Introduction
Post-translational modifications of core histone
proteins play a critical role in the regulation of gene expression
(1). Acetylation of core histones
by histone acetyltransferases is linked to chromatin opening and
transcriptional gene activation; in contrast, histone deacetylases
(HDACs) remove the acetyl group from histones and repress gene
transcription (2). HDACs regulate
many important biological processes, including cell cycle
progression, differentiation and development (3).
Most studies performed to date have reported that,
relative to adjacent normal mucosa, expression of HDAC1, HDAC2,
HDAC3 and HDAC8, which all belong to the class I family of HDACs,
is increased in colorectal carcinoma (4–9).
Class I HDACs are frequently overexpressed in various human
cancers, including colorectal cancer. Furthermore, their
differential expression often correlates with drug resistance and
poor prognosis, which makes them an attractive target for cancer
therapeutics (10,11). Studies have demonstrated a role for
class I HDACs in promoting colon cell proliferation and survival
(4,7). Knockdown of HDAC1, 2 and 3 reduces
the growth of several colon cancer cell lines including HCT-116,
HT-29 and SW-480 (4,7,12).
Mechanistically, the proliferative effects of HDACs in colon cancer
cells have been linked to transcriptional repression of the
cyclin-dependent kinase inhibitor, p21 (4,12).
Knockdown of HDAC1, HDAC2 and HDAC3 induces p21 expression in colon
cancer cell lines, while overexpression of these HDACs represses
HDAC inhibitor-mediated p21 induction (13).
Recently, HDAC inhibitors such as suberoylanilide
hydroxamic acid (SAHA) and trichostatin A (TSA) have emerged as a
promising class of therapeutic drugs. Crystallographic analysis has
shown that SAHA and TSA interact directly with the catalytic site
of an HDAC-like protein and inhibit its enzymatic activity
(14). Inhibition of HDAC activity
by SAHA and related agents alters gene expression, causing cell
cycle arrest and apoptosis in cancer cells, primarily by the
induction of p21 and Bax, a pro-apoptotic protein (15). However, phase I and II studies have
demonstrated that pan-HDAC inhibitors may have numerous side
effects, such as bone marrow depression, diarrhea, disordered
clotting and cardiac arrhythmias (16). Therefore, there is a clinical need
for HDAC inhibitors that are both effective and minimally
toxic.
Runt-related transcription factor 3 (RUNX3) belongs
to the RUNX family of genes, which are important in mammalian
development and neoplasia (17–19).
RUNX3 cooperates with Sma- and Mad-related protein 3 (Smad3) and
Smad4 to activate transforming growth factor-β (TGF-β-dependent
growth inhibition and apoptosis by inducing p21 and Bim (20). The RUNX3 gene is localized to a
locus at chromosome 1p36 and is linked with gastric epithelial
homeostasis and gastric carcinogenesis. The 1p36 region is thought
to harbor at least one tumor suppressor gene because this region
exhibits frequent loss of heterozygosity in colon, gastric, breast
and ovarian cancers (21). In
addition, the introduction of a normal human 1p36 chromosome
fragment into colon cancer cells suppresses their tumorigenicity
(22). The RUNX3 gene is
frequently inactivated through histone modification during the
development of various carcinomas, including gastric and colon
(20,22–25).
Fujii et al demonstrated that enhancer of Zeste Homolog 2, a
histone methytransferase, binds to the RUNX3 promoter, resulting in
the upregulation of H3K27 methylation and concomitant
downregulation of RUNX3 expression in colon cancer (26). Lee et al extended this
finding by demonstrating that hypoxia-induced upregulated
expression of HDAC1 and G9a, a histone methyltransferase, also
causes epigenetic RUNX3 silencing through H3K9 methylation and
decreased histone H3 acetylation at the RUNX3 promoter (27).
A number of saponins have been isolated from ginseng
and their possible antitumor activity has been extensively
investigated in various cancer cell lines (28–30).
Among these saponins,
20-O-(β-D-glucopyranosyl)-20(S)-protopanaxadiol, or Compound K, is
the main metabolite of protopanaxadiol-type ginsenoside, formed in
the intestine after oral administration (31–33).
Our laboratory recently reported that Compound K exhibits
cytotoxicity by inducing apoptosis, arrest of growth at the
G1 phase of the cell cycle and inhibition of telomerase
activity in human leukemia cells (34,35).
In addition, combined treatment with Compound K and gamma
irradiation enhances the death of human lung cancer cells (36) and Compound K induces apoptosis in
MCF-7 breast cancer cells by modulating AMP-activated protein
kinase (37). In addition, we
recently demonstrated that Compound K induces RUNX3 reactivation by
DNA methyltransferase 1 inhibition (38).
The aims of this study were to determine whether
Compound K suppresses HDAC activity and expression and to elucidate
the molecular mechanisms by which Compound K induces cell cycle
arrest and apoptosis. The results demonstrate that Compound K can
downregulate HDAC1, leading to the accessibility of the RUNX3
promoter region in colorectal cancer HT-29 cells via increase of
acetylation of histones H3 and H4. Downregulation of HDACs
represents a novel mechanism underlying the ability of Compound K
to induce cell cycle arrest and apoptosis.
Materials and methods
Cell culture
Human colorectal cancer HT-29 cells were obtained
from the Korean Cell Line Bank (Seoul, Republic of Korea). Cells
were maintained in an incubator at 37°C with a humidified
atmosphere of 5% CO2. The cells were cultured in
RPMI-1640 medium containing 10% fetal calf serum, streptomycin (100
μg/ml) and penicillin (100 U/ml).
Cell proliferation assay
Cell proliferation was determined using the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay. Cells were seeded into a 96-well plate at a density of
1×105 cells/ml and were treated with 8, 16, 32 and 64
μmol/l of Compound K or TSA. After incubating for 48 h, 50
μl of the MTT stock solution (2 mg/ml) was added to each
well to attain a total reaction volume of 250 μl. After 4 h
incubation, the supernatants were aspirated; the formazan crystals
in each well were dissolved in 150 μl dimethylsulfoxide and
absorbance at 540 nm was measured using a scanning multi-well
spectrophotometer (TECAN, Melbourne, Australia).
Reverse transcription polymerase chain
reaction (RT-PCR)
Total RNA was isolated from cells using TRIzol
reagent (Gibco BRL, Grand Island, NY, USA). Complementary DNA
(cDNA) (1 μl) was amplified in a 25 μl reverse
transcription reaction containing primers, dNTPs and 0.5 U of Taq
DNA polymerase. The PCR conditions were 5 min at 94°C for initial
denaturation, followed by 35 cycles of 1 min at 94°C, 1 min at 55°C
and 1 min at 72°C and then a final elongation period of 7 min at
72°C. PCR amplification was carried out in a Perkin-Elmer Cetus
9600 thermal cycler (Roche Molecular Systems Inc., NJ, USA). The
primers used to amplify the RUNX3 and HDAC1 cDNA were as follows:
RUNX3 sense, 5′-GGCAATG ACGAGAACTAC-3′ (located in exon 2); RUNX3
antisense, 5′-GGAGAATGGGTTCAGTTC-3′ (located in exon 5); HDAC1
sense, 5′-AACCTGCCTATGCTGATGCT-3′; HDAC1 antisense,
5′-CAGGCAATTCGTTTGTCAGA-3′; GAPDH sense,
5-GTGGGCCGCCCTAGGCACCAGG-3′; and GAPDH antisense,
5′-GGAGGAAGAGGATGCGGCAGTG-3′. The amplified products were resolved
on 1% agarose gels, which were stained with ethidium bromide and
visualized under ultraviolet light using ImageQuant™ TL analysis
software (Amersham Bioscience, Uppsala, Sweden).
