, et al: Hypoxia-induced
HIF-1α/lncRNA-PMAN inhibits ferroptosis by promoting the
cytoplasmic translocation of ELAVL1 in peritoneal dissemination
from gastric cancer. Redox Biol. 52:1023122022. View Article : Google Scholar
|
|
83
|
Guo S, Miyake M, Liu KJ and Shi H:
Specific inhibition of hypoxia inducible factor 1 exaggerates cell
injury induced by in vitro ischemia through deteriorating cellular
redox environment. J Neurochem. 108:1309–1321. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Zhang Q and Wang Y: MiR-210-3p targets
CELF2 to facilitate progression of lung squamous carcinoma through
PI3K/AKT pathway. Med Oncol. 39:1612022. View Article : Google Scholar
|
|
85
|
Wu JZ, Jiang N, Lin JM and Liu X: STYXL1
promotes malignant progression of hepatocellular carcinoma via
downregulating CELF2 through the PI3K/Akt pathway. Eur Rev Med
Pharmacol Sci. 24:2977–2985. 2020.PubMed/NCBI
|
|
86
|
Shi M, Yang R, Lin J, Wei QI, Chen L, Gong
W, Li Y and Guo X: LncRNA-SNHG16 promotes proliferation and
migration of acute myeloid leukemia cells via PTEN/PI3K/AKT axis
through suppressing CELF2 protein. J Biosci. 46:42021. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Yeung YT, Fan S, Lu B, Yin S, Yang S, Nie
W, Wang M, Zhou L, Li T, Li X, et al: CELF2 suppresses non-small
cell lung carcinoma growth by inhibiting the PREX2-PTEN
interaction. Carcinogenesis. 41:377–389. 2020. View Article : Google Scholar :
|
|
88
|
Zhou B, Liu J, Kang R, Klionsky DJ,
Kroemer G and Tang D: Ferroptosis is a type of autophagy-dependent
cell death. Semin Cancer Biol. 66:89–100. 2020. View Article : Google Scholar
|
|
89
|
Kang R and Tang D: Autophagy and
Ferroptosis-What's the connection? Curr Pathobiol Rep. 5:153–159.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Denton D and Kumar S: Autophagy-dependent
cell death. Cell Death Differ. 26:605–616. 2019. View Article : Google Scholar
|
|
91
|
Gao M, Monian P, Pan Q, Zhang W, Xiang J
and Jiang X: Ferroptosis is an autophagic cell death process. Cell
Res. 26:1021–1032. 2016. View Article : Google Scholar :
|
|
92
|
Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh
HJ III, Kang R and Tang D: Autophagy promotes ferroptosis by
degradation of ferritin. Autophagy. 12:1425–1428. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Park E and Chung SW: ROS-mediated
autophagy increases intracellular iron levels and ferroptosis by
ferritin and transferrin receptor regulation. Cell Death Dis.
10:8222019. View Article : Google Scholar :
|
|
94
|
Gryzik M, Srivastava A, Longhi G, Bertuzzi
M, Gianoncelli A, Carmona F, Poli M and Arosio P: Expression and
characterization of the ferritin binding domain of Nuclear Receptor
Coactivator-4 (NCOA4). Biochim Biophys Acta Gen Subj.
1861:2710–2716. 2017. View Article : Google Scholar
|
|
95
|
Ohshima T, Yamamoto H, Sakamaki Y, Saito C
and Mizushima N: NCOA4 drives ferritin phase separation to
facilitate macroferritinophagy and microferritinophagy. J Cell
Biol. 221:e2022031022022. View Article : Google Scholar :
|
|
96
|
Dowdle WE, Nyfeler B, Nagel J, Elling RA,
Liu S, Triantafellow E, Menon S, Wang Z, Honda A, Pardee G, et al:
Selective VPS34 inhibitor blocks autophagy and uncovers a role for
NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat
Cell Biol. 16:1069–1079. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Ohnstad AE, Delgado JM, North BJ, Nasa I,
Kettenbach AN, Schultz SW and Shoemaker CJ: Receptor-mediated
clustering of FIP200 bypasses the role of LC3 lipidation in
autophagy. EMBO J. 39:e1049482020. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Kuno S, Fujita H, Tanaka YK, Ogra Y and
Iwai K: Iron-induced NCOA4 condensation regulates ferritin fate and
iron homeostasis. EMBO Rep. 23:e542782022. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Goodwin JM, Dowdle WE, DeJesus R, Wang Z,
Bergman P, Kobylarz M, Lindeman A, Xavier RJ, McAllister G, Nyfeler
B, et al: Autophagy-independent lysosomal targeting regulated by
ULK1/2-FIP200 and ATG9. Cell Rep. 20:2341–2356. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Fuhrmann DC, Mondorf A, Beifuß J, Jung M
and Brüne B: Hypoxia inhibits ferritinophagy, increases
mitochondrial ferritin, and protects from ferroptosis. Redox Biol.
