Introduction
Head and neck squamous cell carcinoma (HNSCC) is the
sixth most common cancer in the world and approximately 500,000
cases are diagnosed every year (1). In spite of considerable advances in
multimodality therapy, including surgery, radiotherapy and
chemotherapy, the overall 5-year survival rate for patients with
HNSCC is only approximately 50% (2). Hypopharyngeal SCC (HSCC) comprises
20% of all HNSCC, with an incidence of approximately 10 cases per
million people-years (3). HSCC has
a very poor prognosis compared with other HNSCC, with 5-year
survival rates ranging from 30–35% (4,5).
Local tumor recurrence and distant metastasis after conventional
therapy appear to be major contributing factors for restricted
survival of HSCC patients. Survival rates of HSCC patients have not
markedly improved despite recent advanced combination therapies
(6). Therefore, understanding the
molecular pathways of metastasis accompanying HSCC would help to
improve diagnosis, approaches to therapy and prevention of the
disease.
The discovery of non-coding RNAs (ncRNAs) in the
human genome was an important conceptual breakthrough in the
post-genome sequencing era (7).
Improved understanding of ncRNAs is necessary for continued
progress in cancer research. microRNAs (miRNAs) are endogenous
small ncRNA molecules (18–25 bases in length) that regulate
protein-coding gene expression by repressing translation or
cleaving RNA transcripts in a sequence-specific manner (8). Currently, 2,578 human mature miRNAs
are registered at miRBase release 20.0 (http://www.mirbase.org/). miRNAs are unique in their
ability to regulate multiple protein-coding genes. Bioinformatic
predictions indicate that miRNAs regulate approximately 30–60% of
the protein-coding genes in the human genome (9).
Numerous reports suggest that miRNAs are aberrantly
expressed in many human cancers and that they play significant
roles in the initiation, development, and metastasis of those
cancers (10,11). Some highly expressed miRNAs can
function as oncogenes by repressing tumor suppressors, whereas low
level miRNAs can function as tumor suppressors by negatively
regulating oncogenes (10,12). It is believed that normal
regulatory mechanisms can be disrupted by the aberrant expression
of tumor-suppressive or oncogenic miRNAs in cancer cells.
Therefore, identification of aberrantly expressed miRNAs is an
important first step toward elucidating miRNA-mediated oncogenic
pathways.
Based on these considerations, we have constructed
miRNA expression signatures using HNSCC clinical specimens and
investigated the specific role of miRNAs in HNSCC oncogenesis using
differentially expressed miRNAs (13,14).
Our recent studies demonstrated that miR-1, miR-29s, miR-133a,
miR-218, miR-489 and miR-874 functioned as tumor
suppressors in HNSCC through their targeting of several types of
oncogenic genes (13–23). Our HSCC and esophageal SCC miRNA
expression signatures revealed that miR-504 was
significantly downregulated in cancer tissues, suggesting that this
miRNA is a candidate tumor suppressor in human SCC cells (13,24).
However, several recent reports indicated that miR-504
functions as an oncogene (25,26).
These data contradict our hypothesis that miR-504 functions
as a tumor suppressor. The aim of this study was to investigate the
functional significance of miR-504 in cancer cells and to
identify novel targets regulated by miR-504 in HSCC
cells.
We used two different sources of mature
miR-504 to restore miR-504 function. Transfection of
those miRNAs inhibited cancer cell proliferation. Genome-wide gene
expression analysis of miR-504 transfectants and in
silico database analysis showed that cyclin-dependent kinase 6
(CDK6) was a candidate target of miR-504 in HSCC
cells. Tumor suppressive miR-504-regulated targets provide
new insight into the potential mechanisms of HSCC oncogenesis and
suggest novel therapeutic strategies for treatment of the
disease.
Materials and methods
Clinical specimens
Twenty-three pairs of primary HSCC and corresponding
normal epithelial samples were obtained from patients with HSCC at
Chiba University Hospital (Chiba, Japan) from 2005 to 2013. The
samples considered normal were free of cancer cells as determined
by pathologic examination. The backgrounds and clinicopathological
characteristics of the patient are summarized in Table I. The patients were classified
according to the 2002 Union for International Cancer Control (UICC)
TNM staging criteria prior to treatment. Written consent for tissue
donation for research purposes was obtained from each patient
before tissue collection. The protocol was approved by the
Institutional Review Board of Chiba University. The specimens were
immersed in RNAlater (Qiagen, Valencia, CA, USA) and stored at
−20°C until RNA was extracted.
 | Table I.Patient characteristics. |
Table I.