Methylation specific (MS)-PCR
The bisulfate modification of DNA was carried out
with the Methylamp™ DNA modification kit (Epigentek, Pittsburgh,
PA, USA) according to the manufacturer’s instructions. For analysis
of DNA methylation of RUNX3, MS-PCR carried out using an Epitect
MSP kit (Qiagen, Valencia, CA, USA). The PCR products were
separated on 6% non-denaturing polyacrylamide gels, stained with
ethidium bromide and visualized under UV light. The methylated or
unmethylated RUNX3 primer set is as follows:
5′-TTATGAGGGGTGGTTGTATGTGGG-3′ and 5′-AAA ACAACCAACACAAACACCTCC-3′
for unmethylated RUNX3; 5′-TTACGAGGGGCGGTCGTACGCGGG-3′ and
5′-AAAACGACCGACGCGAACGCCTCC-3′ for methylated RUNX3.
Western blot analysis
Cells were harvested and lysed on ice in 1 ml of
lysis buffer (10 mmol/1 Tris-HCl, pH 7.9, 10 mmol/1 NaCl, 3 mmol/1
MgCl2 and 1% NP-40) for 4 min. After centrifugation for
10 min at 3,000 g, the pellets were re-suspended in 50 μl of
extraction buffer (20 mmol/1 HEPES, pH 7.9, 20% glycerol, 1.5
mmol/1 MgCl2, 0.2 mmol/1 EDTA, 1 mmol/1 DTT and 1 mmol/1
PMSF) and then incubated on ice for 30 min and centrifuged at
13,000 g for 5 min. The protein concentration was measured and
supernatants were stored at −70°C. Aliquots of the lysates (40
μg of protein) were boiled for 5 min and electrophoresed on
a 10% SDS-polyacrylamide gel. Proteins were transferred onto
nitrocellulose membranes, which were subsequently incubated with
primary antibodies. The membranes were further incubated with
secondary immunoglobulin-G-horseradish peroxidase conjugates
(Pierce, Rockford, IL, USA). Protein bands were detected using an
enhanced chemiluminescence western blotting detection kit
(Amersham, Little Chalfont, UK) and were visualized using a
luminescent image analyzer (BMG Labtech, Offenburg, Germany).
HDAC activity
Nuclear extracts were prepared using a nuclear
protein extraction kit (Cayman Chemical, Ann Arbour, MI, USA).
After measuring the protein concentration, the nuclear fractions
were stored at −70°C. HDAC activity was measured using an EpiQuik
HDAC activity assay kit (Epigentek, Brooklyn, NY, USA), according
to the manufacturer’s instructions. In brief, the nuclear extracts
were incubated with a specific substrate for 1 h at 37 °C, followed
by capture antibody for 60 min and then detection antibody for 30
min at room temperature. Absorbance at 450 nm was measured using a
microplate spectrophotometer (TECAN). HDAC activity was calculated
as ng/h/ml = {[optical density (control-blank)-optical density
(sample-blank)] / Slope × h} × sample dilution.
Immunocytochemistry
Cells plated onto coverslips were fixed with 4%
paraformaldehyde for 30 min and permeabilized with 0.1% Triton
X-100 in PBS for 15 min. Cells were treated with blocking medium
(3% bovine serum albumin in PBS) for 1 h and then incubated for 2 h
with the RUNX3 antibody in blocking medium. The primary acetylated
histone H3, acetylated histone H4 and RUNX3 antibodies were
detected by a FITC-conjugated secondary antibody (1:500; Santa Cruz
Biotechnology, Santa Cruz, CA, USA) for 1 h. After washing with
PBS, stained cells were mounted onto microscope slides in mounting
medium with DAPI (Vector, Burlingame, CA, USA) and imaged using the
LSM 510 program on a Zeiss confocal microscope (Carl Zeiss
Microscopy Ltd., Cambridge, UK).
Chromatin immunoprecipitation (chIP)
assay
The ChIP assay was performed using a Simple ChIP™
enzymatic chromatin IP kit (Cell Signaling Technology, Danvers, MA,
USA), according to the manufacturer’s protocol with slight
modifications. Briefly, cells were treated with 20 μg/ml of
Compound K for 48 h and then cross-linked by the addition of 1%
formaldehyde. Chromatin was prepared and digested with nuclease for
12 min at 37°C. ChIP was performed with the acetylated histone H3,
acetylated histone H4 and RUNX3 antibodies [Cell Signaling
Technology and Abeam (Cambridge, UK)] and normal mouse IgG.
Antibodies were added to the chromatin digests and were incubated
with constant rotation at 4°C overnight. ChlP-grade protein G
magnetic beads were then added to capture the immune complexes. The
beads were washed and the immunoprecipitates were eluted with ChIP
elution buffer. The cross-links were reversed by incubation at 65°C
for 30 min. Proteinase K was added and incubated at 65°C for 2 h.
The immunoprecipitated DNA fragments were then purified using spin
columns. DNA recovered from the immunoprecipitated complex was
subjected to 35 cycles of PCR. The primers for the p21 (RUNX3
binding site) and RUNX3 (acetylated H3, H4 binding sites) gene
promoters were as follows: p21 sense,
5′-CACCAGACTTCTCTGAGCCCCAG-3′; p21 antisense,
5′-GCACTGTTAGAATGAGCCCCCTTTC-3′; RUNX3 sense,
5′-GGTTGCAGAAGTCACAGG-3′; and RUNX3 antisense,
5′-AATTTGCTTAGAACGTCCG-3′. The PCR products were separated on 2%
agarose gels and DNA bands were visualized using the Image program
(NIH, Bethesda, MD, USA).
Flow cytometry
A flow cytometric assay was performed to assess the
effects of Compound K or TSA on the cell cycle. Cells were treated
with 32 μmol/l of Compound K or TSA for 48 h. Harvested
cells were then washed twice with PBS and fixed in 70% ethanol for
30 min at 4°C. Subsequently, the cells were incubated in 50 mg/ml
propidium iodide solution and 50 μg/ml RNase A in the dark
for 30 min at 37 °C. Flow cytometric analysis was performed using a
FACSCalibur flow cytometer (Becton-Dickinson, Mountain View, CA,
USA). The cell cycle phases were assessed based on histograms
generated by CellQuest and Mod-Fit software programs.
Nuclear staining with Hoechst 33342
A volume of 1.5 μl of Hoechst 33342 (10
mg/ml), a DNA-specific fluorescent dye, was added to each well and
cells were incubated for 10 min at 37°C. To examine the degree of
nuclear condensation, the stained cells were observed under a
fluorescent microscope, which was equipped with a CoolSNAP-Pro
color digital camera (Carsen Group, Markham, ON, Canada).
Statistical analysis
All measurements were performed in triplicate and
values are expressed as the mean ± standard error of the mean
(SEM). The results were examined using analysis of variance (ANOVA)
and Tukey’s test to determine pairwise differences. P<0.05 was
considered statistically significant.