36:1016702020. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Hara Y, Yanatori I, Tanaka A, Kishi F,
Lemasters JJ, Nishina S, Sasaki K and Hino K: Iron loss triggers
mitophagy through induction of mitochondrial ferritin. EMBO Rep.
21:e502022020. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Zorov DB, Juhaszova M and Sollott SJ:
Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS
release. Physiol Rev. 94:909–950. 2014. View Article : Google Scholar :
|
|
103
|
Rademaker G, Boumahd Y, Peiffer R, Anania
S, Wissocq T, Liégeois M, Luis G, Sounni NE, Agirman F,
Maloujahmoum N, et al: Myoferlin targeting triggers mitophagy and
primes ferroptosis in pancreatic cancer cells. Redox Biol.
53:1023242022. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Basit F, van Oppen LM, Schöckel L,
Bossenbroek HM, van Emst-de Vries SE, Hermeling JC, Grefte S,
Kopitz C, Heroult M, Hgm Willems P and Koopman WJ: Mitochondrial
complex I inhibition triggers a mitophagy-dependent ROS increase
leading to necroptosis and ferroptosis in melanoma cells. Cell
Death Dis. 8:e27162017. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Wei S, Qiu T, Yao X, Wang N, Jiang L, Jia
X, Tao Y, Wang Z, Pei P, Zhang J, et al: Arsenic induces pancreatic
dysfunction and ferroptosis via mitochondrial
ROS-autophagy-lysosomal pathway. J Hazard Mater. 384:1213902020.
View Article : Google Scholar
|
|
106
|
Liu M, Fan Y, Li D, Han B, Meng Y, Chen F,
Liu T, Song Z, Han Y, Huang L, et al: Ferroptosis inducer erastin
sensitizes NSCLC cells to celastrol through activation of the
ROS-mitochondrial fission-mitophagy axis. Mol Oncol. 15:2084–2105.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Xiao B, Deng X, Lim GGY, Xie S, Zhou ZD,
Lim KL and Tan EK: Superoxide drives progression of
Parkin/PINK1-dependent mitophagy following translocation of Parkin
to mitochondria. Cell Death Dis. 8:e30972017. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Gan ZY, Callegari S, Cobbold SA, Cotton
TR, Mlodzianoski MJ, Schubert AF, Geoghegan ND, Rogers KL, Leis A,
Dewson G, et al: Activation mechanism of PINK1. Nature.
602:328–335. 2022. View Article : Google Scholar :
|
|
109
|
Wang C, Liu K, Cao J, Wang L, Zhao Q, Li
Z, Zhang H, Chen Q and Zhao T: PINK1-mediated mitophagy maintains
pluripotency through optineurin. Cell Prolif. 54:e130342021.
View Article : Google Scholar
|
|
110
|
Matsuda N, Sato S, Shiba K, Okatsu K,
Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, et al:
PINK1 stabilized by mitochondrial depolarization recruits Parkin to
damaged mitochondria and activates latent Parkin for mitophagy. J
Cell Biol. 189:211–221. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Li C, Zhang Y, Liu J, Kang R, Klionsky DJ
and Tang D: Mitochondrial DNA stress triggers autophagy-dependent
ferroptotic death. Autophagy. 17:948–960. 2021. View Article : Google Scholar :
|
|
112
|
Zhu L, Wu W, Jiang S, Yu S, Yan Y, Wang K,
He J, Ren Y and Wang B: Pan-cancer analysis of the
Mitophagy-Related protein PINK1 as a biomarker for the
immunological and prognostic role. Front Oncol. 10:5698872020.
View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Zheng Y, Huang C, Lu L, Yu K, Zhao J, Chen
M, Liu L, Sun Q, Lin Z, Zheng J, et al: STOML2 potentiates
metastasis of hepatocellular carcinoma by promoting PINK1-mediated
mitophagy and regulates sensitivity to lenvatinib. J Hematol Oncol.
