Introduction
Based on the updated GLOBOCAN project of the World
Health Organization in 2012, breast, prostate and lung remain the
three most common global cancers (http://globocan.iarc.fr/). The incidence and mortality
rates of lung cancer have increased from 12.7 to 16.7% and 18.2 to
23.2% of all cancers respectively since 2008. Lung cancer is
histologically classified as non-small cell (NSCLC) or small cell
carcinoma (SCLC), and is associated with distinct treatment
implications. The majority (85%) of lung cancer cases are NSCLC,
comprised mostly of adenocarcinoma. Notably, tobacco smoking,
pre-existing lung disease, diet, occupational exposure, exposure to
estrogen, and genetic predisposition are the major causes of lung
cancer (1).
Systemic chemotherapy remains the cornerstone
treatment for advanced or metastatic NSCLC. First-line platinum
doublets with newer agents (docetaxel, gemcitabine, paclitaxel,
pemetrexed or vinorelbine) and salvage monotherapy with docetaxel
or pemetrexed have conferred only a modest survival benefit with
5-year overall survival <5% (2,3).
Emerging molecularly-targeted therapy against epidermal growth
factor receptor or anaplastic lymphoma kinase has provided a
superior treatment option to systemic chemotherapy in patients with
NSCLC driven by actionable targets. Nonetheless development of
acquired drug resistance ~1 year following targeted therapy is
almost inevitable (4). Thus novel
effective treatment for NSCLC is urgently needed.
Arsenic trioxide (ATO), which is now a standard
treatment for acute promyelocytic leukemia, has demonstrated
promising activity in solid tumors including lung cancer (5–8).
Nonetheless the exact mechanisms of action of ATO in NSCLC have not
been fully elucidated. We have recently reported the role of
ATO-induced suppression of thymidylate synthase (TYMS) in 4 lung
adenocarcinoma cell lines with basal expression (9), while ATO might have other effects in
cell lines not expressing TYMS. The role of E2F1 is still not fully
elucidated, therefore, a panel of 7 lung adenocarcinoma cell lines
with basal E2F1 expression was studied. E2F1 is a transcription
factor that controls cell fate including apoptosis (10) and DNA synthesis (11). Depending on specific cancer types,
the E2F1 gene can serve as an oncogene (12) with a prognostic role (13) or a tumor suppressor gene (14). This study aimed to investigate the
action of ATO in lung adenocarcinoma, with an emphasis on
E2F1-mediated pathways and apoptosis.
Materials and methods
Cell lines and reagents
A panel of 7 lung adenocarcinoma cell lines was
obtained from the American Type Culture Collection (Manassas, VA,
USA). Cells were incubated in RPMI-1640 culture medium
(Gibco®, Life Technologies, Carlsbad, CA, USA)
containing 10% fetal bovine serum (FBS) (Gibco) in a humidified
atmosphere at 37°C with 5% CO2. ATO was purchased from
Sigma-Aldrich (St. Louis, MO, USA).
Assay of cell viability
Cell viability following ATO treatment was measured
using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium
bromide (MTT) assay as previously described (9).
Western blot analysis of cell
lysates
Sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) and western blot analysis were carried
out as described (15). Primary
antibodies were purchased from Cell Signaling Technology (Danvers,
MA, USA). β-actin (Sigma-Aldrich) was used as a house-keeping
protein.
E2F1 siRNA knockdown
Cells were cultured for 6 h with a mixture of
transfection reagent and control (sc-37007) or E2F1 (sc-29297)
siRNA (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) in
RPMI-1640 medium. The transfected cells were maintained in 1%
FBS-containing medium for 2 days. Cell viability and E2F1
expression were assessed by MTT assay and western blot analysis,
respectively (9).
Phycoerythrin (PE)-conjugated Annexin V
and 7-(aminoactinomycin D) AAD staining
Phosphatidylserine externalization (PS) (loss of
membrane asymmetry) was examined using the PE-conjugated Annexin V
and 7-AAD staining method as previously described (15).
Measurement of mitochondrial membrane
potential by JC-1 staining
The fluorescent dye JC-1 was employed for the
determination of mitochondrial transmembrane potential. ATO-treated
cells were harvested and re-suspended for 15 min at 37°C in
darkness with RPMI medium containing 2.5 μg/ml JC-1
(Sigma-Aldrich). Flow analysis was performed and signals were
detected by FL-1 (525 nm) and FL-2 (575 nm) channels (Beckman
FC500).
