Introduction
Most patients with malignant tumors do not die of
the primary neoplasia, but of secondary tumors known as metastases.
As early as 1996, Sporn reported that more than 90% of cancer
deaths resulted from the development of metastatic lesions
(1). Metastasis depends upon the
processes of tumor cell migration and invasion. Today, the
molecular and cellular fundamentals of migration and invasion are
recognized as complex multistep biological processes, most probably
regulated by distinct genes and signaling pathways at every step.
The various cytoplasmic proteins and transcription factors
mediating these processes have already been identified (2). It was recently discovered that miRNAs
can affect a wide range of biological functions, including tumor
cell invasion and metastasis (3–7).
MiRNAs are small, endogenous, non-protein-coding single-stranded
RNAs of 20–25 nucleotides that post-transcriptionally regulate the
expression of hundreds of target genes. MiRNAs bind target mRNA
sequences through canonical base pairing between the seed sequence,
which includes nucleotides 2–8 from the 5′-end, and the
complementary sequence found in the 3′UTR of its target mRNA
(8). In mammals, mature miRNAs are
integrated into an RNA-induced silencing complex, which can
suppress translation and occasionally also induce degradation of a
targeted mRNA. MiRNAs are primarily negative gene regulators of
post-transcriptional repression, and targets include tumor
suppressors and oncogenes. For example, miR-21 was found to be
highly expressed in colorectal cancers, and induced metastasis and
invasion by inhibiting the tumor suppressor gene PDCD4 in
colorectal cancer cells (9). Thus,
as an oncogene, miR-21 exhibited regulatory effects. However, when
expressed at lower levels in tumors, miRNAs have also demonstrated
tumor suppressor functions. For example, the action of let-7 miRNA
as an antioncogene was found to be absent in lung cancers by
inhibiting downstream translation of MYC (10).
MiR-125a has been reported to control the
differentiation of the embryonic cancer cell line P19 by negatively
regulating the 3′UTR of lin-28 mRNA (11). It was found to inhibit translation
of the target gene t-trkC and thereby modulating proliferation of
neuroblastomas (12). In addition,
during the process of epithelial-mesenchymal transdifferentiation
(EMT) in ovarian cancer, miR-125a was found to negatively regulate
the target gene ARID3B and inhibit EMT (13).
MiR-125a-3p, a member of the miR-125a family, is
derived from the 3′-end of pre-miR-125a. However, there are few
reports on the function of miR-125a-3p. Our research group has
shown that miR-125a-3p is poorly expressed in non-small cell lung
cancers (NSCLCs) (14). Real-time
polymerase chain reaction (PCR) assays showed that miR-125a-3p
expression was significantly decreased in NSCLCs compared to
corresponding adjacent normal tissue. Therefore, we hypothesized
that miR-125a-3p may act as a tumor suppressor by negatively
regulating some oncogenes. Importantly, Spearman’s correlation
tests demonstrated a negative relationship between miR-125a-3p
expression, pathological tumor stage and lymph node metastasis. In
addition, downregulation of levels of miR-125a-3p expression in
A549 cells resulted in higher rates of invasion and migration.
These previous results led us to hypothesize that miR-125a-3p may
be involved in the migration and invasion of NSCLC tumor cells. To
the best of our knowledge, there have not been any reports on the
mechanisms involved in the regulation of migration and invasion by
miR-125a-3p.
The metastatic sequence of primary tumor cells
includes detachment, local migration and invasion of stromal
tissue, intravasation and transit through blood vessels, capillary
bed arrest and extravasation, further local crawling and invasion,
attachment, formation of micrometastases, survival, perhaps
dormancy and eventually additional proliferation (15,16).
Cellular migration is an essential function of tumor cell invasion
and metastasis. Accordingly, the purpose of this study was to
explore how miR-125a-3p affects the migration of cells of the A549
lung cancer cell line.
Materials and methods
Cell culture
A549 cells were propagated in DMEM (Gibco, Carlsbad,
CA, USA) supplemented with 10% fetal bovine serum (FBS), 100 U/ml
penicillin and 100 U/ml streptomycin. The cells were incubated at
37°C in a 5% CO2 humidified atmosphere until 75%
confluent.