14:162021. View Article : Google Scholar :
|
|
114
|
Chen Y, Chen HN, Wang K, Zhang L, Huang Z,
Liu J, Zhang Z, Luo M, Lei Y, Peng Y, et al: Ketoconazole
exacerbates mitophagy to induce apoptosis by downregulating
cyclooxygenase-2 in hepatocellular carcinoma. J Hepatol. 70:66–77.
2019. View Article : Google Scholar
|
|
115
|
Kung-Chun Chiu D, Pui-Wah Tse A, Law CT,
Ming-Jing Xu I, Lee D, Chen M, Kit-Ho Lai R, Wai-Hin Yuen V,
Wing-Sum Cheu J, Wai-Hung Ho D, et al: Hypoxia regulates the
mitochondrial activity of hepatocellular carcinoma cells through
HIF/HEY1/PINK1 pathway. Cell Death Dis. 10:9342019. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Wu H, Wang T, Liu Y, Li X, Xu S, Wu C, Zou
H, Cao M, Jin G, Lang J, et al: Mitophagy promotes sorafenib
resistance through hypoxia-inducible ATAD3A dependent Axis. J Exp
Clin Cancer Res. 39:2742020. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Lv H and Shang P: The significance,
trafficking and determination of labile iron in cytosol,
mitochondria and lysosomes. Metallomics. 10:899–916. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Rizzollo F, More S, Vangheluwe P and
Agostinis P: The lysosome as a master regulator of iron metabolism.
Trends Biochem Sci. 46:960–975. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Kurz T, Gustafsson B and Brunk UT: Cell
sensitivity to oxidative stress is influenced by ferritin
autophagy. Free Radic Biol Med. 50:1647–1658. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Torii S, Shintoku R, Kubota C, Yaegashi M,
Torii R, Sasaki M, Suzuki T, Mori M, Yoshimoto Y, Takeuchi T, et
al: An essential role for functional lysosomes in ferroptosis of
cancer cells. Biochem J. 473:769–777. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Chen Y, Yang Z, Wang S, Ma Q, Li L, Wu X,
Guo Q, Tao L and Shen X: Boosting ROS-Mediated lysosomal membrane
permeabilization for cancer ferroptosis therapy. Adv Healthc Mater.
12:e22021502023. View Article : Google Scholar
|
|
122
|
Fernández B, Fdez E, Gómez-Suaga P, Gil F,
Molina-Villalba I, Ferrer I, Patel S, Churchill GC and Hilfiker S:
Iron overload causes endolysosomal deficits modulated by
NAADP-regulated 2-pore channels and RAB7A. Autophagy. 12:1487–1506.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Halcrow PW, Lakpa KL, Khan N, Afghah Z,
Miller N, Datta G, Chen X and Geiger JD: HIV-1 gp120-Induced
endolysosome de-Acidification leads to efflux of endolysosome iron,
and increases in mitochondrial iron and reactive oxygen species. J
Neuroimmune Pharmacol. 17:181–194. 2022. View Article : Google Scholar
|
|
124
|
New J, Subramaniam D, Ramalingam S, Enders
J, Sayed AAA, Ponnurangam S, Standing D, Ramamoorthy P, O'Neil M,
Dixon DA, et al: Pleotropic role of RNA binding protein CELF2 in
autophagy induction. Mol Carcinog. 58:1400–1409. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
He Y, She H, Zhang T, Xu H, Cheng L, Yepes
M, Zhao Y and Mao Z: p38 MAPK inhibits autophagy and promotes
microglial inflammatory responses by phosphorylating ULK1. J Cell
Biol. 217:315–328. 2018. View Article : Google Scholar :
|
|
126
|
Trelford CB and Di Guglielmo GM: Canonical
and Non-canonical TGFβ signaling activate autophagy in an
ULK1-Dependent manner. Front Cell Dev Biol. 9:7121242021.
View Article : Google Scholar
|
|
127
|
Keil E, Höcker R, Schuster M, Essmann F,
Ueffing N, Hoffman B, Liebermann DA, Pfeffer K, Schulze-Osthoff K
and Schmitz I: Phosphorylation of Atg5 by the Gadd45β-MEKK4-p38
pathway inhibits autophagy. Cell Death Differ. 20:321–332. 2013.