Patient characteristics.
| No. | Sex | T | N | M | Stage |
Differentiation |
|---|
| 1 | M | 4a | 0 | 0 | IVA | Moderate |
| 2 | M | 3 | 1 | 0 | III | Poor |
| 3 | M | 2 | 2c | 0 | IVA | Moderate |
| 4 | M | 2 | 2b | 0 | IVA | Poor |
| 5 | M | 2 | 2b | 0 | IVA | Poor |
| 6 | F | 4a | 0 | 0 | IVA | Well |
| 7 | M | 2 | 2b | 0 | IVA | Moderate |
| 8 | M | 2 | 0 | 0 | II | Moderate |
| 9 | M | 3 | 2b | 0 | IVA | Moderate |
| 10 | M | 4a | 2b | 0 | IVA | Moderate |
| 11 | M | 3 | 2b | 0 | IVA | Poor |
| 12 | F | 4a | 2c | 0 | IVA | Poor |
| 13 | M | 4a | 2c | 0 | IVA | Well |
| 14 | M | 4b | 2c | 0 | IVB | Moderate |
| 15 | M | 4a | 1 | 0 | IVA | Well |
| 16 | F | 4a | 2c | 0 | IVA | Moderate |
| 17 | M | 4a | 1 | 1 | IVC | Moderate |
| 18 | M | 4a | 2c | 0 | IVA | Poor |
| 19 | M | 2 | 0 | 0 | II | Moderate |
| 20 | M | 4a | 2c | 0 | IVA | Moderate |
| 21 | F | 4a | 1 | 0 | IVA | Poor |
| 22 | M | 4a | 2c | 0 | IVA | Well |
| 23 | M | 4a | 0 | 0 | IVA | Well |
RNA isolation
Total RNA was isolated using TRIzol reagent
(Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s
protocol. RNA concentrations were determined
spectrophotometrically, and molecular integrity was checked by gel
electrophoresis. RNA quality was confirmed using an Agilent 2100
Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA).
Cell culture
The following human cell lines were used: FaDu
(derived from a primary lesion of hypopharyngeal SCC), SAS (derived
from a primary lesion of tongue SCC), HSC3 (derived from lymph node
metastasis of tongue SCC), HCT116 p53+/+ (derived from a
colon adenocarcinoma) and its p53−/− derivative. All
cell lines were grown in Dulbecco’s modified Eagle’s medium (DMEM)
supplemented with 10% fetal bovine serum in a humidified atmosphere
containing 5% CO2 at 37°C.
Quantitative real-time RT-PCR (qPCR)
cDNA synthesis and PCR procedures were described in
our previous reports (13,27). The expression levels of
miR-504 (Assay ID: 002084) were analyzed by TaqMan
quantitative real-time PCR (TaqMan® MicroRNA Assay;
Applied Biosystems) and normalized to RNU48 (Assay ID:
001006). TaqMan® probes and primers for CDK6 (P/N:
Hs01026371_m1), RB1 (P/N: Hs01078066_m1), CDC23 (P/N:
00946641_m1), CCND1 (P/N: 00765553_m1) and GUSB (P/N:
Hs00939627_m1) as an internal control were obtained from Applied
Biosystems (Assay-On-Demand Gene Expression Products). The ΔΔCt
method was adopted and applied to calculate the relative quantities
of subject genes. All reactions were performed in triplicate and
included negative control reactions that lacked cDNA.
Mature miRNA transfection
To perform gain of function studies, we utilized two
different sources of mature miR-504: miR-504-A, Ambion
Pre-miR miRNA Precursor, PM12429 (Applied Biosystems) and
miR-504-T, miRIDIAN Mimic, MIMAT0002875 (Thermo Scientific
Dharmacon, Waltham, MA, USA). Pre-miR Negative Control #2 (AM17111,
Applied Biosystems) was used for negative control experiments. The
miRNA transfection procedures and confirmation of miRNA
transfection efficiency were described in our previous reports
(13,27).
Cell proliferation assay
Cells were transfected with 10 nM miRNA by reverse
transfection and plated in 96-well plates at 3×103 cells
per well. After 72 h, cell proliferation was determined with the
XTT assay using the Cell Proliferation Kit II (Roche Molecular
Biochemicals, Mannheim, Germany) as previously reported (13,27).
Flow cytometry
Cell cycle status was examined using an APC BrdU
Flow kit (BD Bioscience, San Jose, CA, USA) according to the
manufacturer’s protocol. Briefly, SAS and FaDu cells were
transiently transfected with miR-control, miR-504-A or
miR-504-T. Seventy-two hours after transfection, 10
μM BrdU was added to the medium and incubated for 6 h. The
cells were then trypsinized and fixed with paraformaldehyde and
permeabilized with saponin. After DNase treatment, the cells were
stained with anti-BrdU antibodies and 7AAD and analyzed with a
FACSCalibur flow cytometer (BD Bioscience).
Target gene search for miR-504
To identify miR-504 target genes, we used
genome-wide gene expression analysis and in silico analysis.
First, we performed genome-wide gene expression analysis using
miR-504 transfection of SAS and FaDu. SurePrint G3 Human GE
8×60K Microarray (Agilent Technologies) was used for expression
profiling of miR-504 transfectants in comparison with
negative control miRNA transfectants. The genes that were
downregulated in miR-504 transfectant were then categorized
into Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways using
GeneCodis analysis (28)
(http://genecodis.cnb.csic.es/). The
target site for miR-504 was analyzed with TargetScan Release
6.2 (http://www.targetscan.org/).