Tumor growth inhibition in vivo
Tumor xenograft was established by subcutaneous
injection of 10 million H358 cells in PBS into the back of nude
mice (female, 4-week-old, 10–12 g, BALB/cAnN-nu, Charles River
Laboratories, Wilmington, MA, USA). Tumors were allowed to grow for
5 days before mice were randomised to two groups. ATO at 5 mg/kg
(n=8) or PBS as control (n=7), was daily administered
intraperitoneally. Tumor growth was measured using standard
calipers and body weight of mice was recorded on alternate days.
Tumor volume (V) was calculated [V = (length × width × width)/2]
(16). Mice were sacrificed
following completion of ATO treatment. Tumor xenografts were
collected and homogenized to obtain protein lysates for western
blot analysis. The in vivo study was approved by the
Committee on the Use of Live Animals in Teaching and Research
(CULATR) of the University of Hong Kong (CULATR reference no.
2510–11).
Statistical analysis
Data from three individual experiments are shown as
mean ± standard deviation (SD). Comparison between groups was
performed using Student’s two-tailed t-test by Prism (GraphPad
Software, La Jolla, CA, USA). A p-value <0.05 was considered
statistically significant.
Results
In vitro activity of ATO in lung
adenocarcinoma
Incubation with ATO for 48 h reduced cell viability
in different lung adenocarcinoma cell lines, with IC50
values ranging from 1.8 to 16.5 μM (H23, H358, HCC827, H1650,
H1975, HCC2935 and HCC4006 cells: 1.8, 16.1, 2.0, 3.8, 2.6, 12.1
and 9.0 μM, respectively). After 72 h of ATO treatment,
IC50 values were further decreased (H23, H358, HCC827,
H1650, H1975, HCC2935 and HCC4006 cells: 0.5, 7.4, 0.08, 4.0, 1.5,
5.7 and 4.0 μM, respectively).
Downregulation of E2F1 and alteration of
related downstream proteins
ATO reduced expression of E2F1 (Fig. 1A) in a dose-dependent manner, thus
downstream targets of E2F1 were also investigated. Expression of
cyclin A2 (Fig. 1B) was
consistently downregulated by ATO in all cell lines. ATO also
decreased the expression of skp2 (all cell lines except HCC4006
cells) (Fig. 1C), c-myc (H23 and
H1975 cells) (Fig. 1D), thymidine
kinase (TK) (H358, H1650, HCC2935 and HCC4006 cells) (Fig. 1E) and ribonucleotide reductase M1
(RRM1) (all cell lines except HCC827 and H1975 cells) (Fig. 1F). Nevertheless ATO upregulated
p-c-Jun in H23, H358, HCC827 and H1975 cells (Fig. 1G). Representative western blots are
shown in Fig. 1.
E2F1 downregulation reduced cell
viability
The role of E2F1 in lung adenocarcinoma was studied
using siRNA knockdown. With E2F1 protein expression decreased by
50–80% compared with control siRNA treatment, cell viability was
significantly decreased by 60–88% (Fig. 2).
Phosphatidylserine (PS) externalization
and mitochondrial membrane depolarization induced by ATO
ATO caused PS externalization in H23 cells only
(Fig. 3A). Nonetheless ATO
aggravated mitochondrial membrane depolarization in all cell lines
in a dose-dependent manner (Fig.
3B).