View Article : Google Scholar
|
|
128
|
Comes F, Matrone A, Lastella P, Nico B,
Susca FC, Bagnulo R, Ingravallo G, Modica S, Lo Sasso G, Moschetta
A, et al: A novel cell type-specific role of p38alpha in the
control of autophagy and cell death in colorectal cancer cells.
Cell Death Differ. 14:693–702. 2007. View Article : Google Scholar
|
|
129
|
Webber JL and Tooze SA: Coordinated
regulation of autophagy by p38alpha MAPK through mAtg9 and p38IP.
EMBO J. 29:27–40. 2010. View Article : Google Scholar
|
|
130
|
Zhao Y, Wu H, Xing X, Ma Y, Ji S, Xu X,
Zhao X, Wang S, Jiang W, Fang C, et al: CD13 induces autophagy to
promote hepatocellular carcinoma cell chemoresistance through the
P38/Hsp27/CREB/ATG7 pathway. J Pharmacol Exp Ther. 374:512–520.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Choi CH, Lee BH, Ahn SG and Oh SH:
Proteasome inhibition-induced p38 MAPK/ERK signaling regulates
autophagy and apoptosis through the dual phosphorylation of
glycogen synthase kinase 3β. Biochem Biophys Res Commun.
418:759–764. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Xie X, Le L, Fan Y, Lv L and Zhang J:
Autophagy is induced through the ROS-TP53-DRAM1 pathway in response
to mitochondrial protein synthesis inhibition. Autophagy.
8:1071–1084. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Sun T, Li D, Wang L, Xia L, Ma J, Guan Z,
Feng G and Zhu X: c-Jun NH2-terminal kinase activation is essential
for up-regulation of LC3 during ceramide-induced autophagy in human
nasopharyngeal carcinoma cells. J Transl Med. 9:1612011. View Article : Google Scholar :
|
|
134
|
Zhang XY, Wu XQ, Deng R, Sun T, Feng GK
and Zhu XF: Upregulation of sestrin 2 expression via JNK pathway
activation contributes to autophagy induction in cancer cells. Cell
Signal. 25:150–158. 2013. View Article : Google Scholar
|
|
135
|
Li DD, Wang LL, Deng R, Tang J, Shen Y,
Guo JF, Wang Y, Xia LP, Feng GK, Liu QQ, et al: The pivotal role of
c-Jun NH2-terminal kinase-mediated Beclin 1 expression during
anticancer agents-induced autophagy in cancer cells. Oncogene.
28:886–898. 2009. View Article : Google Scholar
|
|
136
|
Wong CH, Iskandar KB, Yadav SK, Hirpara
JL, Loh T and Pervaiz S: Simultaneous induction of non-canonical
autophagy and apoptosis in cancer cells by ROS-dependent ERK and
JNK activation. PLoS One. 5:e99962010. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Byun JY, Yoon CH, An S, Park IC, Kang CM,
Kim MJ and Lee SJ: The Rac1/MKK7/JNK pathway signals upregulation
of Atg5 and subsequent autophagic cell death in response to
oncogenic Ras. Carcinogenesis. 30:1880–1888. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Park JH, Ko J, Park YS, Park J, Hwang J
and Koh HC: Clearance of damaged mitochondria through PINK1
stabilization by JNK and ERK MAPK signaling in Chlorpyrifos-Treated
neuroblastoma cells. Mol Neurobiol. 54:1844–1857. 2017. View Article : Google Scholar
|
|
139
|
Dagda RK, Zhu J, Kulich SM and Chu CT:
Mitochondrially localized ERK2 regulates mitophagy and autophagic
cell stress: Implications for Parkinson's disease. Autophagy.
4:770–782. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Meng Y, Yang Z, Huo T and Jiang H: Realgar
facilitates the Nrf2-Keap1-p62 positive feedback signaling axis via
MAPKs and AKT to interfere with autophagy-induced apoptosis and
oxidative stress in the hippocampus. Biomed Pharmacother.
150:1129642022. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Hosokawa N, Hara T, Kaizuka T, Kishi C,
Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, et
al: Nutrient-dependent mTORC1 association with the
ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell.