Western blotting
Cells were harvested 72 h after transfection and
lysates were prepared. Protein (50 μg) from each lysate was
separated on a Mini-Protean TGX gel (Bio-Rad, Hercules, CA, USA)
and transferred to PVDF membranes. Immunoblotting was performed
with mouse CDK6 antibody (1:500, #3136, Cell Signaling
Technology, Danvers, MA, USA) with GAPDH antibody (1:1,000, ab8245,
Abcam, Cambridge, UK) used as an internal control. The membrane was
washed and incubated with anti-mouse IgG HRP-linked antibody
(#7076, Cell Signaling Technology). Complexes were visualized with
an Immun-Star™ Western Chemiluminescence kit (Bio-Rad), and the
expression levels of these genes were evaluated by ImageJ software
(ver.1.44; http://rsbweb.nih.gov/ij/).
Statistical analysis
The relationships between two groups and the
numerical values obtained by qPCR were analysed using the paired
t-test. Spearman’s rank test was used to evaluate the correlation
between the expression of miR-504 and target genes. The
relationships among more than three variables and numerical values
were analysed using the Bonferroni adjusted Mann-Whitney U test.
All analyses were performed using Expert StatView (version 4, SAS
Institute Inc., Cary, NC, USA).
Results
Expression of miR-504 in HSCC clinical
specimens and cell lines
To validate our past miRNA profiling results, we
evaluated miR-504 expression in 23 clinical HSCC specimens.
The expression levels of miR-504 were significantly lower in
tumor tissues than in corresponding adjacent normal epithelia
(miR-504 expression normalized to RNU48: normal,
0.0026±0.0011; tumor, 0.00070±0.00052, P<0.0001, Fig. 1). The expression levels of
miR-504 in HNSCC cell lines were also lower than those in
normal epithelia (miR-504 expression normalized to
RNU48: FaDu, 0.000026; SAS, 0.00016; HSC3, 0.000025;
Fig. 1).
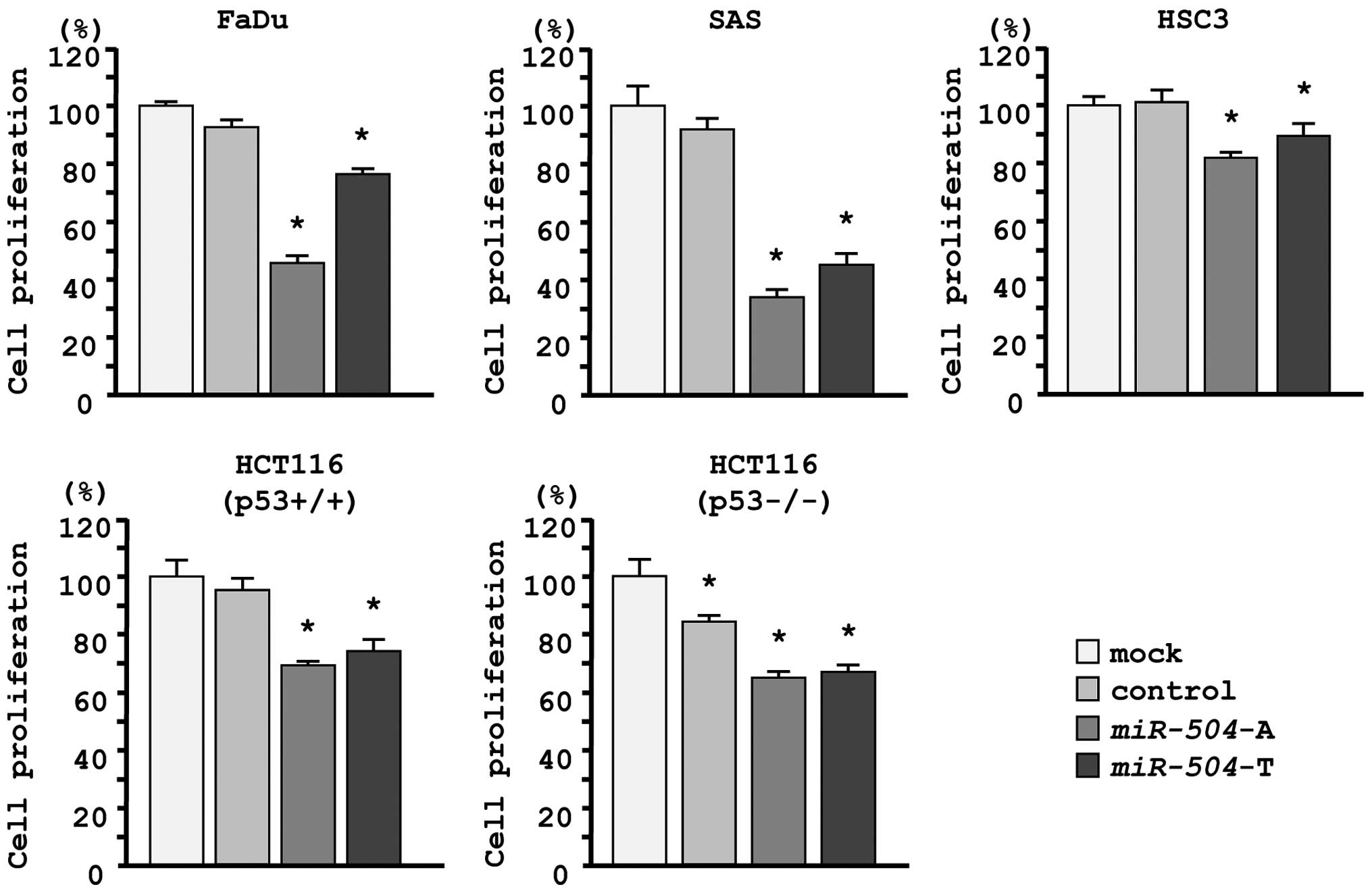
Effects of miR-504 restoration on the
proliferation of HNSCC cell lines
To investigate the role of miR-504, we
performed gain-of-function studies using mature miRNA transfection
of three HNSCC cell lines (FaDu, SAS and HSC3). We utilized two
sources of mature miR-504 (miR-504-A, Ambion;
miR-504-T, Thermo Scientific Dharmacon) to ensure
reproducibility of the data.