Alteration of apoptosis-related factors
by ATO
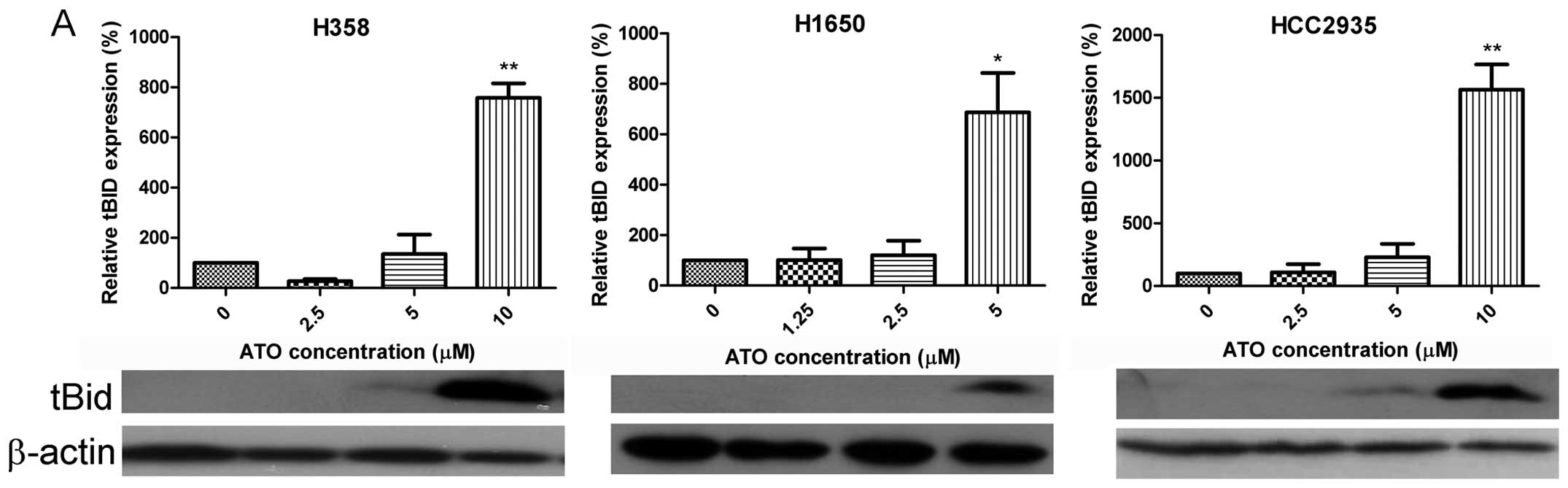
Truncated BID was detected in H358, H1650 and
HCC2935 cells (Fig. 4A) following
treatment with ATO. In contrast, there was a dose-dependent
downregulation of Bcl-2 in all cell lines (Fig. 4B), and upregulation of Bax in H23
cells (Fig. 4C). There was also a
dose-dependent increase in expression of Bak in all cell lines
(Fig. 4D). Expression of cleaved
caspase-9 was elevated in H827 cells (Fig. 4E). On the other hand, cleaved
caspase-3 (CC3) was activated in H23, HCC827, H1975 and HCC4006
cells, but unaltered in H1650 and downregulated in HCC2935 cells.
The expression of CC3 in H358 cells was first elevated when exposed
to 5 μM ATO, then suppressed with 10 μM ATO (Fig. 4F). Caspase-3 expression was
decreased in H358 and H2935 cells upon treatment with ATO (Fig. 4G). The expression of cleaved PARP
was also augmented in H23, H358 and H1975 cells (Fig. 4H). Representative western blots are
shown in Fig. 4.
In vivo effect of ATO on tumor
xenografts
Tumor growth was observed by day 5 following
implantation of H358 cells. Mice were then randomly assigned to two
treatment groups with no significant difference in baseline tumor
volume. Tumor growth was significantly suppressed in the ATO
treatment group compared with controls during 8 days of treatment
(Fig. 5A). As the tumor size had
reached the humane endpoint (a width of 17 mm) in control group,
mice were sacrificed after 8 days of treatment. The relative tumor
volume in the ATO treatment arm was 32% that of the control group
at the end of treatment (p=0.0072). No obvious toxic effect due to
ATO treatment was noted and all the mice were alive following 8
days of treatment. The body weight of mice in the ATO treatment
group and control group was similar during treatment. Based on
western blotting, E2F1 protein was downregulated and truncated BID
and cleaved caspase-3 were upregulated with ATO treatment (Fig. 5B). Histological examination
(H&E staining) of tumor sections revealed prominent apoptosis
(formation of apoptosis bodies) with ATO treatment (Fig. 5C). Immunohistochemical staining
demonstrated nuclear localization of cleaved caspase-3 in the ATO
treatment group (Fig. 5D).
Discussion
In our cell line and xenograft models, ATO has
demonstrated anti-proliferative and cytotoxic activity in lung
adenocarcinoma at least partially mediated via downregulation of
E2F1 and apoptosis. The concentrations of ATO corresponding to the
in vitro IC50 values were within a clinically
reachable plasma level (8.3 μM) (17). The regulatory role of E2F1 in
cellular proliferation in lung adenocarcinoma cell lines was
confirmed using E2F1 siRNA knockdown experiment.