20:1981–1991. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Jung CH, Jun CB, Ro SH, Kim YM, Otto NM,
Cao J, Kundu M and Kim DH: ULK-Atg13-FIP200 complexes mediate mTOR
signaling to the autophagy machinery. Mol Biol Cell. 20:1992–2003.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Kim J, Kundu M, Viollet B and Guan KL:
AMPK and mTOR regulate autophagy through direct phosphorylation of
Ulk1. Nat Cell Biol. 13:132–141. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Nazio F, Strappazzon F, Antonioli M,
Bielli P, Cianfanelli V, Bordi M, Gretzmeier C, Dengjel J,
Piacentini M, Fimia GM and Cecconi F: mTOR inhibits autophagy by
controlling ULK1 ubiquitylation, self-association and function
through AMBRA1 and TRAF6. Nat Cell Biol. 15:406–416. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Ma X, Zhang S, He L, Rong Y, Brier LW, Sun
Q, Liu R, Fan W, Chen S, Yue Z, et al: MTORC1-mediated NRBF2
phosphorylation functions as a switch for the class III PtdIns3K
and autophagy. Autophagy. 13:592–607. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Kim YM, Jung CH, Seo M, Kim EK, Park JM,
Bae SS and Kim DH: mTORC1 phosphorylates UVRAG to negatively
regulate autophagosome and endosome maturation. Mol Cell.
57:207–218. 2015. View Article : Google Scholar :
|
|
147
|
Koren I, Reem E and Kimchi A: DAP1, a
novel substrate of mTOR, negatively regulates autophagy. Curr Biol.
20:1093–1098. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Yang C, Li Y, Hu W, Wang X, Hu J, Yuan C,
Zhou C, Wang H, Du J, Wang Y and Tong X: TEOA promotes autophagic
cell death via ROS-Mediated inhibition of mTOR/p70S6k signaling
pathway in pancreatic cancer cells. Front Cell Dev Biol.
9:7348182021. View Article : Google Scholar :
|
|
149
|
Nàger M, Sallán MC, Visa A, Pushparaj C,
Santacana M, Macià A, Yeramian A, Cantí C and Herreros J:
Inhibition of WNT-CTNNB1 signaling upregulates SQSTM1 and
sensitizes glioblastoma cells to autophagy blockers. Autophagy.
14:619–636. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Petherick KJ, Williams AC, Lane JD,
Ordóñez-Morán P, Huelsken J, Collard TJ, Smartt HJ, Batson J, Malik
K, Paraskeva C and Greenhough A: Autolysosomal β-catenin
degradation regulates Wnt-autophagy-p62 crosstalk. EMBO J.
32:1903–1916. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Fan B, Su B, Song G, Liu X, Yan Z, Wang S,
Hu F and Yang J: miR-363-3p induces EMT via the Wnt/β-catenin
pathway in glioma cells by targeting CELF2. J Cell Mol Med.
25:10418–10429
|
|
152
|
Wei H, Tang X, Chen Q, Yue T and Dong B:
An endoplasmic reticulum-targeting fluorescent probe for the
visualization of the viscosity fluctuations during ferroptosis in
live cells. Anal Chim Acta. 1232:3404542022. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Song W, Zhang W, Yue L and Lin W:
Revealing the effects of endoplasmic reticulum stress on
ferroptosis by Two-Channel Real-Time Imaging of pH and viscosity.
Anal Chem. 94:6557–6565. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Zhao C, Yu D, He Z, Bao L, Feng L, Chen L,
Liu Z, Hu X, Zhang N, Wang T and Fu Y: Endoplasmic reticulum
stress-mediated autophagy activation is involved in cadmium-induced
ferroptosis of renal tubular epithelial cells. Free Radic Biol Med.
175:236–248. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
He Z, Shen P, Feng L, Hao H, He Y, Fan G,
Liu Z, Zhu K, Wang Y, Zhang N, et al: Cadmium induces liver
dysfunction and ferroptosis through the endoplasmic
stress-ferritinophagy axis. Ecotoxicol Environ Saf. 245:1141232022.
View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Fu F, Wang W, Wu L, Wang W, Huang Z, Huang
Y, Wu C and Pan X: Inhalable biomineralized liposomes for cyclic
Ca2+-Burst-Centered endoplasmic reticulum stress
enhanced lung cancer ferroptosis therapy. ACS Nano. 17:5486–5502.
2023. View Article : Google Scholar
|
|
157
|
Chen PH, Wu J, Xu Y, Ding CC, Mestre AA,
Lin CC, Yang WH and Chi JT: Zinc transporter ZIP7 is a novel
determinant of ferroptosis. Cell Death Dis. 12:1982021. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Chen Y, Mi Y, Zhang X, Ma Q, Song Y, Zhang
L, Wang D, Xing J, Hou B, Li H, et al: Dihydroartemisinin-induced
unfolded protein response feedback attenuates ferroptosis via
PERK/ATF4/HSPA5 pathway in glioma cells. J Exp Clin Cancer Res.