The XTT assay demonstrated that cell proliferation
was significantly inhibited in miR-504 transfectants in
comparison with the mock or miR-control transfectant cells.
Specifically, we observed the following growth, expressed as a
percentage of the mock: i) FaDu-mock, 100.0±1.0; miR control,
92.6±2.7; miR-504-A, 45.5±2.3; miR-504-T, 76.4±1.9;
ii) SAS-mock, 100.0±6.8; miR control, 92.0±3.9; miR-504-A,
34.0±2.4; miR-504-T, 45.1±3.3; iii) HSC3-mock, 100.0±3.7;
miR control, 103.4±3.9; miR-504-A, 81.6±2.7;
miR-504-T, 89.6±4.9, with P<0.0083 (Fig. 2).
Because miR-504 has been reported to promote
tumorigenicity by regulating TP53, we evaluated functional
effects of miR-504 in HCT116 p53+/+ cells and its
p53−/− derivative cell line. The XTT assay showed that
cell proliferation was significantly inhibited by miR-504
transfection in both cell lines, suggesting that miR-504
functioned as a tumor suppressor regardless of p53 status. We
observed the following growth, expressed as a percentage of the
mock: i) HCT116 p53+/+-mock, 100.0±3.7; miR control,
94.5±3.8; miR-504-A, 68.7±2.2; miR-504-T, 74.9±3.8;
ii) HCT116 p53−/−-mock, 100.0±4.4; miR control,
82.8±2.9; miR-504-A, 63.8±2.1; miR-504-T, 65.9±2.1,
with P<0.0083 (Fig. 2).
Effects of miR-504 restoration on cell
cycle status in HNSCC cell lines
To study whether miR-504 affected the cell
cycle status of cancer cells, we performed flow cytometric analysis
of cells stained with anti-BrdU and 7AAD allowing the
discrimination of cell fractions that resided in G0/G1, S or G2/M
phases of the cell cycle (Fig.
3A). The fraction of FaDu cells in the G0/G1 phase was
significantly larger in miR-504 transfectants in comparison
with the miR control transfectants, whereas the fraction of SAS
cells in G2/M phase was significantly larger in miR-504
transfectants (Fig. 3B).
Identification of candidate target genes
regulated by miR-504 in HNSCC cells
To identify miR-504 target genes, we used
genome-wide gene expression analysis and in silico analysis.
First, we performed genome-wide gene expression analysis using two
cancer cell lines (FaDu and SAS) and selected genes downregulated
by miR-504 transfection compared with miR control
transfection. In this analysis, 810 genes and 1,145 genes were
recognized as downregulated genes (log2 ratio <−0.5)
in FaDu and SAS, respectively. Entries from the microarray data
were approved by the Gene Expression Omnibus (GEO) and were
assigned GEO accession no. GSE37119.
Next, genes downregulated in
miR-504-transfectants were categorized into KEGG pathways
using GeneCodis analysis and 24 pathways were identified as
significantly enriched in both lines (Tables II and III). Among these pathways, we focused on
the ‘cell cycle’ pathway because this pathway has been implicated
in cancer cell proliferation. A total of 19 genes were identified
in this pathway and four genes (RB1, CDK6, CDC23 and
CCND1) had putative miR-504 target sites predicted by
the TargetScan database (Table
IV).
| Table II.Significantly enriched annotations
among downregulated genes by miR-504 transfection in
FaDu. |
Table II.