Transfection
All transfections were carried out using
Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the
manufacturer’s instructions. The following 2′-O-methyl
oligonucleotides were synthesized by Integrated DNA Technologies
and purchased from Shanghai GeneChem: 2′-O-Me-sense-125a-3p: 5′-ACA
GGU GAG GUU CUU GGG AGCC-3′, 2′-O-Me-antisense-125a-3p: 5′-GGC UCC
CAA GAA CCU CAC CUGU-3′, and 2′-O-Me-scramble-125a-3p: 5′-GGU CGG
UGC UCG AUG CAG GUAA-3′. Twenty-four hours prior to transfection,
the cells were plated into 6-well plates for RNA extraction and
into 10-cm dishes for protein extraction. A549 cells were
transfected with 20 μM of 2′-O-methyl oligo-nucleotides, and
RNA and protein were extracted 24 h after transfection. In
addition, RhoA small interfering RNA (siRNA) or the negative
control was used in co-transfection with or without
antisense-125a-3p. The sequences of the RhoA siRNAs were:
5′-GAAGUCAAGCAUUUCUGUCTT-3′ (sense) and 5′-GACAGAAAUGCUUGACUUCTT-3′
(antisense) (17).
To block the function of RhoA, the Rho inhibitor
CT04 (Cytoskeleton, Denver, CO, USA), was added to the culture
medium at a final concentration of 2.0 μg/ml, as described
previously (18). Migration was
investigated in the presence of CT04 using a Transwell chamber as
described in the following section.
Cell migration assay
A549 cells (5×104 cells/chamber) were
trypsinized, washed, resuspended in serum-free DMEM, and placed in
the top portion of the chamber. The lower portion of the chamber
contained 10% FBS as a chemoattractant. The chambers were incubated
at 37°C, 5% CO2 for 12 h, and then the cells remaining
on the membrane were washed with PBS, fixed in 100% methanol,
stained with haematoxylin, photographed and counted. Five random
fields were selected randomly, then analyzed for each chamber. Each
of 3 independent experiments was carried out in duplicate.
Luciferase assays
The plasmid used for these assays contained a
full-length RhoA 3′UTR coupled to a luciferase reporter, and was a
kind gift from Dr William Kong and Dr Jin Q. Cheng (H. Lee Moffitt
Cancer Center and Research Institute, Tampa, FL, USA). The
following primers were used to amplify the 3′UTR of the RhoA gene
from human cDNA (NM_001664.2): forward primer 5′-GAC TAG TCA ATC
TGG GTG CCT TGT CTTG-3′, reverse primer 5′-CCC AAG CTT GGG TGC CTT
TAT TCT ATT AGT AGT TGG AAA-3′. The PCR fragment were digested and
cloned into the pMIR-Report vector (Ambion) at the SpeI and
HindIII sites. The mutant plasmid containing a full-length
RhoA 3′UTR mutant coupled to a luciferase reporter was constructed
by Sauer Biotechnology Inc. (China).
A549 cells were seeded onto 24-well plates the day
prior to transfection. They were transfected with 0.4 μg of
the luciferase expression construct, 20 μM 2′-O-methyl sense
or scramble oligonucleotides, and 0.02 μg of the Renilla
luciferase vector pRL-TK (Promega) for normalization. Mock
transfected cells were transfected with the luciferase constructs
alone. After 24 h, luciferase assays were performed using the
Dual-Luciferase Reporter Assay System (Promega). Firefly luciferase
activity was normalized to Renilla luciferase activity. All
transfection experiments were conducted in triplicate.
RNA isolation and quantitative real-time
polymerase chain reaction (qRT-PCR)
Total RNA was prepared using TRIzol (Invitrogen).
RhoA cDNA was generated by PrimerScript RT reagent kit (Takara,
Dalian, China) and amplified using RhoA primers with SYBR Premix EX
Taq II (Takara). The primer sequences for RhoA were: 5′-CGA CAG CCC
TGA TAG TTT A-3′ (forward) and 5′-GTG CTC ATC ATT CCG AAG A-3′
(reverse), and the primer sequences for β-actin were F:
5′-AGCACAGAGCCTCGCCTTTG-3′ (forward), R: 5′-ACATGCCGGAGCCGTTGT-3′
(reverse). The relative quantity of RhoA, normalized to β-actin,
calculated based on the following equation RQ =
2−ΔΔCt.
Western blot analysis
Cells were washed twice with ice-cold
phosphate-buffered saline (PBS) and lysed using the M-PER reagent
(Pierce Biotechnology) containing 1 mM PMSF and phosphatase
inhibitors for 1 h at 4°C. The supernatants were centrifuged at
12,000 × g for 30 min at 4°C, and collected for analysis. Aliquots
of supernatant containing 80 μg of total protein were
separated on 12 or 10% SDS-PAGE gels and transferred to PVDF
membranes. The membranes were blocked using 5% fat-free milk, and
incubated with mouse anti-RhoA (1:200, Santa Cruz Biotechnology,
Santa Cruz, CA, USA), or mouse anti-GAPDH (1:1,000, Santa Cruz
Biotechnology). Membranes were incubated at 4°C overnight and
incubated with corresponding secondary antibodies (1:4,000,
Chemicon, Temecula, CA, USA) at room temperature for 1 h.