Pi Shuang is notoriously poisonous and has
been paradoxically used in traditional Chinese medicine to treat
various conditions, including cancers. The active ingredient of
Pi Shuang is now known to be arsenic trioxide
(As2O3 or ATO). ATO has been shown to induce
apoptosis (at 0.5–2 μM) and promote cellular differentiation (at
0.1–0.5 μM) in acute promyelocytic leukemia (APL) cells (18). Its mechanisms of action in leukemia
have been extensively investigated in the past decade, and involve
alteration or activation of Bcl-2, cytochrome c, caspase-9,
-3 and reactive oxygen species (19), p73, XIAP, cIAP2, Bcl-xL and
survivin (20), DNA mutation and
apoptosis (21), tubulin assembly
disarrangement and microtubule depolymerization (22), survivin and telomerase (7). An intravenous formulation of ATO has
received approval from the US Food and Drug Administration in the
treatment of APL. In recent years, our institution has developed an
oral liquid form of ATO that is more convenient for clinical use
with a better safety profile (5).
The role of ATO in the treatment of NSCLC has been less
well-defined, though some preclinical data have suggested potential
activity. We therefore aimed to further investigate the activity
and mechanisms of action of ATO in preclinical models of lung
adenocarcinoma.
The role of E2F1 in cancer appears to be a
double-edged sword, with oncogenic or tumor suppressive properties,
depending on the specific cancer type. In human breast cancer: E2F1
mRNA expression was lower with more advanced tumor stage in
malignant breast tissue (23),
nonetheless E2F1 was shown to promote proliferation in breast
cancer cells (24). In NSCLC, E2F1
was reported to be oncogenic (12)
and associated with an adverse prognosis (13) that is also observed in thyroid
(25), liver (26) and pancreatic (27) cancers.
E2F1 consists of a cyclin A binding domain, DNA
binding domain, pocket protein binding domain, nuclear export
signal and nuclear localization signal (11). It is a transcription factor that
controls apoptosis, cell cycle, senescence and tumor growth
(10), as well as DNA damage,
repair, synthesis and replication (11). The E2F1 pathway is frequently
deregulated in cancers. As a consequence, amplification of cyclin
A2 (28), c-myc (29), thymidylate synthase (TYMS)
(30) and skp2 (31) is commonly found in various tumors,
serving as important cell cycle regulators that are essential for
cell proliferation. Thymidine kinase (TK) overexpression is
associated with a higher incidence of clinical disease recurrence
and mortality in breast cancer (32), while a lower level of expression of
ribonucleotide reductase M1 (RRM1) predicts a longer time to
progression in lung cancer with chemotherapy treatment (33). Activation of c-Jun is correlated
with CHOP upregulation and induction of apoptosis by AW00178 in
human H1299 lung carcinoma cells (34) and apoptosis activation by
6-(7-nitro-2,1,3-benzoxadiazol-4-ylthio)hexanol in
multidrug-resistant small cell lung cancer H69AR cells (35). These important molecular signals
are ultimately controlled by E2F1. In this study, ATO has been
shown to suppress E2F1 expression with alteration of its downstream
targets. Notably TYMS (9), TK and
RRM1 were downregulated, leading to inhibition of DNA synthesis. In
addition, decreased expression of other proliferation factors
(cyclin A2, c-myc, skp2) may also have contributed to the observed
antiproliferative effect of ATO.
While E2F1 has been reported as an oncogene
(12), its functional role in lung
adenocarcinoma was demonstrated by specific E2F1 siRNA knockdown in
our cell line model. Upon E2F1 knockdown by 50–80%, cell viability
was significantly reduced by 60–88%, in support of its critical
role in cell survival. The same phenomenon was recently reported in
other lung cancer cell lines (36), nonetheless neither downstream
targets of E2F1 nor other possible mechanisms were studied. In our
study, E2F1 and its downstream targets were downregulated with ATO
treatment, while the pro-apoptotic factor p-c-Jun was upregulated.
As an executioner of apoptosis, expression of cleaved caspase-3
(CC3) after E2F1 knockdown was investigated. By simply knocking
down E2F1, expression of CC3 was increased in HCC2935 cells only
(data not shown), suggesting that E2F1 is mainly responsible for
cell proliferation rather than apoptosis.
Although the induction of cell death by ATO has been
investigated extensively in different cancer models, only a few
reports have shown ATO-induced PS externalization (37–41).
To our knowledge, this is the first report of PS externalization in
an ATO-treated lung cancer cell line (H23). Nonetheless flow
analysis did reveal that more cells became susceptible to
mitochondrial membrane depolarization across different cell lines
in our model with treatment of increasing ATO concentration,
similar to previous reports in both lung cancer (8,42,43)
and other cancer cell lines (44–46).