Significantly enriched annotations
among downregulated genes by miR-504 transfection in
FaDu.
| No. of genes | P-value | Annotations |
|---|
| 19 | 2.16E-08 | RNA transport |
| 15 | 1.28E-06 | Cell cycle |
| 14 | 1.70E-08 | Ribosome biogenesis
in eukaryotes |
| 14 | 1.49E-07 | Systemic lupus
erythematosus |
| 13 | 2.78E-05 | Spliceosome |
| 12 | 1.44E-03 | Purine
metabolism |
| 11 | 7.99E-05 | Pyrimidine
metabolism |
| 11 | 1.37E-03 | Ubiquitin mediated
proteolysis |
| 9 | 3.74E-03 | Oocyte meiosis |
| 9 | 9.33E-03 | Measles |
| 8 | 3.69E-03 |
Progesterone-mediated oocyte
maturation |
| 7 | 3.93E-04 | Nucleotide excision
repair |
| 7 | 3.56E-03 | p53 signaling
pathway |
| 7 | 5.44E-03 | Chronic myeloid
leukemia |
| 6 | 3.58E-04 | RNA polymerase |
| 6 | 1.22E-03 | DNA
replication |
| 6 | 1.83E-03 | Aminoacyl-tRNA
biosynthesis |
| 6 | 4.62E-03 | Mineral
absorption |
| 6 | 5.72E-03 | Arginine and
proline metabolism |
| 5 | 1.38E-03 | Mismatch
repair |
| 5 | 1.83E-03 | Homologous
recombination |
| 5 | 3.46E-03 | Citrate cycle (TCA
cycle) |
| 4 | 3.96E-03 | One carbon pool by
folate |
| 3 | 6.43E-03 | Valine, leucine and
isoleucine biosynthesis |
| Table III.Significantly enriched annotations
among downregulated genes by miR-504 transfection in
SAS. |
Table III.
Significantly enriched annotations
among downregulated genes by miR-504 transfection in
SAS.
| No. of genes | P-value | Annotations |
|---|
| 24 | 9.36E-04 | Pathways in
cancer |
| 21 | 2.54E-06 | Protein processing
in endoplasmic reticulum |
| 20 | 8.23E-05 | Focal adhesion |
| 18 | 2.10E-06 | Lysosome |
| 17 | 7.50E-04 | Huntington’s
disease |
| 15 | 1.36E-04 | Cell cycle |
| 15 | 1.93E-03 | Alzheimer’s
disease |
| 15 | 6.78E-03 | Endocytosis |
| 14 | 6.72E-03 | Tuberculosis |
| 13 | 5.94E-05 | Small cell lung
cancer |
| 13 | 2.17E-03 | Oxidative
phosphorylation |
| 12 | 4.03E-05 | p53 signaling
pathway |
| 12 | 2.00E-03 | Oocyte meiosis |
| 12 | 6.67E-03 | Ubiquitin mediated
proteolysis |
| 10 | 9.11E-04 | Melanoma |
| 10 | 3.60E-03 | Prostate
cancer |
| 9 | 3.55E-04 | Lysine
degradation |
| 9 | 3.65E-03 | Chronic myeloid
leukemia |
| 8 | 2.30E-03 | Non-small cell lung
cancer |
| 8 | 5.70E-03 | Glioma |
| 8 | 9.15E-03 | Pancreatic
cancer |
| 7 | 6.46E-03 | Mineral
absorption |
| 6 | 2.91E-03 | Citrate cycle (TCA
cycle) |
| 5 | 5.46E-03 | Protein export |