Results
Compound K inhibits HDAC activity and
expression in HT-29 cells
We recently demonstrated that Compound K acts as a
DNA methyltrasferase inhibitor in human colorectal cancer cells
(38); the present study provides
evidence that Compound K also inhibits HDACs in human colorectal
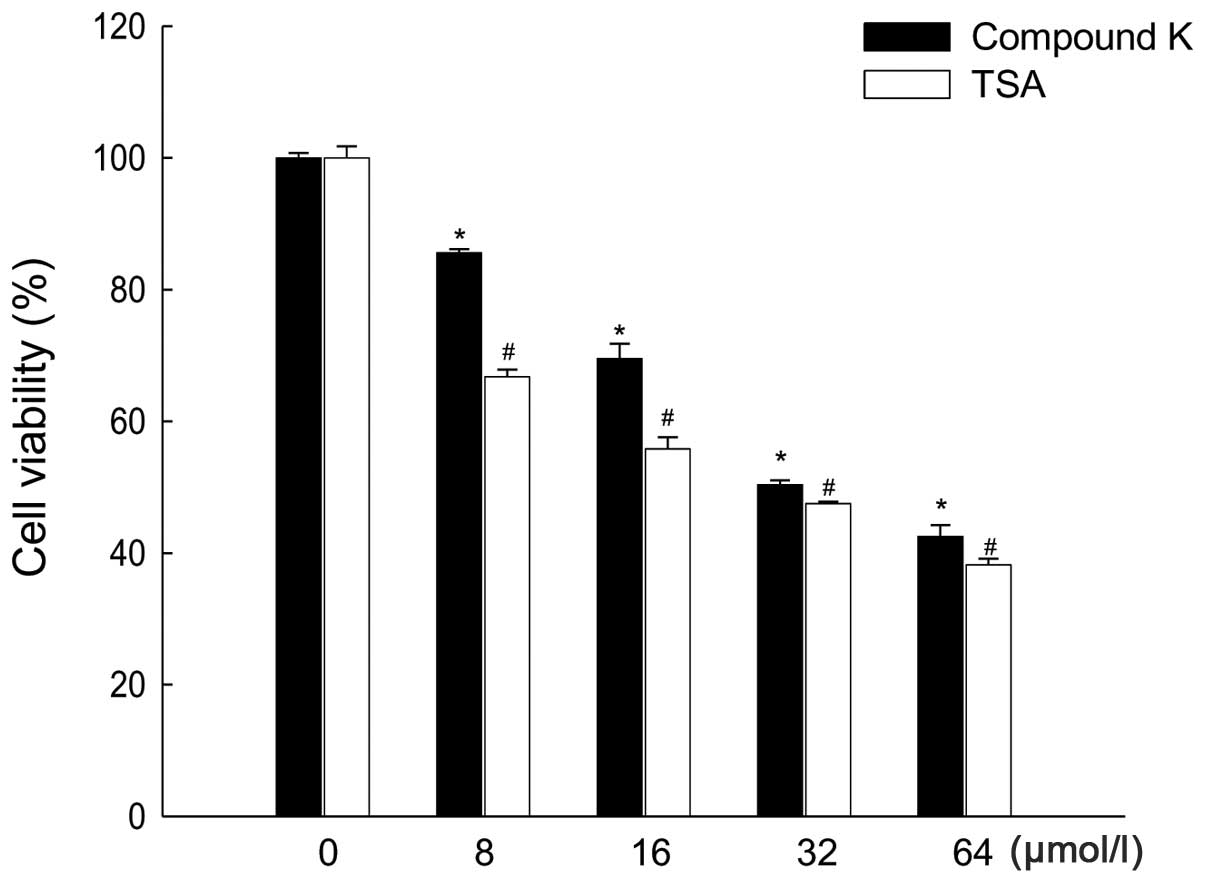
cancer HT-29 cells. Compound K and TSA inhibited HT-29 cell growth
in a dose-dependent manner at concentrations between 8 and 64
μmol/l; the concentration that yielded 50% growth inhibition
(IC50) was 32 μmol/l for Compound K and 24
μmol/1 for TSA (Fig. 1). As
a result, 32 and 24 μmol/1 were chosen as the optimal doses
of Compound K and TSA, respectively. Treatment of cells with the
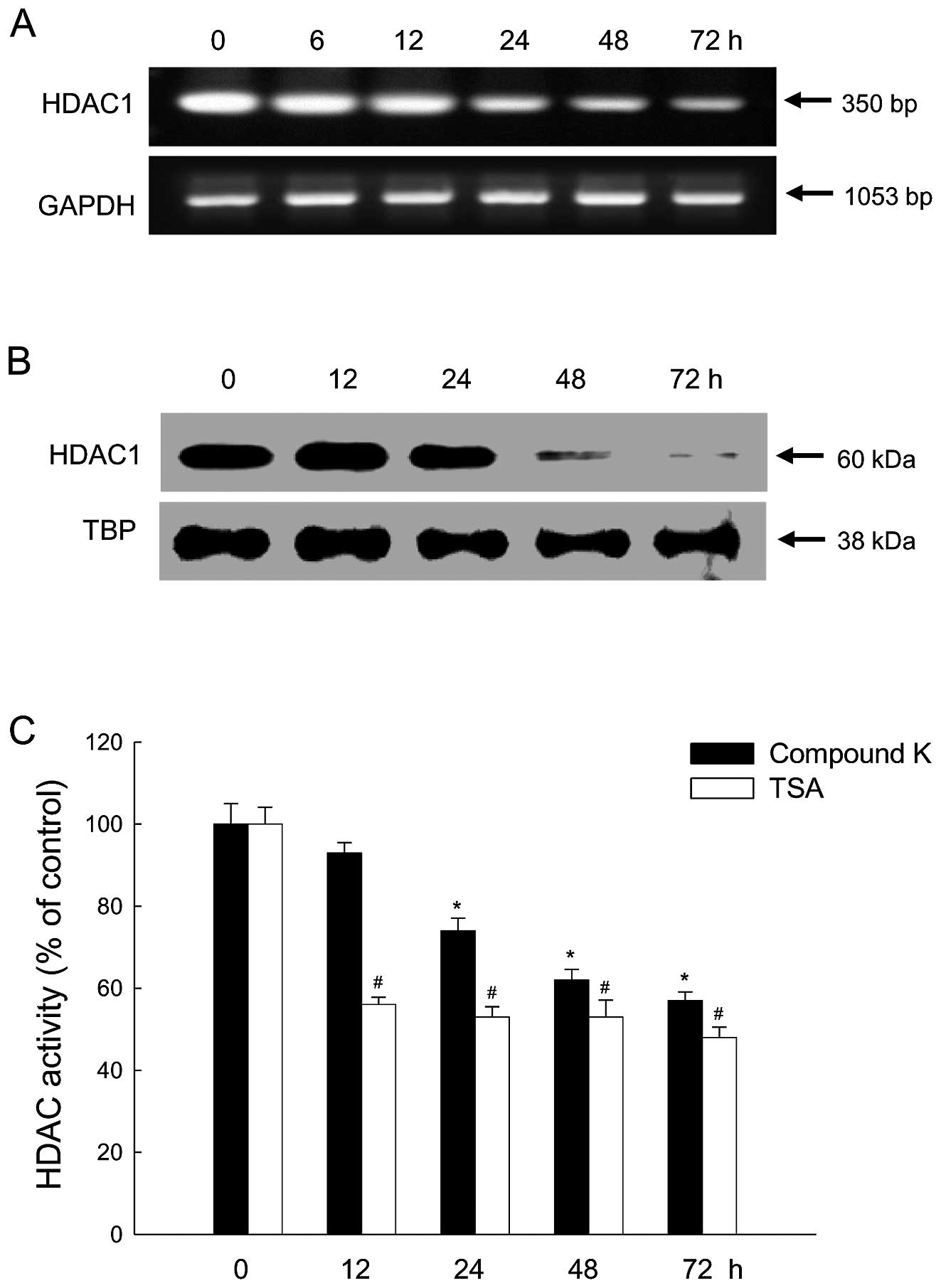
optimal dose of Compound K decreased HDAC1 mRNA and protein
expression in a time-dependent manner (Fig. 2A and B). HDAC enzyme activity was
reduced at all time-points in cells treated with TSA and enzyme
activity was also decreased in a time-dependent manner in cells
treated with Compound K (Fig.