Theoretically, truncation of BID can increase the
expression of Bax and Bak. Together with reduction in the
expression of Bcl-2, an anti-apoptotic factor, truncated BID can
direct the activation of caspase-9 and -3. The activation of
caspase-3 may then cleave PARP leading to apoptosis. Thus the key
apoptotic factors related to mitochondrial pathway were
investigated in our lung adenocarcinoma cell line model with ATO
treatment.
The expression of Bcl-2 was frequently inhibited by
ATO in other lung cancer cell lines (8,47,48).
In accordance with previous reports, we have demonstrated
downregulation of Bcl-2 expression in our panel of ATO-treated lung
adenocarcinoma cell lines. Upregulation of Bax was induced by ATO
in H23 cells, while a similar phenomenon was only reported in small
cell lung carcinoma (49).
Nonetheless expression of Bak was elevated across different lung
adenocarcinoma cell lines with ATO treatment. This is the first
report to date of BID truncation and Bak upregulation in
ATO-treated lung cancer cell lines.
Although there are reports of cleaved caspase-9
upregulation by ATO in other cancer cell lines (50,51),
our similar observation in HCC827 cells is the first report in a
lung cancer model. Caspase-3 activation was shown in ATO-treated
A549 cells (52), Calu-6 cells
(8) and SCLC cell lines (49). This study has reinforced these
findings in a panel of lung adenocarcinoma cell lines.
Interestingly, the expression of CC3 in H358 cells was first
increased when exposed to 5 μM ATO and then decreased with 10 μM
ATO, whereas, CC3 expression decreased in a dose-dependent manner
in HCC2935 cells when incubated with ATO. A similar observation was
reported with prolonged incubation of ATO in lymphocytic leukemia
cells (53). This paradoxical
result was due to the direct suppression of caspase-3 expression by
ATO in H358 and HCC2935 cells, and has been previously reported
(53). ATO-induced cleavage of
PARP has been reported in the H1355 NSCLC cell line (54) and in SCLC cell lines (49). We have provided further evidence of
PARP cleavage in lung adenocarcinoma cell lines with ATO
treatment.
Apart from promising in vitro activity in our
lung adenocarcinoma model, the in vivo effect of ATO was
confirmed using a nude mouse xenograft model. E2F1 downregulation
was observed in tumor xenografts in keeping with the
antiproliferative effect of ATO. Moreover, formation of apoptotic
bodies and upregulation of truncated Bid and CC3 were also observed
in treated tumor xenografts. Translocation of CC3 from the
cytoplasm to the nucleus was shown by IHC staining. Pro-caspase-3
is located predominantly in the cytoplasm of cells. Caspase-3 is
activated by upstream caspases and its active form (CC3) is then
translocated into the nucleus. The substrates in the nucleus, e.g.,
PARP, are then cleaved. Eventually, chromatin condensation, DNA
fragmentation and nuclear disruption occur and cells are directed
to apoptosis (55). Our findings
have provided evidence that apoptosis is induced by ATO in a lung
adenocarcinoma xenograft model.
In conclusion, the anticancer effect of ATO was
demonstrated through antiproliferation (E2F1 downregulation) and
cell death (apoptosis) in both in vitro and in vivo
lung adenocarcinoma models. Our novel finding of E2F1 suppression
by ATO provides an additional mechanism to explain the activity of
ATO in lung adenocarcinoma. Future potential clinical applications
of ATO in lung adenocarcinoma treatment should be explored.
Acknowledgements
This study was supported by the Simon K.Y. Lee
Foundation research fund and the University of Hong Kong small
project funding.
References
|
1
|
Yano T, Haro A, Shikada Y, Maruyama R and
Maehara Y: Non-small cell lung cancer in never smokers as a
representative ‘non-smoking-associated lung cancer’: epidemiology
and clinical features. Int J Clin Oncol. 16:287–293. 2011.
|
|
2
|
Favaretto AG, Pasello G and Magro C:
Second and third line treatment in advanced non-small cell lung
cancer. Discov Med. 8:204–209. 2009.PubMed/NCBI
|
|
3
|
Goldstraw P, Crowley J, Chansky K, et al:
The IASLC Lung Cancer Staging Project: proposals for the revision
of the TNM stage groupings in the forthcoming (seventh) edition of
the TNM classification of malignant tumours. J Thorac Oncol.