38:4022019. View Article : Google Scholar :
|
|
159
|
Wei R, Zhao Y, Wang J, Yang X, Li S, Wang
Y, Yang X, Fei J, Hao X, Zhao Y, et al: Tagitinin C induces
ferroptosis through PERK-Nrf2-HO-1 signaling pathway in colorectal
cancer cells. Int J Biol Sci. 17:2703–1277. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Zheng X, Liu B, Liu X, Li P, Zhang P, Ye
F, Zhao T, Kuang Y, Chen W, Jin X and Li Q: PERK regulates the
sensitivity of hepatocellular carcinoma cells to High-LET carbon
ions via either apoptosis or ferroptosis. J Cancer. 13:669–680.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Zhao R, Lv Y, Feng T, Zhang R, Ge L, Pan
J, Han B, Song G and Wang L: ATF6α promotes prostate cancer
progression by enhancing PLA2G4A-mediated arachidonic acid
metabolism and protecting tumor cells against ferroptosis.
Prostate. 82:617–629. 2022. View Article : Google Scholar :
|
|
162
|
Hwang J and Qi L: Quality control in the
endoplasmic reticulum: Crosstalk between ERAD and UPR pathways.
Trends Biochem Sci. 43:593–605. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Krshnan L, van de Weijer ML and Carvalho
P: Endoplasmic reticulum-associated protein degradation. Cold
Spring Harb Perspect Biol. 14:a0412472022. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Lopata A, Kniss A, Löhr F, Rogov VV and
Dötsch V: Ubiquitination in the ERAD process. Int J Mol Sci.
21:53692020. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Haynes CM, Titus EA and Cooper AA:
Degradation of misfolded proteins prevents ER-derived oxidative
stress and cell death. Mol Cell. 15:767–776. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Liu Q, Yang X, Long G, Hu Y, Gu Z,
Boisclair YR and Long Q: ERAD deficiency promotes mitochondrial
dysfunction and transcriptional rewiring in human hepatic cells. J
Biol Chem. 295:16743–16753. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Zhou Z, Torres M, Sha H, Halbrook CJ, Van
den Bergh F, Reinert RB, Yamada T, Wang S, Luo Y, Hunter AH, et al:
Endoplasmic reticulum-associated degradation regulates
mitochondrial dynamics in brown adipocytes. Science. 368:54–60.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Wang X, Wang QC, Sun Z, Li T, Yang K, An
C, Guo C and Tang TS: ER stress mediated degradation of
diacylglycerol acyltransferase impairs mitochondrial functions in
TMCO1 deficient cells. Biochem Biophys Res Commun. 512:914–920.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Takashi Y, Tomita K, Kuwahara Y, Roudkenar
MH, Roushandeh AM, Igarashi K, Nagasawa T, Nishitani Y and Sato T:
Mitochondrial dysfunction promotes aquaporin expression that
controls hydrogen peroxide permeability and ferroptosis. Free Radic
Biol Med. 161:60–70. 2020. View Article : Google Scholar :
|
|
170
|
Sereti E, Tsimplouli C, Kalaitsidou E,
Sakellaridis N and Dimas K: Study of the Relationship between sigma
receptor expression levels and some common sigma ligand activity in
cancer using human cancer cell lines of the NCI-60 cell line panel.
Biomedicines. 9:382021. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Oyer HM, Sanders CM and Kim FJ:
Small-molecule modulators of sigma1 and Sigma2/TMEM97 in the
context of cancer: Foundational concepts and emerging themes. Front
Pharmacol. 10:11412019. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Gueguinou M, Crottès D, Chantôme A,
Rapetti-Mauss R, Potier-Cartereau M, Clarysse L, Girault A, Fourbon
Y, Jézéquel P, Guérin-Charbonnel C, et al: The SigmaR1 chaperone
drives breast and colorectal cancer cell migration by tuning
SK3-dependent Ca2+ homeostasis. Oncogene. 36:3640–3647.