| Table IV.Candidate target genes of
miR-504 in the cell cycle pathway. |
Table IV.
Candidate target genes of
miR-504 in the cell cycle pathway.
| Gene | Log2
ratio (miR-504/miR-control)
| miR-504
target site |
|---|
| FaDu | SAS | Average |
|---|
| ANAPC10 | −1.40 | −1.49 | −1.44 | 0 |
| PCNA | −0.97 | −1.26 | −1.12 | 0 |
| RB1 | −0.55 | −1.37 | −0.96 | 1 |
| CCND2 | −0.84 | −1.04 | −0.94 | 0 |
| ANAPC11 | −0.66 | −1.17 | −0.91 | 0 |
| CDK6 | −0.61 | −0.94 | −0.77 | 1 |
| ANAPC5 | −0.51 | −0.93 | −0.72 | 0 |
| CDKN2D | −0.02 | −1.26 | −0.64 | 0 |
| CDK4 | −0.75 | −0.52 | −0.63 | 0 |
| CCNE1 | −0.53 | −0.56 | −0.54 | 0 |
| HDAC1 | −0.25 | −0.72 | −0.49 | 0 |
| ANAPC4 | −0.15 | −0.70 | −0.43 | 0 |
| CCNH | −0.72 | −0.06 | −0.39 | 0 |
| CDC23 | −0.56 | −0.22 | −0.39 | 1 |
| CCND1 | −0.14 | −0.63 | −0.39 | 1 |
| E2F3 | −0.41 | −0.11 | −0.26 | 0 |
| BUB1 | −0.54 | 0.06 | −0.24 | 0 |
| MAD2L1 | −0.63 | 0.20 | −0.22 | 0 |
| CCNE2 | 0.28 | −0.50 | −0.11 | 0 |
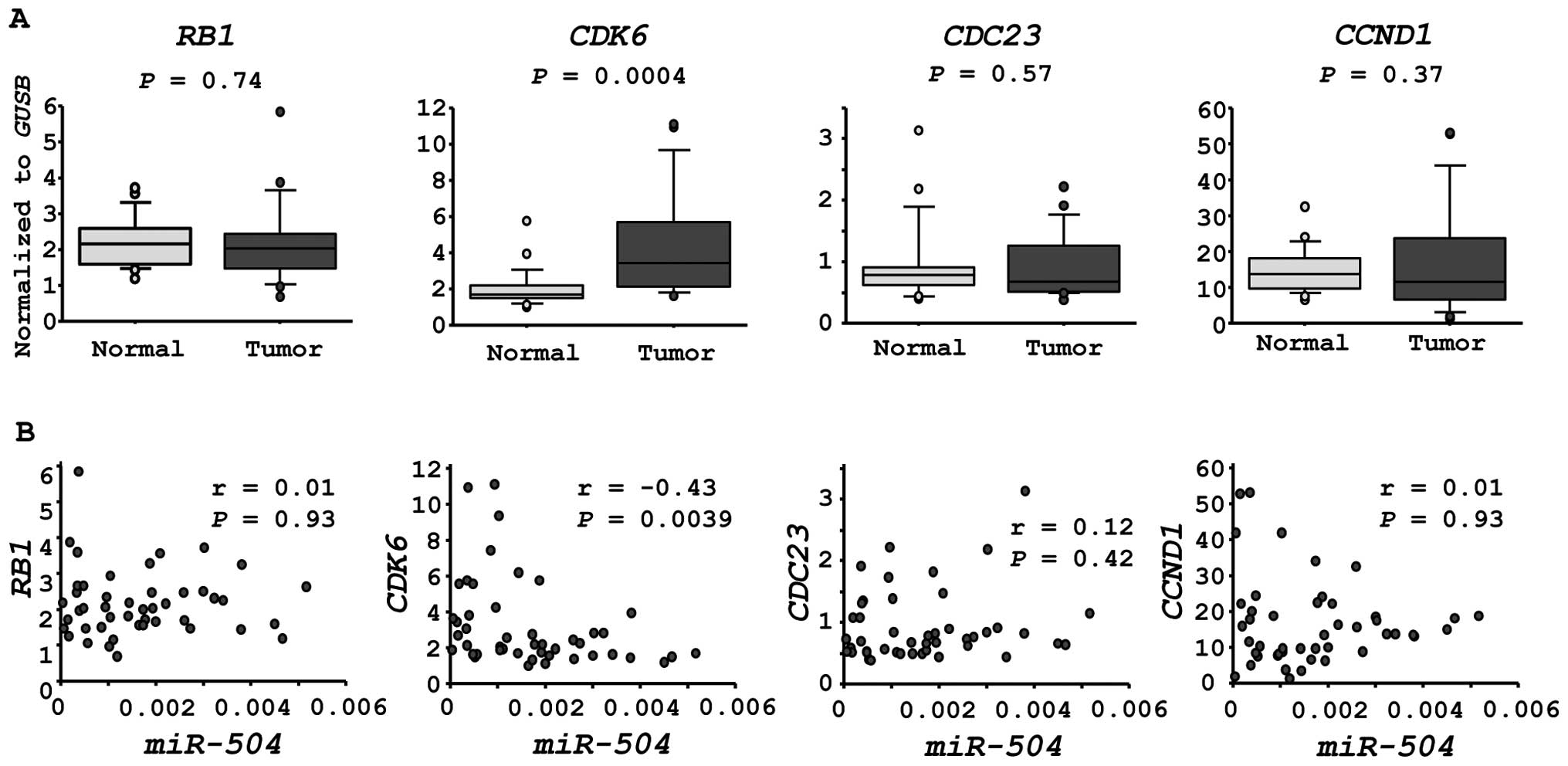
CDK6 is a candidate of miR-504 regulation
in HNSCC cells
We investigated the expression levels of four
candidate genes in HSCC clinical specimens. CDK6 was
significantly upregulated in cancer tissues (P=0.0004, Fig. 4A). Furthermore, the expression of
CDK6 was inversely correlated with that of miR-504 in
HSCC specimens (r=−0.43, P=0.0039, Fig. 4B).
We performed qPCR and western blotting in FaDu and
SAS to investigate whether CDK6 expression was downregulated
by restoration of miR-504. CDK6 mRNA expression was
significantly repressed by miR-504 transfection of FaDu
cells, while no changes were observed in SAS cells (Fig. 5A). The expression levels of CDK6
protein were repressed in miR-504 transfectants in
comparison with mock or miR-control transfectants in both FaDu and
SAS cells (Fig. 5B).
Discussion
Aberrant expression of miRNAs can disrupt the
tightly regulated system by which miRNA regulates protein-coding
RNA networks in cancer cells (10,12).
Therefore, studies of differentially expressed miRNAs in cancer
cells provide important information regarding the molecular
mechanisms underlying oncogenesis and metastasis. To elucidate the
molecular mechanisms underlying HNSCC, we have identified
tumor-suppressive miRNAs, focusing on their regulated molecular
targets and novel cancer pathways based on HNSCC expression
signatures (13–24,27).