Immunoreactive bands were identified using Super ECL reagent
(Pierce Biotechnology), according to the manufacturer’s protocol.
Specific bands for RhoA, F-actin and GAPDH were identified using
prestained protein molecular weight markers (SM0441, MBI
Fermentas). The ECL Imaging System (UVP Inc.) was used to visualize
the specific bands and the optical density of each band was
measured using Image J software.
Wound healing assay
When cell confluence was less than 90% at 48 h after
transfection, wounds were created in confluent cells using a 200-ml
pipette tip. The cells were then rinsed with medium to remove any
free-floating cells and debris. Medium was then added, and culture
plates were incubated at 37°C. Wound healing within the scrape line
was observed at different time points, and representative scrape
lines for each cell line were photographed. Duplicate wells for
each condition were examined for each experiment, and each
experiment was repeated 3 times. The optical the distance of wound
were measured using Image J software.
Rho-GTP pull-down assay
The Rho-GTP pull-down assay was performed using an
RhoA/Rac1/Cdc42 Activation Assay Combo Kit (STA-405, Cell Biolabs
Inc., San Diego, CA, USA). Medium was aspirated off cells cultured
to approximately 80 to 90% confluence, and the cells were rinsed
twice with ice-cold PBS before being treated with ice-cold lysis
buffer (0.5–1 ml per 100-mm tissue culture plate). The cell culture
plates were subsequently placed on ice for 10–20 min. The cells
were centrifugated at 14,000 × g for 10 min at 4°C, and then 20
μl of 0.5 M EDTA was added to each aliquot sample. The
positive and negative controls received 10 μl of 100X GTPγS
and 10 μl of 100X GDP, respectively. All tubes were
incubated with agitation for 30 min at 30°C. Reactions were stopped
using 65 μl of 1.0 M MgCl2. Rhotekin RBD or PAK
PBD Agarose beads were added to the cell lysates and incubated for
1 h at 4°C. After centrifugation for 10 sec at 14,000 × g, the
beads were washed with lysis buffer and Rho-GTP was eluted in
Laemmli sample buffer. Antibody against RhoA was used to analyze
eluted sample on western blots.
Immunofluorescence
When cell confluence was less than 5×105
cells at 48 h after transfection, cells were serum-starved for 4 h
and fixed in 2% formaldehyde/PBS for 7 min at room temperature.
After permeabilization with 0.1% Triton X-100 and blocking with
1.5% normal goat serum, the cells were stained with
rhodamine-phalloidin for 30 min at 37°C to label F-actin, and
mounted using Gelvatol mounting medium.
Statistical analysis
All data are presented as mean ± SD of 3 independent
experiments. A p-value ≤0.05 was considered significant. All
statistical analyses were performed using the SPSS13.0 software
package.
Results
RhoA is a potential miR-125a-3p target
gene
To study how miR-125a-3p might regulate migration,
we first proceeded to identify potential targets known to play a
role in cell mobility. Among the candidates surveyed previously
(14), we found that the 3′UTR of
the RhoA gene contains highly conserved regions that may serve as
binding sites for miR-125a-3p, as determined at microrna.org (Fig.
1).