2C).
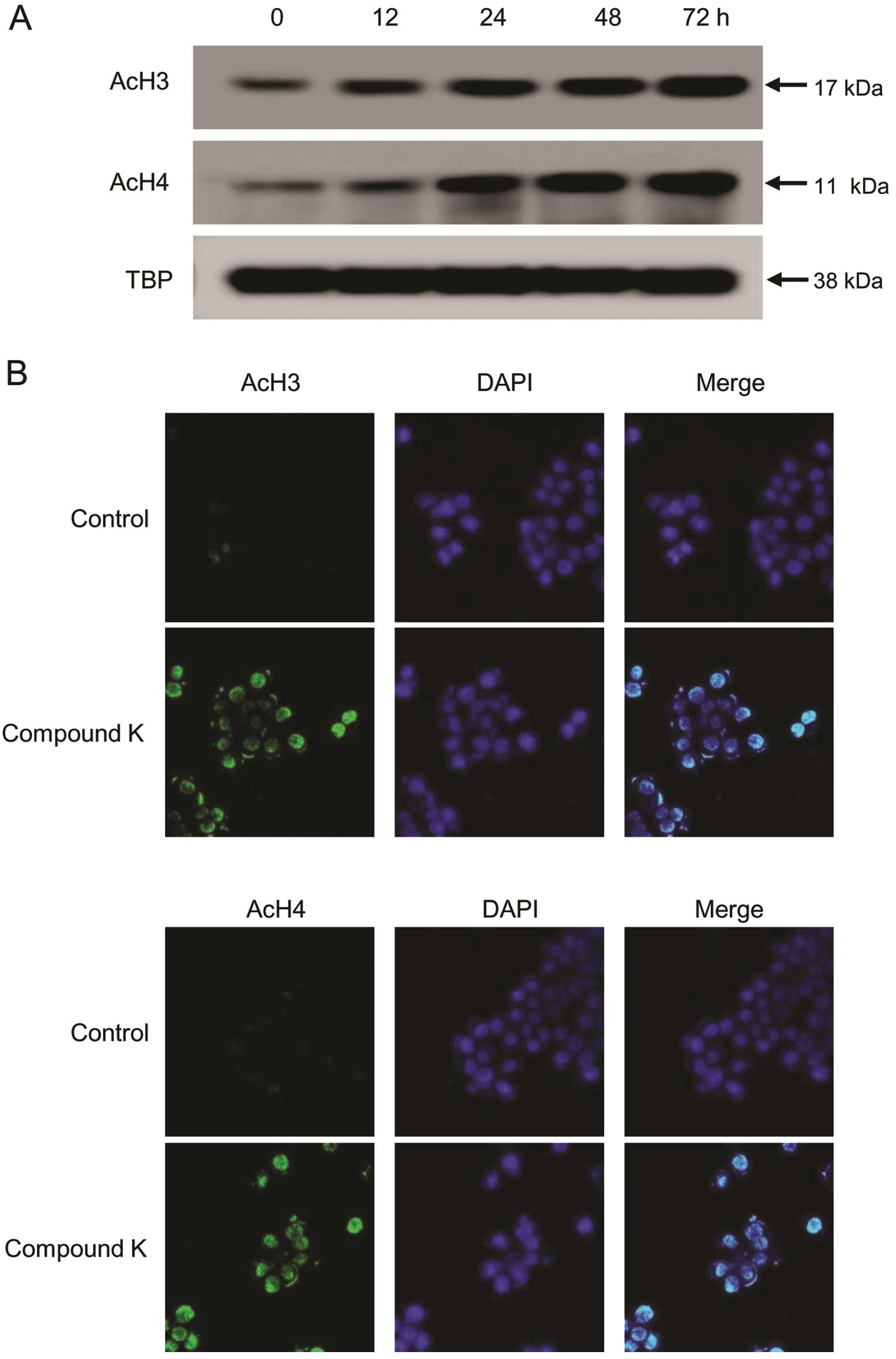
Compound K increases histone
acetylation
We then determined whether the decrease in HDAC
expression and activity induced by Compound K affects histone
acetylation. Compound K induced a significant increase in the
acetylation of histones H3 and H4 in a time-dependent manner
(Fig. 3A). Furthermore, direct
immunofluorescence analysis revealed that acetylated histones H3
and H4 are present in the nuclei of HT-29 cells following treatment
with Compound K (Fig. 3B).
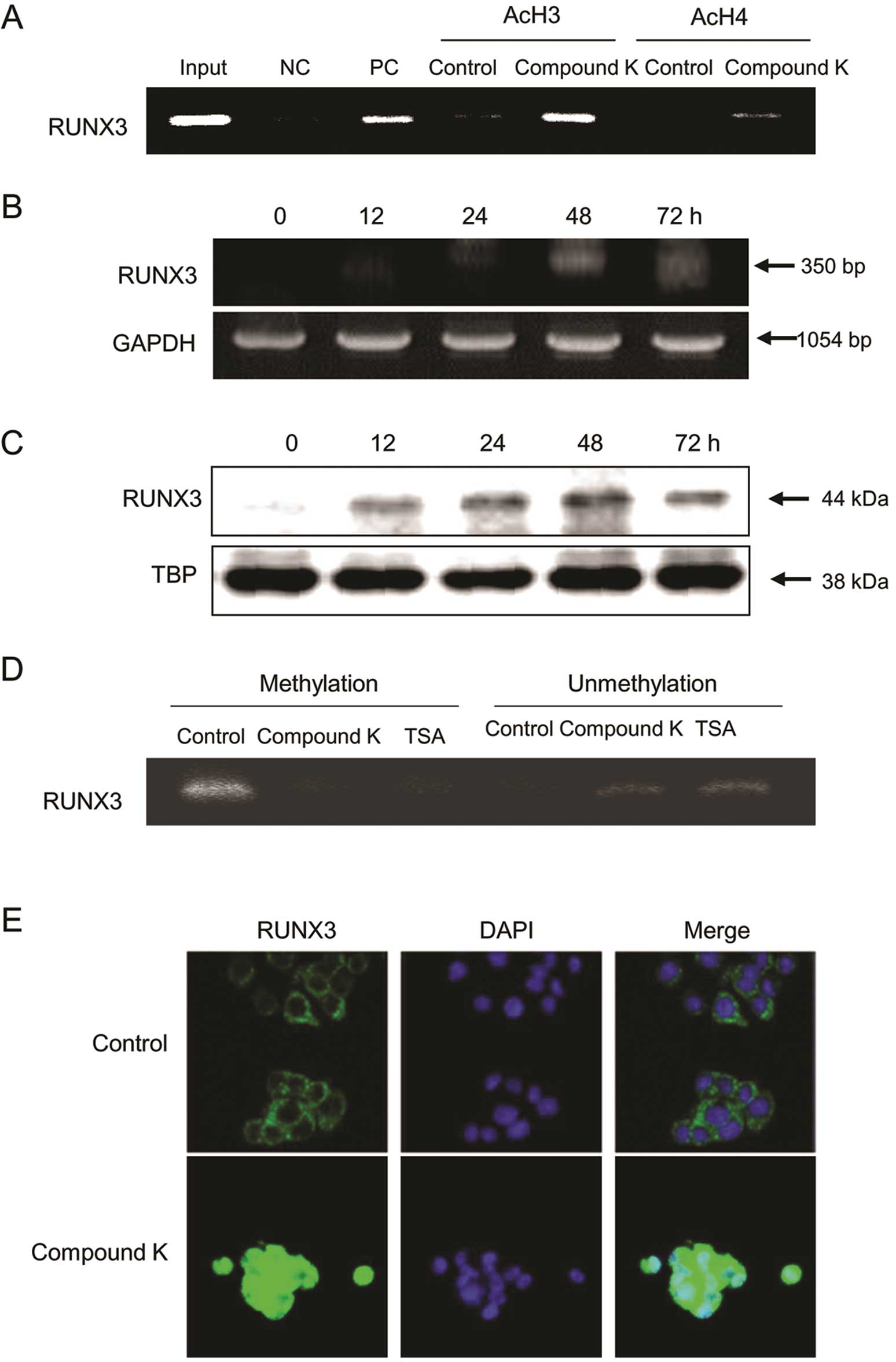
Compound K reactivates RUNX3 via
inhibition of HDAC
The RUNX3 gene in HT-29 cells is reportedly
epigenetically silenced (39–43).