Our recent studies of miRNA expression signatures of
HSCC and esophageal SCC showed that miR-504 was
significantly reduced in cancer tissues compared to normal tissues
(13,24). Those results suggested that
miR-504 was a candidate tumor suppressor. In glioblastoma,
miR-504 expression was reported to be downregulated and
functioned as a tumor suppressor by regulating mesenchymal genes
(29). This finding is consistent
with our results. However, miR-504 has also been reported to
have oncogenic functions. For example, a recent study showed that
miR-504 was a negative regulator of human TP53 and
directly bound to its 3′-UTR region (25). Overexpression of miR-504
induced TP53 silencing and caused inhibition of p53-mediated
apoptosis and cell cycle arrest in response to stress (25). Another report showed that ectopic
expression of miR-504 increased migration and invasion in an
oral cancer cell line by targeting FOXP1, a member of
forkhead transcriptional factors (26).
We conducted two analyses to test the conclusions of
the above reports. First, we restored function using two different
sources of mature miR-504 in several cancer cell lines. In
HNSCC cell lines, restoration of both types of miR-504
significantly inhibited cancer cell proliferation. Similar results
were observed in HPV16- and HPV18-positive cervical-SCC cell lines
(data not shown). Furthermore, we investigated the
anti-proliferative effects of miR-504 and p53 status using
HCT116 p53+/+ and HCT116 p53−/− cells. Our
data demonstrated that the anti-proliferative effect was not
affected by the p53 status. In this study, our data indicated that
miR-504 had a tumor-suppressive function, particularly
promotion of cell cycle arrest.
A unique aspect of miRNAs is that one miRNA
regulates many protein-coding genes. Thus, it is important to
elucidate the molecular targets and pathways regulated by a single
tumor suppressive molecule, miR-504, in cancer cells. To
solve this problem, we performed genome-wide gene expression
analysis using miR-504-transfectants. We categorized
differentially expressed genes of miR-504-transfectants into
KEGG pathways. Several pathways were enriched in this analysis and
we focused on ‘cell cycle’ pathways because
miR-504-transfectants underwent cell cycle arrest. Finally,
CDK6 was chosen as a miR-504 target oncogenic gene
that met several conditions. These included the following: i) mRNA
sequence contained a putative miR-504 binding site, ii)
inhibition of its expression in miR-504 transfects, and iii)
overexpression in HSCC clinical specimens.
It is well known that CDK-cyclin complexes are
deregulated in cancer cells, resulting in either continued
proliferation or unscheduled re-entry into the cell cycle (30). Several CDK4/CDK6 inhibitors
have been shown to induce G1 arrest and inhibit proliferation of
tumor cells (31,32). Our data indicated that restoration
of miR-504 repressed CDK6 and induced G1 arrest in
FaDu cell. On the other hand, in SAS cells, restoration of
miR-504 induced G2 arrest. We have no reasonable data to
explain this phenomenon but it is possible that some G2 phase
related genes were affected by miR-504 in SAS cells. Thus,
further study is needed.
In conclusion, downregulation of miR-504 was
frequently observed in HSCC clinical specimens. Restoration of
miRNA significantly inhibited cancer cell proliferation, suggesting
that miR-504 functioned as a tumor suppressor in HSCC cells.
To the best of our knowledge, this is the first report
demonstrating that tumor-suppressive miR-504 regulated ‘cell
cycle’ pathways and that CDK6 was a putative target. The
identification of target oncogenes regulated by miR-504
might lead to a better understanding of HSCC oncogenesis and the
development of new therapeutic strategies to treat this
disease.
Acknowledgements
This study was supported by JSPS
KAKENHI Grant nos. 23592505, 24592590, 25462676 and 25861528.
References
|
1.
|
Jemal A, Siegel R, Xu J and Ward E: Cancer
statistics, 2010. CA Cancer J Clin. 60:277–300. 2010. View Article : Google Scholar
|
|
2.
|
Leemans CR, Braakhuis BJ and Brakenhoff
RH: The molecular biology of head and neck cancer. Nat Rev Cancer.
11:9–22. 2011. View Article : Google Scholar
|
|
3.
|
Davies L and Welch HG: Epidemiology of
head and neck cancer in the United States. Otolaryngol Head Neck
Surg. 135:451–457. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Hoffman HT, Karnell LH, Shah JP, et al:
Hypopharyngeal cancer patient care evaluation. Laryngoscope.
107:1005–1017. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Bova R, Goh R, Poulson M and Coman WB:
Total pharyngolaryngectomy for squamous cell carcinoma of the
hypopharynx: a review. Laryngoscope. 115:864–869. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Godballe C, Jorgensen K, Hansen O and
Bastholt L: Hypopharyngeal cancer: results of treatment based on
radiation therapy and salvage surgery. Laryngoscope. 112:834–838.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Carthew RW and Sontheimer EJ: Origins and
mechanisms of miRNAs and siRNAs. Cell. 136:642–655. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Bartel DP: MicroRNAs: genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Filipowicz W, Bhattacharyya SN and
Sonenberg N: Mechanisms of post-transcriptional regulation by
microRNAs: are the answers in sight? Nat Rev Genet. 9:102–114.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Esquela-Kerscher A and Slack FJ: Oncomirs
- microRNAs with a role in cancer. Nat Rev Cancer. 6:259–269. 2006.