Effects of miR-125a-3p on RhoA
expression
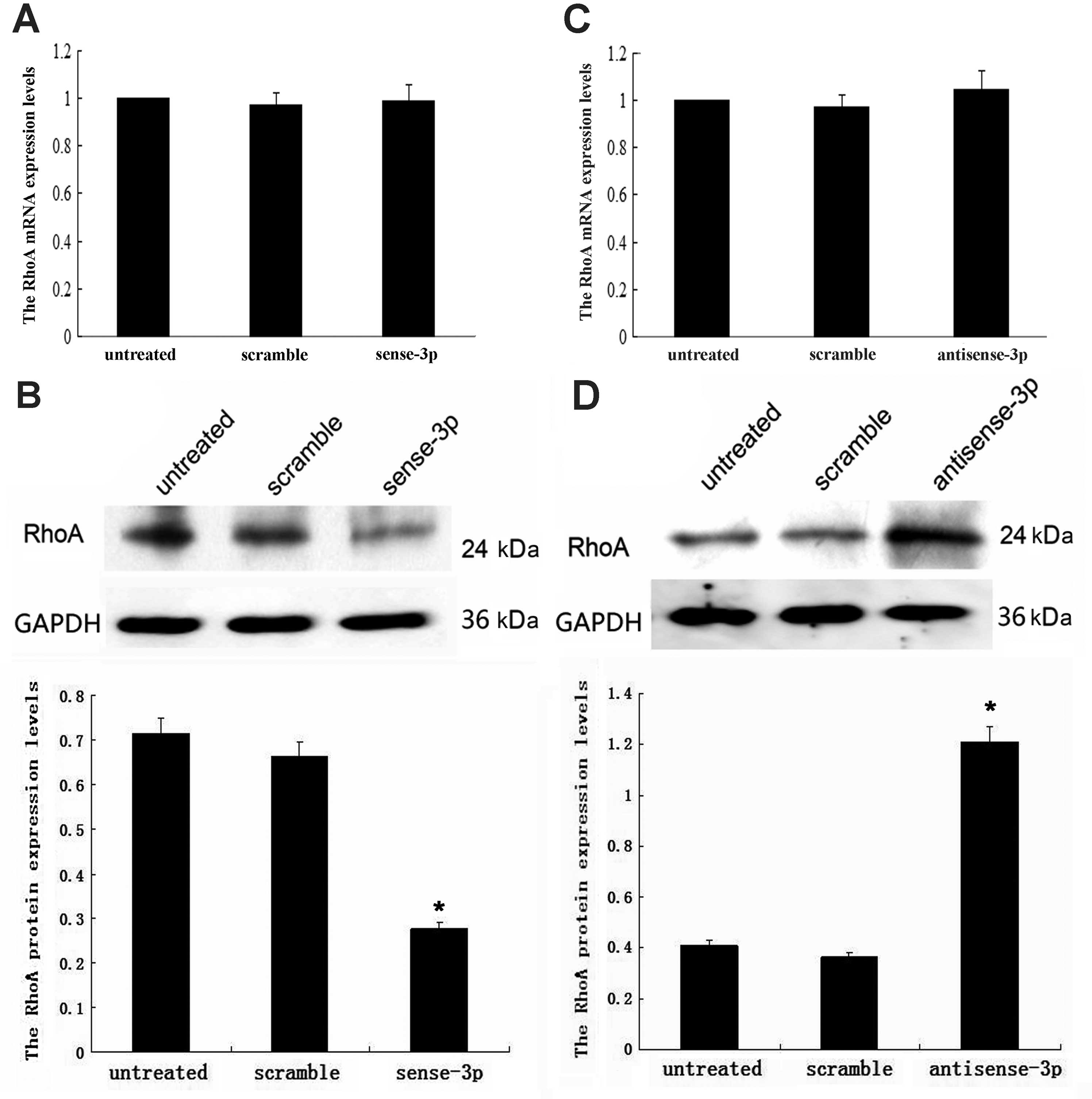
Since results of our previous study showed that
miR-125a-3p expressed moderately in A549 NSCLC cells, we chose A549
cells for this study. To confirm whether suppression of migration
by miR-125a-3p is associated with changes in RhoA expression, we
measured the level of both RhoA mRNA and protein using qRT-PCR and
western blot analysis in the presence of upregulation and
downregulation of miR-125a-3p. Upregulation of miR-125a-3p was
achieved using transfected sense-miR-125a-3p; while RhoA mRNA
remained unchanged (p=0.309, Fig.
2A), the RhoA protein concentration decreased (p<0.001,
Fig. 2B). Downregulation of
miR-125a-3p was achieved using transfected antisense-miR-125a-3;
while RhoA mRNA remained unchanged (p=0.942, Fig. 2C), the RhoA protein concentration
increased (p<0.001, Fig. 2D).
These results indicate that miR-125a-3p may post-transcriptionally
regulate RhoA expression.
MiR-125a-3p directly regulates the
expression of the target gene RhoA
To further demonstrate that RhoA is a potential
target of miR-125a-3p, we generated luciferase reporters that
contained the 3′UTR of the RhoA gene (Fig. 3A). Results from 3 independent
experiments showed that reporter activity was reduced by the
ectopic expression of miR-125a-3p (p<0.001, Fig. 3B). We also generated luciferase
reporters that contained a mutated sequence within the predicted
target sites of the 3′UTR of the RhoA gene (Fig. 3A) to further demonstrate the
interaction between miR-125a-3p and the 3′UTR of RhoA. Data showed
that reporter activity was not reduced by the ectopic expression of
miR-125a-3p (Fig. 3B). Taken
together, these results indicated that in A549 cells, the RhoA gene
was a functional target of miR-125a-3p.