2017. View Article : Google Scholar
|
|
173
|
Bai T, Lei P, Zhou H, Liang R, Zhu R, Wang
W, Zhou L and Sun Y: Sigma-1 receptor protects against ferroptosis
in hepatocellular carcinoma cells. J Cell Mol Med. 23:7349–7359.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Bai T, Wang S, Zhao Y, Zhu R, Wang W and
Sun Y: Haloperidol, a sigma receptor 1 antagonist, promotes
ferroptosis in hepatocellular carcinoma cells. Biochem Biophys Res
Commun. 491:919–925. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Zou Y, Li H, Graham ET, Deik AA, Eaton JK,
Wang W, Sandoval-Gomez G, Clish CB, Doench JG and Schreiber SL:
Cytochrome P450 oxidoreductase contributes to phospholipid
peroxidation in ferroptosis. Nat Chem Biol. 16:302–309. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Acharya P, Liao M, Engel JC and Correia
MA: Liver cytochrome P450 3A endoplasmic reticulum-associated
degradation: A major role for the p97 AAA ATPase in cytochrome P450
3A extraction into the cytosol. J Biol Chem. 286:3815–3828. 2011.
View Article : Google Scholar :
|
|
177
|
Prochazka L, Tesarik R and Turanek J:
Regulation of alternative splicing of CD44 in cancer. Cell Signal.
26:2234–2239. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Lai S, Wang Y, Li T, Dong Y, Lin Y, Wang
L, Weng S, Zhang X and Lin C: N6-methyladenosine-mediated CELF2
regulates CD44 alternative splicing affecting tumorigenesis via
ERAD pathway in pancreatic cancer. Cell Biosci. 12:1252022.
View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Wang Y, Zhao G, Condello S, Huang H,
Cardenas H, Tanner EJ, Wei J, Ji Y, Li J, Tan Y, et al: Frizzled-7
identifies platinum-tolerant ovarian cancer cells susceptible to
ferroptosis. Cancer Res. 81:384–399. 2021. View Article : Google Scholar :
|
|
180
|
Nuñez F, Bravo S, Cruzat F, Montecino M
and De Ferrari GV: Wnt/β-catenin signaling enhances
cyclooxygenase-2 (COX2) transcriptional activity in gastric cancer
cells. PLoS One. 6:e185622011. View Article : Google Scholar
|
|
181
|
Wang H, Zhang H, Chen Y, Wang H, Tian Y,
Yi X, Shi Q, Zhao T, Zhang B, Gao T, et al: Targeting Wnt/β-Catenin
signaling exacerbates ferroptosis and increases the efficacy of
melanoma immunotherapy via the regulation of MITF. Cells.
11:35802022. View Article : Google Scholar
|
|
182
|
Chen QF, Shi F, Huang T, Huang C, Shen L,
Wu P and Li W: ASTN1 is associated with immune infiltrates in
hepatocellular carcinoma, and inhibits the migratory and invasive
capacity of liver cancer via the Wnt/β-catenin signaling pathway.
Oncol Rep. 44:1425–1440. 2020.PubMed/NCBI
|
|
183
|
Tu B, Ma TT, Peng XQ, Wang Q, Yang H and
Huang XL: Targeting of COX-2 expression by recombinant adenovirus
shRNA attenuates the malignant biological behavior of breast cancer
cells. Asian Pac J Cancer Prev. 15:8829–8836. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Wang Y, Zheng L, Shang W, Yang Z, Li T,
Liu F, Shao W, Lv L, Chai L, Qu L, et al: Wnt/beta-catenin
signaling confers ferroptosis resistance by targeting GPX4 in
gastric cancer. Cell Death Differ. 29:2190–2202. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Liu W, Zhou Y, Duan W, Song J, Wei S, Xia
S, Wang Y, Du X, Li E, Ren C, et al: Glutathione peroxidase
4-dependent glutathione high-consumption drives acquired platinum
chemoresistance in lung cancer-derived brain metastasis. Clin
Transl Med. 11:e5172021. View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Krejci P, Aklian A, Kaucka M, Sevcikova E,
Prochazkova J, Masek JK, Mikolka P, Pospisilova T, Spoustova T,
Weis M, et al: Receptor tyrosine kinases activate canonical
WNT/β-catenin signaling via MAP kinase/LRP6 pathway and direct
β-catenin phosphorylation. PLoS One. 7:e358262012. View Article : Google Scholar
|
|
187
|
Červenka I, Wolf J, Mašek J, Krejci P,
Wilcox WR, Kozubík A, Schulte G, Gutkind JS and Bryja V:
Mitogen-activated protein kinases promote WNT/beta-catenin
signaling via phosphorylation of LRP6. Mol Cell Biol. 31:179–189.