ChIP analysis revealed acetylation of the RUNX3 promoter in
Compound K-treated HT-29 cells (Fig.
4A), which resulted in the time-dependent re-expression of
RUNX3 mRNA (Fig. 4B) and protein
(Fig. 4C). We then investigated
whether the status of DNA methylation in the RUNX3 promoter region
changed after treatment of Compound K and resulted in restoration
of RUNX3 gene expression. It was shown that unmethylation of the
RUNX3 promoter region induced in HT-29 cells by treatment of
Compound K, as well as TSA (Fig.
4D). Mislocalization of nuclear RUNX3 protein to the cytoplasm
was also observed in HT-29 cells; however, treatment of cells with
Compound K increased the nuclear localization of the RUNX3 protein
(Fig. 4E).
Compound K induces RUNX3-mediated
expression of p21
RUNX3 cooperates with Smad3 and Smad4 to activate
TGF-β-dependent growth inhibition and also causes apoptosis by
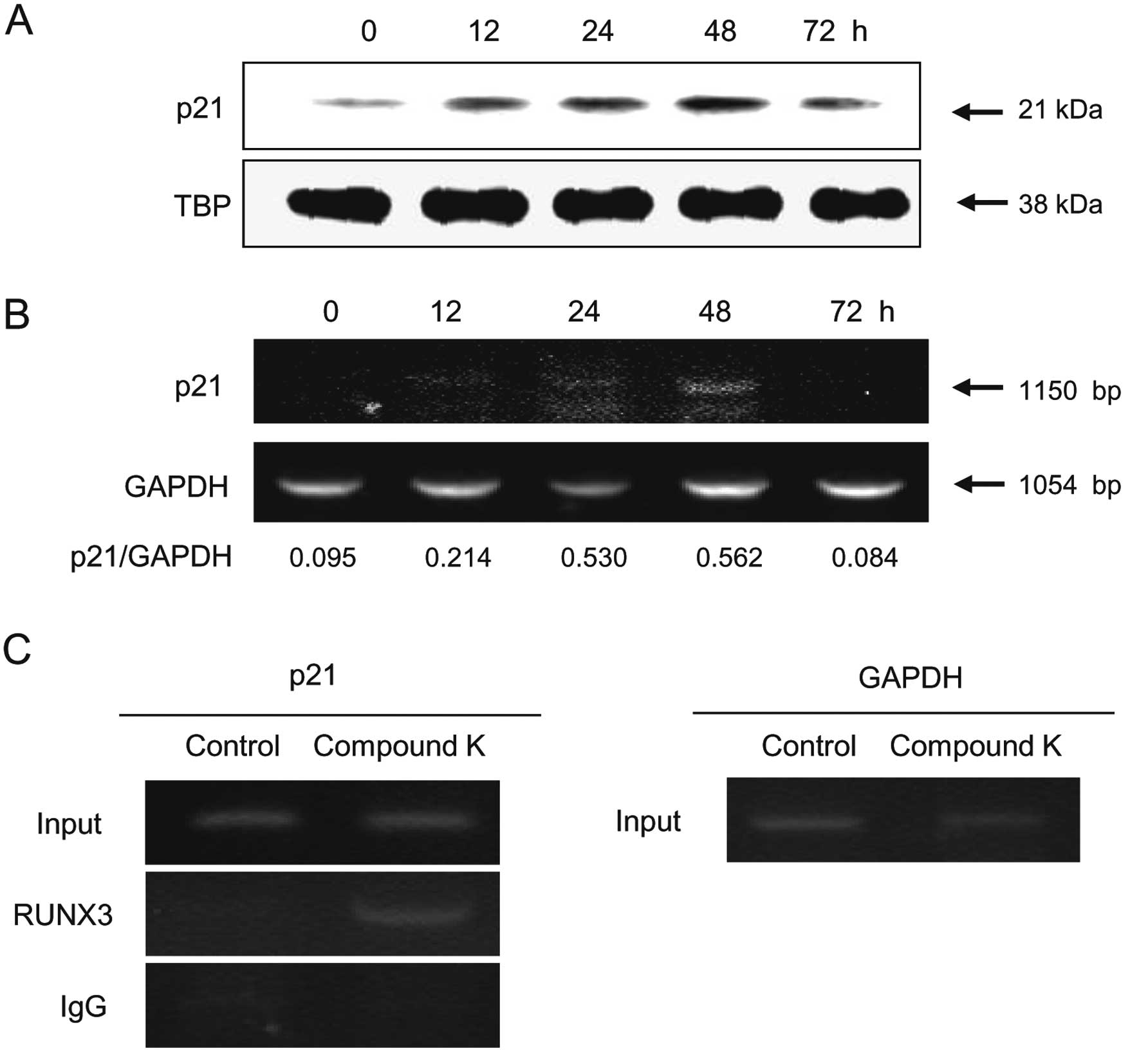
inducing p21 and Bim (20,41). Enhanced p21 mRNA (Fig. 5A) and protein (Fig. 5B) expression was observed in HT-29
cells following Compound K treatment. Additionally, ChIP assay data
showed that RUNX3 interacted with p21 (Fig. 5C); therefore, Compound K induced
p21 expression via the binding of RUNX3 to the p21 promoter
region.