MiR-125a-3p inhibits migration via the
Rho-dependent pathway
Previously we found that downregulation of
miR-125a-3p induced increased migration of A549 cells in transwell
experiments (14). Fig. 4A shows that the number of migrating
cells decreased after transfection with sense miR-125a-3p. In
contrast, the number of migrating cells increased after
transfection with antisense miR-125a-3p. A similar trend was seen
in the wound healing assays (Fig.
5A). To further validate whether miR-125a-3p inhibits migration
in a Rho-dependent manner, we blocked RhoA activity using the Rho
inhibitor CT04 at a final concentration of 2.0 μg/ml. The
number of migrating cells was significantly decreased after
treatment with CT04 compared with the untreated cells in a
Transwell experiment without matrigel (p<0.001, Fig. 4B and C), and as seen in the wound
healing assay (p<0.001, Fig. 5B and
C). Moreover, after treatment with CT04, the migratory ability
of A549 cells transfected with the antisense-miR-125a-3p or the
sense miR-125a-3p was neither decreased nor increased (p<0.001,
Figs. 4B, C, 5B and C). These results suggest that
miR-125a-3p cannot regulate RhoA when the Rho pathway is
blocked.
Downregulation of RhoA by the inhibitor CT04 may
directly affect the function of RhoA, independent of the
concentration of protein. To rule out this effect, siRNA was used
for specific downregulation of the expression of RhoA to confirm
that miR-125a-3p mediated NSCLC cell migration through RhoA. The
knockdown efficacy was identified using western blot analysis
(p<0.001, Fig. 6A). Knockdown
of the expression of RhoA (p<0.001, Fig. 6B), abolished the effect of
antisense-miR-125a-3p on cell migration (Fig. 6C and D).
Effects of RhoA expression on activated
RhoA (RhoA-GTP) and intracellular actomyosin
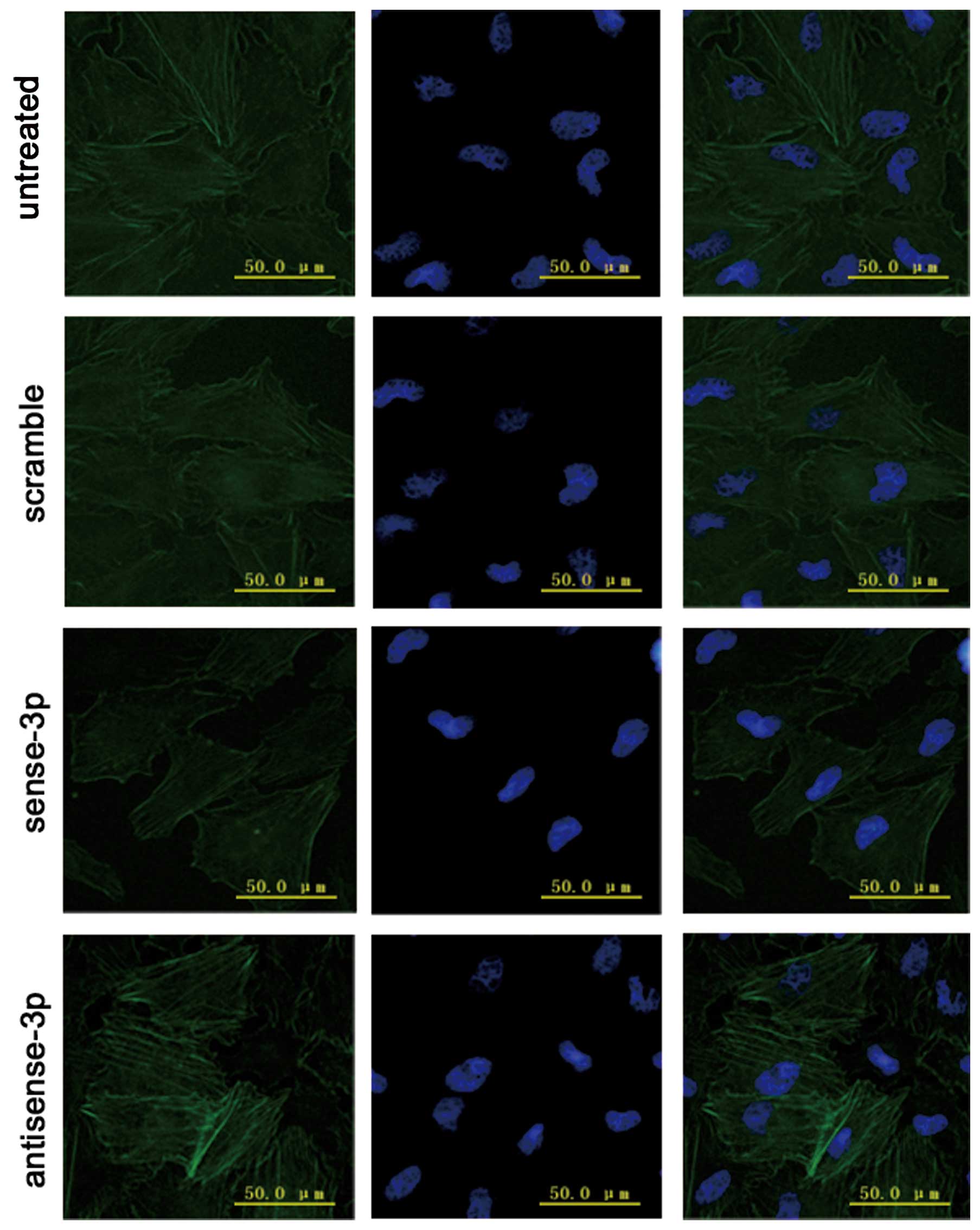
We investigated whether RhoA expression affected the
migration of A549 cells via organization of the actin cytoskeleton,
and also affected the levels of RhoA-GTP. The levels of RhoA-GTP
were quantified using a pull-down assay after transfecting A549
cells with sense-miR-125a-3p or antisense-miR-125a-3p. The results
showed that the levels of RhoA-GTP were decreased (p<0.001,
Fig. 7) along with decreased
expression of RhoA when sense-miR-125a-3p was transfected. In
addition immunofluorescence microscopy demonstrated that
accumulative actin filaments were lower (Fig. 8). By contrast, the levels of
RhoA-GTP increased (Fig. 7) with
increase in RhoA expression when antisense-miR-125a-3p was
transfected. Moreover, immunofluorescence microscopy results
demonstrated that the accumulative actin filaments had grown
(Fig. 8).
Discussion
MiRNAs negatively control downstream targeted genes