2:706–714. 2007. View Article : Google Scholar
|
|
4
|
Suda K, Onozato R, Yatabe Y and Mitsudomi
T: EGFR T790M mutation: a double role in lung cancer cell survival?
J Thorac Oncol. 4:1–4. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Au WY, Kumana CR, Kou M, et al: Oral
arsenic trioxide in the treatment of relapsed acute promyelocytic
leukemia. Blood. 102:407–408. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chien CW, Yao JH, Chang SY, Lee PC and Lee
TC: Enhanced suppression of tumor growth by concomitant treatment
of human lung cancer cells with suberoylanilide hydroxamic acid and
arsenic trioxide. Toxicol Appl Pharmacol. 257:59–66. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ghaffari SH, Momeny M, Bashash D, Mirzaei
R, Ghavamzadeh A and Alimoghaddam K: Cytotoxic effect of arsenic
trioxide on acute promyelocytic leukemia cells through suppression
of NFkbeta-dependent induction of hTERT due to down-regulation of
Pin1 transcription. Hematology. 17:198–206. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Han YH, Kim SZ, Kim SH and Park WH:
Arsenic trioxide inhibits the growth of Calu-6 cells via inducing a
G2 arrest of the cell cycle and apoptosis accompanied with the
depletion of GSH. Cancer Lett. 270:40–55. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lam SK, Mak JC, Zheng CY, Li YY, Kwong YL
and Ho JC: Downregulation of thymidylate synthase with arsenic
trioxide in lung adenocarcinoma. Int J Oncol. 44:2093–2102.
2014.PubMed/NCBI
|
|
10
|
Slee EA and Lu X: Requirement for
phosphorylation of P53 at Ser312 in suppression of chemical
carcinogenesis. Sci Rep. 3:31052013.PubMed/NCBI
|
|
11
|
Tsantoulis PK and Gorgoulis VG:
Involvement of E2F transcription factor family in cancer. Eur J
Cancer. 41:2403–2414. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Huang CL, Liu D, Nakano J, et al: E2F1
overexpression correlates with thymidylate synthase and survivin
gene expressions and tumor proliferation in non small-cell lung
cancer. Clin Cancer Res. 13:6938–6946. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gorgoulis VG, Zacharatos P, Mariatos G, et
al: Transcription factor E2F-1 acts as a growth-promoting factor
and is associated with adverse prognosis in non-small cell lung
carcinomas. J Pathol. 198:142–156. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liontos M, Niforou K, Velimezi G, et al:
Modulation of the E2F1-driven cancer cell fate by the DNA damage
response machinery and potential novel E2F1 targets in
osteosarcomas. Am J Pathol. 175:376–391. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li YY, Lam SK, Mak JC, Zheng CY and Ho JC:
Erlotinib-induced autophagy in epidermal growth factor receptor
mutated non-small cell lung cancer. Lung Cancer. 81:354–361. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kousparou CA, Yiacoumi E, Deonarain MP and
Epenetos AA: Generation of a selectively cytotoxic fusion protein
against p53 mutated cancers. BMC Cancer. 12:3382012. View Article : Google Scholar
|
|
17
|
Yamauchi KIY, Ogata K, Hara S, Tamura K,
Suzumiya J, Ishitsuka K and Ono N: Pharmacokinetics of arsenic
trioxide in Japanese patients with acute promyelocytic leukemia and
adult T-cell leukemia lymphoma. Jpn J Pharmaceut Health Care Sci.
30:492–496. 2004. View Article : Google Scholar
|
|
18
|
Chen GQ, Shi XG, Tang W, et al: Use of
arsenic trioxide (As2O3) in the treatment of
acute promyelocytic leukemia (APL): I. As2O3
exerts dose-dependent dual effects on APL cells. Blood.
89:3345–3353. 1997.PubMed/NCBI
|
|
19
|
Bustamante J, Nutt L, Orrenius S and
Gogvadze V: Arsenic stimulates release of cytochrome c from
isolated mitochondria via induction of mitochondrial permeability
transition. Toxicol Appl Pharmacol. 207:110–116. 2005.PubMed/NCBI
|
|
20
|
Momeny M, Zakidizaji M, Ghasemi R, et al:
Arsenic trioxide induces apoptosis in NB-4, an acute promyelocytic
leukemia cell line, through up-regulation of p73 via suppression of
nuclear factor kappa B-mediated inhibition of p73 transcription and
prevention of NF-kappaB-mediated induction of XIAP, cIAP2, BCL-XL
and survivin. Med Oncol. 27:833–842. 2010.