Compound K induces cell cycle arrest and
apoptosis
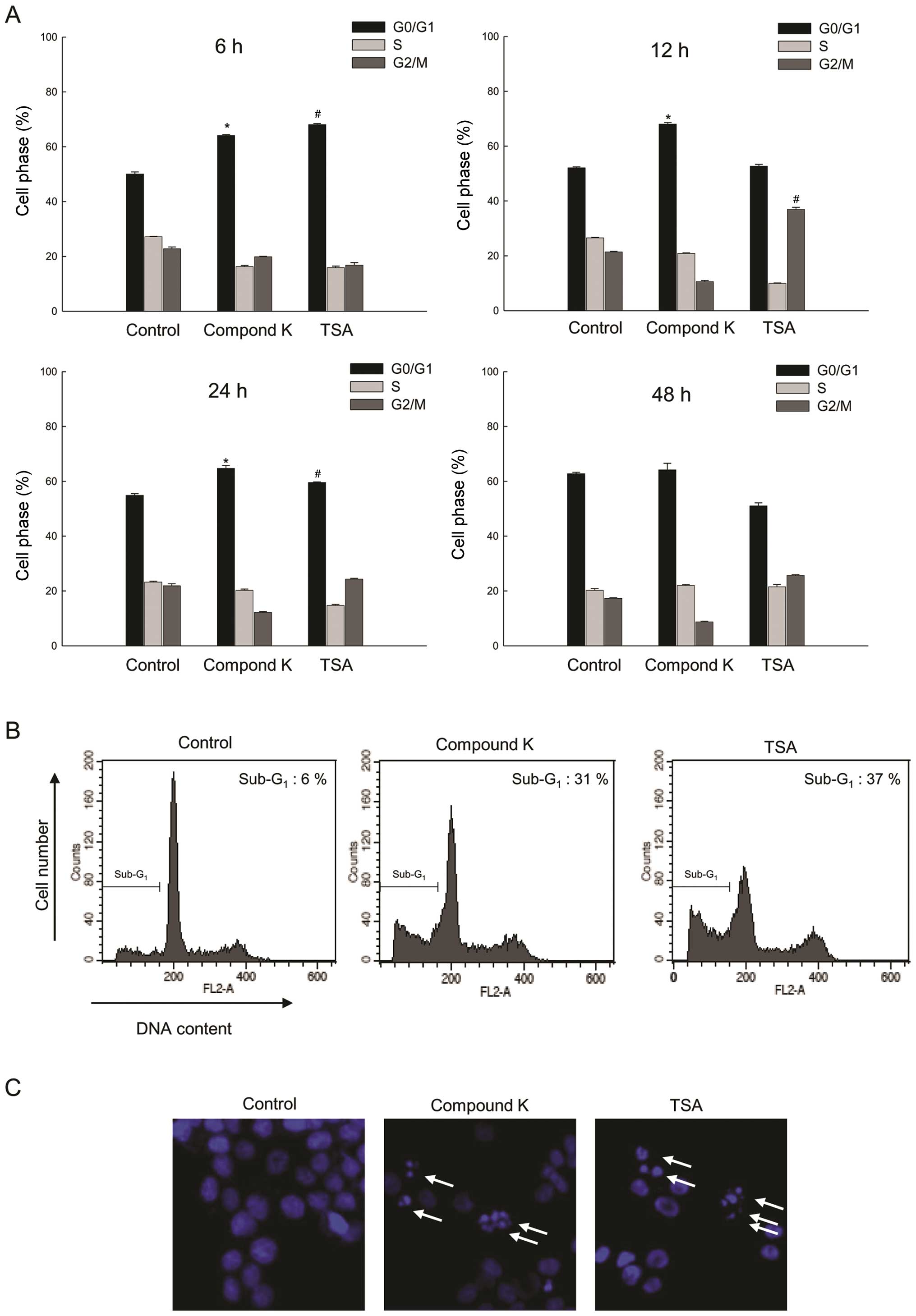
Analysis of DNA content by flow cytometry revealed
that Compound K and TSA caused a marked increase in the percentage
of cells in the G0/G1 phases of the cell
cycle. Following Compound K treatment of HT-29 cells, the cell
population in the G0/G1 phase at 6 and 12 h
was 64 and 68% comparing to 50 and 52% cell population in the
G0/G1 phase of control group; following TSA
treatment, 69% was in the G0/G1 phase at 6 h
and 37% in the G2/M phase at 12 h compared to 50% in the
G0/G1 phase at 6 h and 21% in the
G2/M phase at 12 h of control group (Fig. 6A). Furthermore, the
sub-G1 population (Fig.
6B) and apoptotic body formation (Fig. 6C) were increased in Compound
K-treated cells compared with control cells.
Discussion
Epigenetic gene regulation plays a crucial role in
the etiology of cancer. Post-translational histone modifications
are important epigenetic events in the regulation of gene
expression and maintenance of cellular function, which may
contribute to cancer development (44–46).
Abnormal histone modification is a hallmark of cancer that often
causes silencing of tumor suppressor genes, which in turn leads to
cancer development and progression. The anti-carcinogenic effects
and preventative action of Compound K, the main metabolite of
ginseng saponin, against colorectal cancer have been extensively
studied in various tumor models (35–37,47);
however, less attention has been given to the epigenetic
modifications induced by Compound K and their potential role in
reducing the risk of developing colorectal cancer. In this study,
Compound K induced the transcription of RUNX3 and p21 transcription
factors and reduced DNA methylation of RUNX3. Also, this Compound K
increased acetylation of histones H3 and H4, leading to cell cycle
arrest and apoptosis.
Class I HDACs are overexpressed in human colorectal
cancer specimens and colorectal cancer cell lines (5,48);
specifically, HDAC1, HDAC2 and HDAC3 are highly expressed in
colorectal cancer (11).
Overexpression of HDAC1 in prostate cancer cells causes increased
cell proliferation and a reduction in cell differentiation markers;
HDAC1 may therefore represent a putative therapeutic target for
cancer (49,50). HDACs form multi-subunit
transcriptional co-repressor complexes that are recruited to
promoter regions by sequence-specific transcription factors
(51). There are reportedly
several co-repressor complexes for distinct promoters, which
recruit specific HDAC isoforms that silence their target genes.
Class I HDACs are specifically responsible for deacetylation of the
catalytic core of different co-repressor complexes, resulting in
transcriptional repression. For example, HDAC1 and HDAC2 are
present in the CoREST, mi2/NURD and Sin3 complexes and HDAC3 is
responsible for the catalytic activities of the N-CoR and SRMT
co-repressor complexes (51).
Silencing of RUNX3 by HDACs plays an important role in the
regulation of tumor suppression during gastric and colon
carcinogenesis (26,27).
It is important to note that Compound K affects
HDAC1 and therefore has profound effects on cancer cells; however,
the specific mechanisms underlying these effects requires further
clarification, specifically with regard to understanding the
differential response of colorectal cancer cells to Compound K.
Repression of tumor suppressor genes involves deacetylation of
various transcription factors by class I HDACs (32,33).
In addition, evidence to date indicates that specific HDACs are
consistently overexpressed in colon cancer. The effect of
overexpression and depletion of multiple class I HDACs on the
growth and survival of colon cancer cells has been investigated
in vitro in a number of studies; for example, deficiency of
HDAC1, 2 and 3 reduces the growth of several colon cancer cell
lines (4,7,12).
In this study, we observed a differential response of Compound
K-mediated cell death, probably due to significant inhibition of
class I HDACs at the protein and mRNA levels. The results provide
additional evidence that Compound K inhibits class I HDACs by
altering the acetylated histone status and enhancing the expression
of tumor suppressor genes and pro-apoptotic proteins. These effects
were accompanied by corresponding increases in cell growth
inhibition and apoptosis.
Tumor suppressor gene repression involves
deacetylation of various transcription factors by class I HDACs.
Class I HDAC-mediated deacetylation has been shown to decrease the
DNA-binding activity of sequence-specific transcription factors.
For example, covalent modifications of several transcription
factors, including E2F, SP1/ SP3, p53, GATA1, TFIIF and RUNX3 by
class I HDACs, have been reported (51–53).
HDAC inhibitors reduce HDAC activity, altering the dynamic balance
between HDAC and histone acetyl-transferase activity, which results
in increased acetylation of non-histone proteins, including RUNX3.
Deacetylation by HDAC1 and HDAC2 results in the silencing of RUNX3
and HDAC inhibitors have been shown to reverse this process
(54,55). Inhibition of HDACs results in the
acetylation of RUNX3, thereby increasing both its half-life and
binding to the p21 promoter (54).
Our chromatin immunoprecipitation assays confirmed an increase in
the expression of acetylated histones H3 and H4 associated with the
RUNX3 promoter in the nuclei of HT-29 cells.
In conclusion, this study demonstrates that Compound
K can decrease HDAC enzyme activity and may be effective in
inhibiting cancer cell growth. The degree of HDAC inhibition and
the alterations in downstream gene expression induced by Compound K
were similar to those induced by TSA, a pharmacological inhibitor
of HDACs. Our data suggest that epigenetic regulation of tumor
suppressor genes by Compound K may play a role in colorectal cancer
chemoprevention or therapy. These findings are important for
understanding the anticancer mechanisms and clinical applications
of Compound K.