to interfere with cell activities. Therefore, the functions of
miRNAs are very important, especially with regard to their
modulation of target genes. In this study, we first used
bioinformatics to determine that RhoA is closely associated with
cell migration and is a target gene regulated by miR-125a-3p. By
manipulating miR-125a-3p expression levels in A549 NSCLC cells, we
found that miR-125a-3p can post-transcriptionally regulate the
expression of RhoA mRNA and repress expression of the RhoA protein.
Luciferase reporter assays using reporter constructs containing the
RhoA 3′UTR or specific mutated sites of RhoA 3′UTR indicated that
miR-125a-3p directly targets the RhoA 3′UTR in A549 cells.
RhoA was the first identified member of the Rho
family of GTPases (19). It
shuttles between an inactive GDP-bound state and an active
GTP-bound state and exhibits intrinsic GTPase activities, mainly
involved in forming stress fibers and assembling focal adhesion
complexes. Results of research using stable transfectants of active
RhoA have demonstrated that the RhoA-actomyosin pathway plays a
pivotal role in transmigration (20). RhoA regulates the signal
transduction from cell surface receptors to intracellular target
molecules and is involved in a variety of biological processes,
including cell morphogenesis (21), motility (22), cytokinesis (23,24)
and tumor progression (25,26).
The inconsistency of the degree of RhoA transcription and levels of
protein expression suggest that RhoA may be subject to
post-transcriptional regulation by microRNAs. Fritz et al
first detected the expression of Rho GTPases in human tumor
tissues, including lung cancer. They proposed that RhoA may be
overexpressed in certain tumors (27). RhoA has also been demonstrated to
play an important role in tumor invasion and metastasis by its
mediation of the cytoskeletal reorganization of key regulatory
proteins. Results of our previous and current Transwell experiments
suggest that miR-125a-3p directly targets RhoA, and may be involved
in migration of NSCLC cells.
Thus, miR-125a-3p may act as an anti-oncogene by
suppressing cell migration via the post-transcriptional repression
of RhoA. To the best of our knowledge, this is the first study to
demonstrate that miR-125a-3p impacts migration by regulating the
3′UTR of RhoA in human lung cancer A549 cells. After we blocked
RhoA activity with the Rho inhibitor CT04, we found that
miR-125a-3p could not regulate RhoA. In addition, the expression of
RhoA was knocked down using siRNA. We found that downregulation of
RhoA expression by siRNA significantly abrogated
antisense-miR-125a-3p-induced cell migration. These results further
validated the possibility that miR-125a-3p inhibited migration via
a Rho-dependent pathway.