View Article : Google Scholar
|
|
11.
|
Tran N, O’Brien CJ, Clark J and Rose B:
Potential role of micro-RNAs in head and neck tumorigenesis. Head
Neck. 32:1099–1111. 2010. View Article : Google Scholar
|
|
12.
|
Caldas C and Brenton JD: Sizing up miRNAs
as cancer genes. Nat Med. 11:712–714. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Kikkawa N, Hanazawa T, Fujimura L, et al:
miR-489 is a tumour-suppressive miRNA target PTPN11 in
hypopharyngeal squamous cell carcinoma (HSCC). Br J Cancer.
103:877–84. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Nohata N, Hanazawa T, Kikkawa N, et al:
Tumour suppressive microRNA-874 regulates novel cancer networks in
maxillary sinus squamous cell carcinoma. Br J Cancer. 105:833–841.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Kinoshita T, Hanazawa T, Nohata N, et al:
Tumor suppressive microRNA-218 inhibits cancer cell migration and
invasion through targeting laminin-332 in head and neck squamous
cell carcinoma. Oncotarget. 3:1386–1400. 2012.PubMed/NCBI
|
|
16.
|
Kinoshita T, Nohata N, Fuse M, et al:
Tumor suppressive microRNA-133a regulates novel targets: moesin
contributes to cancer cell proliferation and invasion in head and
neck squamous cell carcinoma. Biochem Biophys Res Commun.
418:378–383. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Kinoshita T, Nohata N, Watanabe-Takano H,
et al: Actin-related protein 2/3 complex subunit 5 (ARPC5)
contributes to cell migration and invasion and is directly
regulated by tumor-suppressive microRNA-133a in head and neck
squamous cell carcinoma. Int J Oncol. 40:1770–1778. 2012.PubMed/NCBI
|
|
18.
|
Kinoshita T, Nohata N, Yoshino H, et al:
Tumor suppressive microRNA-375 regulates lactate dehydrogenase B in
maxillary sinus squamous cell carcinoma. Int J Oncol. 40:185–193.
2012.PubMed/NCBI
|
|
19.
|
Mutallip M, Nohata N, Hanazawa T, et al:
Glutathione S-transferase P1 (GSTP1) suppresses cell apoptosis and
its regulation by miR-133alpha in head and neck squamous cell
carcinoma (HNSCC). Int J Mol Med. 27:345–352. 2011.PubMed/NCBI
|
|
20.
|
Nohata N, Hanazawa T, Kikkawa N, et al:
Caveolin-1 mediates tumor cell migration and invasion and its
regulation by miR-133a in head and neck squamous cell carcinoma.
Int J Oncol. 38:209–217. 2011.PubMed/NCBI
|
|
21.
|
Nohata N, Hanazawa T, Kikkawa N, et al:
Identification of novel molecular targets regulated by tumor
suppressive miR-1/miR-133a in maxillary sinus squamous cell
carcinoma. Int J Oncol. 39:1099–1107. 2011.PubMed/NCBI
|
|
22.
|
Nohata N, Hanazawa T, Kinoshita T, et al:
Tumour-suppressive microRNA-874 contributes to cell proliferation
through targeting of histone deacetylase 1 in head and neck
squamous cell carcinoma. Br J Cancer. 108:1648–1658. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Nohata N, Sone Y, Hanazawa T, et al: miR-1
as a tumor suppressive microRNA targeting TAGLN2 in head and neck
squamous cell carcinoma. Oncotarget. 2:29–42. 2011.PubMed/NCBI
|
|
24.
|
Kano M, Seki N, Kikkawa N, et al: miR-145,
miR-133a and miR-133b: tumor-suppressive miRNAs target FSCN1 in
esophageal squamous cell carcinoma. Int J Cancer. 127:2804–2814.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Hu W, Chan CS, Wu R, et al: Negative
regulation of tumor suppressor p53 by microRNA miR-504. Mol Cell.
38:689–699. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Yang MH, Lin BR, Chang CH, et al:
Connective tissue growth factor modulates oral squamous cell
carcinoma invasion by activating a miR-504/FOXP1 signalling.
Oncogene. 31:2401–2411. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Ichimi T, Enokida H, Okuno Y, et al:
Identification of novel microRNA targets based on microRNA
signatures in bladder cancer. Int J Cancer. 125:345–352. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Tabas-Madrid D, Nogales-Cadenas R and
Pascual-Montano A: GeneCodis3: a non-redundant and modular
enrichment analysis tool for functional genomics. Nucleic Acids
Res. 40:W478–W483. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Ma X, Yoshimoto K, Guan Y, et al:
Associations between microRNA expression and mesenchymal marker
gene expression in glioblastoma. Neuro Oncol. 14:1153–1162. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Malumbres M and Barbacid M: Cell cycle,
CDKs and cancer: a changing paradigm. Nat Rev Cancer. 9:153–166.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Musgrove EA, Caldon CE, Barraclough J,
Stone A and Sutherland RL: Cyclin D as a therapeutic target in
cancer. Nat Rev Cancer. 11:558–572. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Shapiro GI: Cyclin-dependent kinase
pathways as targets for cancer treatment. J Clin Oncol.
24:1770–1783. 2006. View Article : Google Scholar : PubMed/NCBI
|