Acknowledgements
This study was supported by a grant
from the National R&D Program for Cancer Control, Ministry for
Health and Welfare, Republic of Korea (1120340).
References
|
1.
|
Kouzarides T: Chromatin modifications and
their function. Cell. 128:693–705. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Strahl BD and Allis CD: The language of
covalent histone modifications. Nature. 403:41–45. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Gallinari P, Di Marco S, Jones P, Pallaoro
M and Steinkühler C: HDACs, histone deacetylation and gene
transcription: from molecular biology to cancer therapeutics. Cell
Res. 17:195–211. 2007.PubMed/NCBI
|
|
4.
|
Wilson AJ, Byun DS, Popova N, Murray LB,
L’Italien K, Sowa Y, Arango D, Velcich A, Augenlicht LH and
Mariadason JM: Histone deacetylase 3 (HDAC3) and other class I
HDACs regulate colon cell maturation and p21 expression and are
deregulated in human colon cancer. J Biol Chem. 281:13548–13558.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Nakagawa M, Oda Y, Eguchi T, Aishima S,
Yao T, Hosoi F, Basaki Y, Ono M, Kuwano M, Tanaka M and Tsuneyoshi
M: Expression profile of class I histone deacetylases in human
cancer tissues. Oncol Rep. 18:769–774. 2007.PubMed/NCBI
|
|
6.
|
Ishihama K, Yamakawa M, Semba S, Takeda H,
Kawata S, Kimura S and Kimura W: Expression of HDAC1 and CBP/p300
in human colorectal carcinomas. J Clin Pathol. 60:1205–1210. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Zhu P, Martin E, Mengwasser J, Schlag P,
Janssen KP and Göttlicher M: Induction of HDAC2 expression upon
loss of APC in colorectal tumorigenesis. Cancer Cell. 5:455–463.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Huang BH, Laban M, Leung CH, Lee L, Lee
CK, Salto-Tellez M, Raju GC and Hooi SC: Inhibition of histone
deacetylase 2 increases apoptosis and p21(Cipl/WAFl) expression,
independent of histone deacetylase 1. Cell Death Differ.
12:395–404. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Giannini R and Cavallini A: Expression
analysis of a subset of coregulators and three nuclear receptors in
human colorectal carcinoma. Anticancer Res. 25:4287–4292.
2005.PubMed/NCBI
|
|
10.
|
Weichert W, Röske A, Niesporek S, Noske A,
Buckendahl AC, Dietel M, Gekeler V, Boehm M, Beckers T and Denkert
C: Class I histone deacetylase expression has independent
prognostic impact in human colorectal cancer: specific role of
class I histone deacetylases in vitro and in vivo. Clin Cancer Res.
14:1669–1677. 2008. View Article : Google Scholar
|
|
11.
|
Beckers T, Stephan C, Jung K, Fritzsche
FR, Niesporek S, Denkert C, Dietel M and Kristiansen G: Histone
deacetylases 1, 2 and 3 are highly expressed in prostate cancer and
HDAC2 expression is associated with shorter PSA relapse time after
radical prostatectomy. Br J Cancer. 98:604–610. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Spurling CC, Godman CA, Noonan EJ,
Rasmussen TP, Rosenberg DW and Giardina C: HDAC3 overexpression and
colon cancer cell proliferation and differentiation. Mol Carcinog.
47:137–147. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Archer SY, Meng S, Shei A and Hodin RA:
p21WAF-1 is required for butyrate-mediated growth
inhibition of colon cancer cells. Proc Natl Acad Sci USA.
95:6791–6796. 1998.
|
|
14.
|
Finnin MS, Donigian JR, Cohen A, Richon
VM, Rifkind RA, Marks PA, Breslow R and Pavletich NP: Structures of
a histone deacetylase homologue bound to the TSA and SAHA
inhibitors. Nature. 401:188–193. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Myzak MC and Dashwood RH: Histone
deacetylases as targets for dietary cancer preventive agents:
lessons learned with butyrate, diallyl disulfide and sulforaphane.
Curr Drug Targets. 7:443–445. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Bruserud Ø, Stapnes C, Ersvaer E, Gjertsen
BT and Ryningen A: Histone deacetylase inhibitors in cancer
treatment: a review of the clinical toxicity and the modulation of
gene expression in cancer cells. Curr Pharm Biotechnol. 8:388–400.
2007.PubMed/NCBI
|
|
17.
|
Levanon D, Brenner O, Negreanu V, Bettoun
D, Woolf E, Eilam R, Lotem J, Gat U, Otto F, Speck N and Groner Y:
Spatial and temporal expression pattern of Runx3 (Aml2) and Runxl
(Amll) indicates non-redundant functions during mouse
embryogenesis. Mech Dev. 109:413–417. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Woolf E, Xiao C, Fainaru O, Lotem J, Rosen
D, Negreanu V, Bernstein Y, Goldenberg D, Brenner O, Berke G,
Levanon D and Groner Y: Runx3 and Runxl are required for CD8 T cell
development during thymopoiesis. Proc Natl Acad Sci USA.
100:7731–7736. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Choi JK, Lee YH, Kim HM, Li LS, Kim H,
Chang J, Ito Y, Lee KY and Bae SC: Transcriptional silencing of the
RUNX3 gene by CpG hypermethylation is associated with lung cancer.
Biochem Biophys Res Commun. 314:223–228. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Chuang LS and Ito Y: RUNX3 is
multifunctional in carcinogenesis of multiple solid tumors.
Oncogene. 29:2605–2615. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Ragnarsson G, Eiriksdottir G,
Johannsdottir JT, Jonasson JG, Egilsson V and Ingvarsson S: Loss of
heterozygosity at chromosome 1p in different solid human tumours:
association with survival. Br J Cancer. 79:1468–1474. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Tanaka K, Yanoshita R, Konishi M, Oshimura
M, Maeda Y, Mori T and Miyaki M: Suppression of tumourigenicity in
human colon carcinoma cells by introduction of normal chromosome
1p36 region. Oncogene. 8:2253–2258. 1993.PubMed/NCBI
|
|
23.
|
Herman JG and Baylin SB: Gene silencing in
cancer in association with promoter hypermethylation. N Engl J Med.
349:2042–2054. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Li QL, Ito K, Sakakura C, Fukamachi H,
Inoue Ki, Chi XZ, Lee KY, Nomura S, Lee CW, Han SB, Kim HM, Kim WJ,
Yamamoto H, Yamashita N, Yano T, Ikeda T, Itohara S, Inazawa J, Abe
T, Hagiwara A, Yamagishi H, Ooe A, Kaneda A, Sugimura T, Ushijima
T, Bae SC and Ito Y: Causal relationship between the loss of RUNX3
expression and gastric cancer. Cell. 109:113–124. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Goel A, Arnold CN, Tassone P, Chang DK,
Niedzwiecki D, Dowell JM, Wasserman L, Compton C, Mayer RJ,
Bertagnolli MM and Boland CR: Epigenetic inactivation of RUNX3 in
microsatellite unstable sporadic colon cancers. Int J Cancer.