The effects of RhoA protein levels on activated RhoA
have been reported. There is a report (28) that the stable RhoA transfectant of
the ovarian cancer cell line SKOV3 resulted in overexpression of
RhoA, which did not alter proliferative activity, but significantly
increased invasiveness. The invasiveness was suppressed by the
addition of a Rho inhibitor. A nude mouse model was used to show
that the frequency of dissemination and the number of disseminated
lesions were significantly increased in mice inoculated with RhoA
transfectants compared tocontrol mice. The investigators also
revealed that overexpression of RhoA mRNA and protein were
associated with the spread of ovarian cancer, and believed that
overexpression of RhoA had facilitated the accumulation of RhoA
protein in the cell membrane and further to activate RhoA (28). A similar conclusion was also
demonstrated from work using a rat liver cell model of MM1
(29). Overexpression of RhoA had
also resulted in increased levels of activated RhoA, and further
enhanced tumor cell motility.
The results of our study led us to conclusions
similar to these earlier reports. Upregulated expression of
miR-125a-3p negatively inhibited the expression of RhoA protein
because of incomplete complementation to nucleotides within the
3′UTR of the target gene RhoA. At the same time, the level of
RhoA-GTP decreased in A549 cells, along with decreasing
accumulative actin filaments, as shown by immunofluorescence.
Downregulation of miR-125a-3p expression led to increased levels of
RhoA-GTP and increased expression of RhoA protein. In addition,
accumulative actin filaments increased.
We suggest that in the lung cancer cell line A549,
miR-125a-3p directly regulates the target gene RhoA by incomplete
complementation with the 3′UTR, ultimately inhibiting the
expression levels of the RhoA protein. The level of RhoA-GTP is
directly correlated with the level of RhoA protein, as well as the
accumulation of actin filaments in A549 cells, which affects cell
motility. MiR-125a-3p may indirectly affect the level of RhoA-GTP
by direct regulation of RhoA, and thereby controls the migration of
cells.
In this study, we demonstrated that miR-125a-3p
post-transcriptionally regulates the 3′-UTR of RhoA mRNA and
inhibits expression of RhoA protein, which indirectly decreases the
levels of RhoA-GTP. Furthermore, downregulation of miR-125a-3p led
to increasing accumulation of actin filaments, ultimately leading
to increased migratory capacity of A549 cells. It is likely that
miR-125a-3p is involved in the RhoA-actomyosin pathway, which
affects the migration of the lung cancer cell line A549.
Acknowledgements
We are very grateful to Dr William
Kong and Dr Jin Q. Cheng for providing the plasmid containing the
full-length RhoA 3′UTR used in this study. We are very grateful to
Dr Baoshen Zhou from the Department of Epidemiology, School of
Public Health, China Medical University for statistical analysis.
We also thank Nan Liu for technical assistance, and the members of
our lab for useful suggestions. This study was supported by grants
from the National Natural Science Foundation of China (no.
30972967), Specialized Research Fund for the Doctoral Program of
Higher Education (no. 20092104110018), Program for Liaoning
Excellent Talents in University, Liaoning Provincial Natural
Science Foundation (no. 20102122), and Shenyang Science and
Technology Program (F10-149-9-41).
References
|
1.
|
Sporn MB: The war on cancer. Lancet.
347:1377–1381. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Nguyen DX, Bos PD and Massague J:
Metastasis: from dissemination to organ-specific colonization. Nat
Rev Cancer. 9:274–284. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Crawford M, Brawner E, Batte K, et al:
MicroRNA-126 inhibits invasion in non-small cell lung carcinoma
cell lines. Biochem Biophys Res Commun. 373:607–612. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Xiong BH, Cheng Y, Ma L and Zhang CQ:
Mir-21 regulates biological behavior through the PTEN/PI3K/AKT
signaling pathway in human colorectal cancer cells. Int J Oncol.
42:219–228. 2013.PubMed/NCBI
|
|
5.
|
Tavazoie SF, Alarcón C, Oskarsson T, et
al: Endogenous human microRNAs that suppress breast cancer
metastasis. Nature. 451:147–152. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Nicoloso MS, Spizzo R, Shimizu M, et al:
MicroRNAs - the micro steering wheel of tumour metastases. Nat Rev
Cancer. 9:293–302. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Valastyan S, Reinhardt F, Benaich N, et
al: A pleiotropically acting microRNA, miR-31, inhibits breast
cancer metastasis. Cell. 137:1032–1046. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Bartel DP: MicroRNAs: target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Asangani IA, Rasheed SA, Nikolova DA, et
al: MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor
suppressor Pdcd4 and stimulates invasion, intravasation and
metastasis in colorectal cancer. Oncogene. 27:2128–2136. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
He XY, Chen JX and Zhang Z: The let-7a
microRNA protects from growth of lung carcinoma by suppression of
k-Ras and c-Myc in nude mice. J Cancer Res Clin Oncol.