2011. View Article : Google Scholar
|
|
188
|
Siddharth S, Mohapatra P, Preet R, Das D,
Satapathy SR, Choudhuri T and Kundu CN: Induction of apoptosis by
4-(3-(tert-butylamino)imidazo[1,2-α]pyridine-2-yl) benzoic acid in
breast cancer cells via upregulation of PTEN. Oncol Res. 21:1–13.
2013. View Article : Google Scholar
|
|
189
|
Khare V, Dammann K, Asboth M, Krnjic A,
Jambrich M and Gasche C: Overexpression of PAK1 promotes cell
survival in inflammatory bowel diseases and colitis-associated
cancer. Inflamm Bowel Dis. 21:287–296. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Ji H, Wang J, Nika H, Hawke D, Keezer S,
Ge Q, Fang B, Fang X, Fang D, Litchfield DW, et al: EGF-induced ERK
activation promotes CK2-mediated disassociation of alpha-Catenin
from beta-Catenin and transactivation of beta-Catenin. Mol Cell.
36:547–559. 2009. View Article : Google Scholar :
|
|
191
|
Gao C, Cao W, Bao L, Zuo W, Xie G, Cai T,
Fu W, Zhang J, Wu W, Zhang X and Chen YG: Autophagy negatively
regulates Wnt signalling by promoting Dishevelled degradation. Nat
Cell Biol. 12:781–790. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Nasiri-Aghdam M, Garcia-Garduño TC and
Jave-Suárez LF: CELF family proteins in cancer: Highlights on the
RNA-binding protein/noncoding RNA regulatory axis. Int J Mol Sci.
22:110562021. View Article : Google Scholar : PubMed/NCBI
|
|
193
|
Zhang Y, Yin C, Wei C, Xia S, Qiao Z,
Zhang XW, Yu B, Zhou J and Wang R: Exosomal miR-625-3p secreted by
cancer-associated fibroblasts in colorectal cancer promotes EMT and
chemotherapeutic resistance by blocking the CELF2/WWOX pathway.
Pharmacol Res. 186:1065342022. View Article : Google Scholar
|
|
194
|
Zhao Y, Zhou H and Dong W: LncRNA
RHPN1-AS1 promotes the progression of nasopharyngeal carcinoma by
targeting CELF2 expression. Exp Mol Pathol. 122:1046712021.
View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Wang J, Liu L, Sun Y, Xue Y, Qu J, Pan S,
Li H, Qu H, Wang J and Zhang J: miR-615-3p promotes proliferation
and migration and inhibits apoptosis through its potential target
CELF2 in gastric cancer. Biomed Pharmacother. 101:406–413. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Xu H, Wang F and Wang L: Suppression of
miR-106a-5p expression inhibits tumorigenesis via increasing CELF-2
expression in spinal cord glioma. Oncol Lett. 22:6272021.
View Article : Google Scholar : PubMed/NCBI
|
|
197
|
Ge L, Zhou F, Nie J, Wang X and Zhao Q:
Hypoxic colorectal cancer-secreted exosomes deliver miR-210-3p to
normoxic tumor cells to elicit a protumoral effect. Exp Biol Med
(Maywood). 246:1895–1906. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Fan HN, Zhao XY, Liang R, Chen XY, Zhang
J, Chen NW and Zhu JS: CircPTK2 inhibits the tumorigenesis and
metastasis of gastric cancer by sponging miR-134-5p and activating
CELF2/PTEN signaling. Pathol Res Pract. 227:1536152021. View Article : Google Scholar
|
|
199
|
Xie SC, Zhang JQ, Jiang XL, Hua YY, Xie
SW, Qin YA and Yang YJ: LncRNA CRNDE facilitates epigenetic
suppression of CELF2 and LATS2 to promote proliferation, migration
and chemoresistance in hepatocellular carcinoma. Cell Death Dis.
11:6762020. View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Subramaniam D, Ramalingam S, Linehan DC,
Dieckgraefe BK, Postier RG, Houchen CW, Jensen RA and Anant S: RNA
binding protein CUGBP2/CELF2 mediates curcumin-induced mitotic
catastrophe of pancreatic cancer cells. PLoS One. 6:e169582011.
View Article : Google Scholar : PubMed/NCBI
|