|
|
21
|
Meng R, Zhou J, Sui M, Li Z, Feng G and
Yang B: Arsenic trioxide promotes mitochondrial DNA mutation and
cell apoptosis in primary APL cells and NB4 cell line. Sci China
Life Sci. 53:87–93. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Baek JH, Moon CH, Cha SJ, et al: Arsenic
trioxide induces depolymerization of microtubules in an acute
promyelocytic leukemia cell line. Korean J Hematol. 47:105–112.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Worku D, Jouhra F, Jiang GW, Patani N,
Newbold RF and Mokbel K: Evidence of a tumour suppressive function
of E2F1 gene in human breast cancer. Anticancer Res. 28:2135–2139.
2008.PubMed/NCBI
|
|
24
|
Xu F, You X, Liu F, et al: The oncoprotein
HBXIP up-regulates Skp2 via activating transcription factor E2F1 to
promote proliferation of breast cancer cells. Cancer Lett.
333:124–132. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Onda M, Nagai H, Yoshida A, et al:
Up-regulation of transcriptional factor E2F1 in papillary and
anaplastic thyroid cancers. J Hum Genet. 49:312–318. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Farra R, Dapas B, Pozzato G, et al:
Effects of E2F1-cyclin E1–E2 circuit down regulation in
hepatocellular carcinoma cells. Dig Liver Dis. 43:1006–1014.
2011.
|
|
27
|
Yamazaki K, Yajima T, Nagao T, et al:
Expression of transcription factor E2F-1 in pancreatic ductal
carcinoma: an immunohistochemical study. Pathol Res Pract.
199:23–28. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yam CH, Fung TK and Poon RY: Cyclin A in
cell cycle control and cancer. Cell Mol Life Sci. 59:1317–1326.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hilbe W, Dirnhofer S, Greil R and Woll E:
Biomarkers in non-small cell lung cancer prevention. Eur J Cancer
Prev. 13:425–436. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ceppi P, Rapa I, Lo Iacono M, et al:
Expression and pharmacological inhibition of thymidylate synthase
and Src kinase in nonsmall cell lung cancer. Int J Cancer.
130:1777–1786. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hung WC, Tseng WL, Shiea J and Chang HC:
Skp2 overexpression increases the expression of MMP-2 and MMP-9 and
invasion of lung cancer cells. Cancer Lett. 288:156–161. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Huang ZH, Tian XS, Li R, et al: Elevated
thymidine kinase 1 in serum following neoadjuvant chemotherapy
predicts poor outcome for patients with locally advanced breast
cancer. Exp Ther Med. 3:331–335. 2012.
|
|
33
|
Wang L, Meng L, Wang XW, Ma GY and Chen
JH: Expression of RRM1 and RRM2 as a novel prognostic marker in
advanced non-small cell lung cancer receiving chemotherapy. Tumour
Biol. 35:1899–1906. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ryu BJ, Hwang MK, Park M, Lee K and Kim
SH: Thiourea compound AW00178 sensitizes human H1299 lung carcinoma
cells to TRAIL-mediated apoptosis. Bioorg Med Chem Lett.
22:3862–3865. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Filomeni G, Turella P, Dupuis ML, et al:
6-(7-Nitro-2,1,3-benzoxadiazol-4-ylthio)hexanol, a specific
glutathione S-transferase inhibitor, overcomes the multidrug
resistance (MDR)-associated protein 1-mediated MDR in small cell
lung cancer. Mol Cancer Ther. 7:371–379. 2008. View Article : Google Scholar
|
|
36
|
Walker T, Nolte A, Steger V, et al: Small
interfering RNA-mediated suppression of serum response factor,
E2-promotor binding factor and survivin in non-small cell lung
cancer cell lines by non-viral transfection. Eur J Cardiothorac
Surg. 43:624–633. 2013.
|
|
37
|
Hu T, Shi J, Jiao X, Zhou J and Yin X:
Measurement of Annexin V uptake and lactadherin labeling for the
quantification of apoptosis in adherent Tca8113 and ACC-2 cells.
Braz J Med Biol Res. 41:750–757. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Korper S, Nolte F, Thiel E, Schrezenmeier
H and Rojewski MT: The role of mitochondrial targeting in arsenic
trioxide-induced apoptosis in myeloid cell lines. Br J Haematol.