136:1023–1028. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Wu L and Belasco JG: Micro-RNA regulation
of the mammalian lin-28 gene during neuronal differentiation of
embryonal carcinoma cells. Mol Cell Biol. 25:9198–9208. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Laneve P, Di Marcotullio L, Gioia U, et
al: The interplay between microRNAs and the neurotrophin receptor
tropomyosin-related kinase C controls proliferation of human
neuroblastoma cells. Proc Natl Acad Sci USA. 104:7957–7962. 2007.
View Article : Google Scholar
|
|
13.
|
Cowden Dahl KD, Dahl R, Kruichak JN, et
al: The epidermal growth factor receptor responsive miR-125a
represses mesenchymal morphology in ovarian cancer cells.
Neoplasia. 11:1208–1215. 2009.PubMed/NCBI
|
|
14.
|
Jiang L, Huang Q, Zhang S, et al:
Hsa-125a-3p and hsa-miR-125a-5p are downregulated in non-small cell
lung cancer and have inverse effects on invasion and migration of
lung cancer cells. BMC Cancer. 10:3182010. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Chambers AF, Groom AC and MacDonald IC:
Dissemination and growth of cancer cells in metastatic sites. Nat
Rev Cancer. 2:563–572. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Woodhouse EC, Chuaqui RF and Liotta LA:
General mechanisms of metastasis. Cancer. 80(Suppl 8): 1529–1537.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Ho TT, Merajver SD, Lapiere CM, et al:
RhoA-GDP regulates RhoB protein stability. J Biol Chem.
283:21588–21598. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Ming J, Liu N, Gu Y, et al: PRL-3
facilitates angiogenesis and metastasis by increasing ERK
phosphorylation and up-regulating the levels and activities of
Rho-A/C in lung cancer. Pathology. 41:118–126. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Etienne-Manneville S and Hall A: Rho
GTPases in cell biology. Nature. 420:629–635. 2002. View Article : Google Scholar
|
|
20.
|
Yoshioka K, Matsumura F and Akedo H: Small
GTP-binding protein Rho stimulates the actomyosin system, leading
to invasion of tumor cells. J Biol Chem. 273:5146–5154. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Paterson HF, Self AJ, Garrett MD, et al:
Microinjection of recombinant p21rho induces rapid changes in cell
morphology. J Cell Biol. 111:1001–1007. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Takaishi K, Kikuchi A, Kuroda S, et al:
Involvement of rho p21 and its inhibitory GDP/GTP exchange protein
(rho GDI) in cell motility. Mol Cell Biol. 13:72–79.
1993.PubMed/NCBI
|
|
23.
|
Kishi K, Sasaki T, Kuroda S, et al:
Regulation of cytoplasmic division of Xenopus embryo by rho
p21 and its inhibitory GDP/GTP exchange protein (rho GDI). J Cell
Biol. 120:1187–1195. 1993.PubMed/NCBI
|
|
24.
|
Mabuchi I, Hamaguchi Y, Fujimoto H, et al:
A rho-like protein is involved in the organisation of the
contractile ring in dividing sand dollar eggs. Zygote. 1:325–331.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Perona R, Esteve P, Jimenez B, et al:
Tumorigenic activity of rho genes from Aplysia californica.
Oncogene. 8:1285–1292. 1993.PubMed/NCBI
|
|
26.
|
Prendergast GC, Khosravi-Far R, Solski P,
et al: Critical role of Rho in cell transformation by oncogenic
Ras. Oncogene. 10:2289–2296. 1995.PubMed/NCBI
|
|
27.
|
Fritz G, Just I and Kaina B: Rho GTPases
were over-expressed in human tumors. Int J Cancer. 81:682–687.
1990. View Article : Google Scholar
|
|
28.
|
Horiuchi A, Kikuchi N, Osada R, et al:
Overexpression of RhoA enhances peritoneal dissemination: RhoA
suppression with Lovastatin may be useful for ovarian cancer.
Cancer Sci. 12:2532–2539. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Yoshioka K, Nakamori S and Itoh K:
Overexpression of small GTP-binding protein RhoA promotes invasion
of tumor cells. Cancer Res. 59:2004–2010. 1999.PubMed/NCBI
|