112:754–759. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Fujii S, Ito K, Ito Y and Ochiai A:
Enhancer of zeste homologue 2 (EZH2) down-regulates RUNX3 by
increasing histone H3 methylation. J Biol Chem. 283:17324–17332.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Lee SH, Kim J, Kim WH and Lee YM: Hypoxic
silencing of tumor suppressor RUNX3 by histone modification in
gastric cancer cells. Oncogene. 28:184–194. 2009. View Article : Google Scholar
|
|
28.
|
Hong SY, Cho JY and Seo DW: Ginsenoside
Rp1 inhibits proliferation and migration of human lung cancer
cells. Biomole Ther. 19:411–418. 2011. View Article : Google Scholar
|
|
29.
|
Park JW, Lee JC, Ann S, Seo DW, Choi WS,
Yoo YH, Park SK, Choi JY, Um SH, Ahn SH and Han JW: A fermented
ginseng extract, BST204, inhibits proliferation and motility of
human colon cancer cells. Biomole Ther. 19:211–217. 2011.
View Article : Google Scholar
|
|
30.
|
Seo EY and Kim WK: Red ginseng extract
reduced metastasis of colon cancer cells in vitro and in vivo. J
Gins Res. 35:315–324. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Akao T, Kanaoka M and Kobashi K:
Appearance of Compound K, a major metabolite of ginsenoside Rb1 by
intestinal bacteria, in rat plasma after oral
administration-measurement of Compound K by enzyme immunoassay.
Biol Pharm Bull. 21:245–249. 1998. View Article : Google Scholar
|
|
32.
|
Hasegawa H, Sung JH and Huh JH: Ginseng
intestinal bacterial metabolite IH901 as a new anti-metastatic
agent. Arch Pharm Res. 20:539–544. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Quan LH, Cheng LQ, Kim HB, Kim JH, Son NR,
Kim SY, Jin HO and Yang DC: Bioconversion of Ginsenoside Rd into
Compound K by Lactobacillus pentosus DC101 isolated from
Kimchi. J Gins Res. 34:288–295. 2010. View Article : Google Scholar
|
|
34.
|
Kang KA, Kim YW, Kim SU, Chae S, Koh YS,
Kim HS, Choo MK, Kim DH and Hyun JW: G1 phase arrest of the cell
cycle by a ginseng metabolite, Compound K, in U937 human monocytic
leukamia cells. Arch Pharm Res. 28:685–690. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Kang KA, Lim HK, Kim SU, Kim YW, Kim WT,
Chung HS, Choo MK, Kim DH, Kim HS, Shim MJ, Chung MH and Hyun JW:
Induction of apoptosis by ginseng saponin metabolite in U937 human
monocytic leukemia cells. J Food Biochem. 29:27–40. 2005.
View Article : Google Scholar
|
|
36.
|
Chae S, Kang KA, Chang WY, Kim MJ, Lee SJ,
Lee YS, Kim HS, Kim DH and Hyun JW: Effect of Compound K, a
metabolite of ginseng saponin, combined with gamma-ray radiation in
human lung cancer cells in vitro and in vivo. J Agric Food Chem.
57:5777–5782. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Kim AD, Kang KA, Zhang R, Lim CM, Kim HS,
Kim DH, Jeon YJ, Lee CH, Park J, Chang WY and Hyun JW: Ginseng
saponin metabolite induces apoptosis in MCF-7 breast cancer cells
through the modulation of AMP-activated protein kinase. Environ
Toxicol Pharmacol. 30:134–140. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Kang KA, Kim HS, Kim DH and Hyun JW: The
role of a ginseng saponin metabolite as a DNA methyltransferase
inhibitor in colorectal cancer cells. Int J Oncol. 43:228–236.
2013.PubMed/NCBI
|
|
39.
|
Ito K, Lim AC, Salto-Tellez M, Motoda L,
Osato M, Chuang LS, Lee CW, Voon DC, Koo JK, Wang H, Fukamachi H
and Ito Y: RUNX3 attenuates beta-catenin/T cell factors in
intestinal tumorigenesis. Cancer Cell. 14:226–237. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Ku JL, Kang SB, Shin YK, Kang HC, Hong SH,
Kim IJ, Shin JH, Han IO and Park JG: Promoter hypermethylation
downregulates RUNX3 gene expression in colorectal cancer cell
lines. Oncogene. 23:6736–6742. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Yano T, Ito K, Fukamachi H, Chi XZ, Wee
HJ, Inoue K, Ida H, Bouillet P, Strasser A, Bae SC and Ito Y: The
RUNX3 tumor suppressor upregulates Bim in gastric epithelial cells
undergoing transforming growth factor beta-induced apoptosis. Mol
Cell Biol. 26:4474–4488. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Tong DD, Jiang Y, Li M, Kong D, Meng XN,
Zhao YZ, Jin Y, Bai J, Fu SB and Geng JS: RUNX3 inhibits cell
proliferation and induces apoptosis by TGF-beta-dependent and
-independent mechanisms in human colon carcinoma cells.
Pathobiology. 76:163–169. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Kodach LL, Jacobs RJ, Heijmans J, van
Noesel CJ, Langers AM, Verspaget HW, Hommes DW, Offerhaus GJ, van
den Brink GR and Hardwick JC: The role of EZH2 and DNA methylation
in the silencing of the tumour suppressor RUNX3 in colorectal
cancer. Carcinogenesis. 31:1567–1575. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Jones PA and Baylin SB: The fundamental
role of epigenetic events in cancer. Nat Rev Genet. 3:415–428.
2002.PubMed/NCBI
|
|
45.
|
Jones PA: DNA methylation and cancer.
Oncogene. 21:5358–5360. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Counts JL and Goodman JI: Alterations in
DNA methylation may play a variety of roles in carcinogenesis.
Cell. 83:13–15. 1995. View Article : Google Scholar
|
|
47.
|
Shibata S: Chemistry and cancer preventing
activities of ginseng saponins and some related triterpenoid
compounds. J Korean Med Sci. 16:S28–S37. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
48.
|
Wang L, Zou X, Berger AD, Twiss C, Peng Y,
Li Y, Chiu J, Guo H, Satagopan J, Wilton A, Gerald W, Basch R, Wang
Z, Osman I and Lee P: Increased expression of histone deacetylaces
(HDACs) and inhibition of prostate cancer growth and invasion by
HDAC inhibitor SAHA. Am J Transl Res. 1:62–71. 2009.PubMed/NCBI
|
|
49.
|
Patra SK, Patra A and Dahiya R: Histone
deacetylase and DNA methyltransferase in human prostate cancer.
Biochem Biophys Res Commun. 287:705–713. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
50.
|
Halkidou K, Gaughan L, Cook S, Leung HY,
Neal D and Robson CN: Upregulation and nuclear recruitment of HDAC1
in hormone refractory prostate cancer. Prostate. 59:177–189. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
51.
|
Yang XJ and Seto E: Collaborative spirit
of histone deacetylases in regulating chromatin structure and gene
expression. Curr Opin Genet Dev. 13:143–153. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
52.
|
Kim DH, Kim M and Kwon HJ: Histone
deacetylase in carcinogenesis and its inhibitors as anti-cancer
agents. J Biochem Mol Biol. 36:110–119. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
53.
|
Buchwald M, Krämer OH and Heinzel T: HDACi
- targets beyond chromatin. Cancer Lett. 280:160–167. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
54.
|
Mellert HS, Stanek TJ, Sykes SM, Rauscher
FJ III, Schultz DC and McMahon SB: Deacetylation of the DNA-binding
domain regulates p53-mediated apoptosis. J Biol Chem.
286:4264–4270. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
55.
|
Oh ET, Park MT, Choi BH, Ro S, Choi EK,
Jeong SY and Park HJ: Novel histone deacetylase inhibitor CG200745
induces clonogenic cell death by modulating acetylation of p53 in
cancer cells. Invest New Drugs. 30:435–442. 2012. View Article : Google Scholar : PubMed/NCBI
|