124:186–189. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Shen ZY, Shen J, Cai WJ, Hong C and Zheng
MH: The alteration of mitochondria is an early event of arsenic
trioxide induced apoptosis in esophageal carcinoma cells. Int J Mol
Med. 5:155–158. 2000.PubMed/NCBI
|
|
40
|
Wang Y, Xu Y, Wang H, et al: Arsenic
induces mitochondria-dependent apoptosis by reactive oxygen species
generation rather than glutathione depletion in Chang human
hepatocytes. Arch Toxicol. 83:899–908. 2009. View Article : Google Scholar
|
|
41
|
Xu Y, Wang H, Wang Y, Zheng Y and Sun G:
Effects of folate on arsenic toxicity in Chang human hepatocytes:
involvement of folate antioxidant properties. Toxicol Lett.
195:44–50. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Jin HO, Seo SK, Woo SH, et al: A
combination of sulindac and arsenic trioxide synergistically
induces apoptosis in human lung cancer H1299 cells via c-Jun
NH2-terminal kinase-dependent Bcl-xL phosphorylation. Lung Cancer.
61:317–327. 2008. View Article : Google Scholar
|
|
43
|
Jin HO, Yoon SI, Seo SK, et al:
Synergistic induction of apoptosis by sulindac and arsenic trioxide
in human lung cancer A549 cells via reactive oxygen
species-dependent down-regulation of survivin. Biochem Pharmacol.
72:1228–1236. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Qin DB, Chen JP and Wang SQ: Mechanism of
apoptosis of NB4 cells induced by arsenic trioxide and
cyclooxygenase-2 expression. Zhongguo Shi Yan Xue Ye Xue Za Zhi.
19:648–651. 2011.(In Chinese).
|
|
45
|
Su Y, Wang X, Xu W, et al: Arsenic
trioxide increases the sensitivity of 786–0 renal carcinoma cells
to radiotherapy. Cancer Invest. 30:114–118. 2012.PubMed/NCBI
|
|
46
|
Tung JN, Cheng YW, Hsu CH, et al:
Normoxically overexpressed hypoxia inducible factor 1-alpha is
involved in arsenic trioxide resistance acquisition in
hepatocellular carcinoma. Ann Surg Oncol. 18:1492–1500. 2011.
View Article : Google Scholar
|
|
47
|
Li HC, Wang CX, Huang C, et al: Effect and
mechanism of arsenic trioxide on chemosensitivity of human lung
adenocarcinoma cells. Zhonghua Jie He He Hu Xi Za Zhi. 26:689–692.
2003.(In Chinese).
|
|
48
|
Shi Y, Liu Y, Huo J and Gao G: Arsenic
trioxide induced apoptosis and expression of p53 and bcl-2 genes in
human small cell lung cancer cells. Zhonghua Jie He He Hu Xi Za
Zhi. 25:665–666. 2002.PubMed/NCBI
|
|
49
|
Pettersson HM, Pietras A, Munksgaard
Persson M, et al: Arsenic trioxide is highly cytotoxic to small
cell lung carcinoma cells. Mol Cancer Ther. 8:160–170. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yuan Z, Wang F, Zhao Z, et al:
BIM-mediated AKT phosphorylation is a key modulator of arsenic
trioxide-induced apoptosis in cisplatin-sensitive and -resistant
ovarian cancer cells. PLoS One. 6:e205862011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Potin S, Bertoglio J and Breard J:
Involvement of a Rho-ROCK-JNK pathway in arsenic trioxide-induced
apoptosis in chronic myelogenous leukemia cells. FEBS Lett.
581:118–124. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Walker AM, Stevens JJ, Ndebele K and
Tchounwou PB: Arsenic trioxide modulates DNA synthesis and
apoptosis in lung carcinoma cells. Int J Environ Res Public Health.
7:1996–2007. 2010. View Article : Google Scholar
|
|
53
|
Bairey O, Vanichkin A and Shpilberg O:
Arsenic-trioxide-induced apoptosis of chronic lymphocytic leukemia
cells. Int J Lab Hematol. 32:e77–85. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Cheng Y, Chang LW and Tsou TC:
Mitogen-activated protein kinases mediate arsenic-induced
down-regulation of survivin in human lung adenocarcinoma cells.
Arch Toxicol. 80:310–318. 2006. View Article : Google Scholar
|
|
55
|
Luo M, Lu Z, Sun H, et al: Nuclear entry
of active caspase-3 is facilitated by its p3-recognition-based
specific cleavage activity. Cell Res. 20:211–222. 2010. View Article : Google Scholar : PubMed/NCBI
|