Introduction
Breast cancer is one of the most common cancers in
women. In recent years, major advances in breast cancer therapy
have been achieved. Nevertheless, because cancer cells may present
a number of resistance mechanisms reducing the effectiveness of
chemotherapeutic drugs, new and more effective strategies for
treatment and prevention are still much needed. Breast cancer
encompasses a heterogeneous group of diseases at the molecular
level and can be classified into at least five distinct subtypes by
gene-expression profiling: luminal A, luminal B, normal
breast-like, human epidermal receptor 2 (HER2), and basal-like.
Recent studies based on neoadjuvant clinical trials for breast
cancer suggested that chemosensitivity depends on its molecular
subtype (1).
There is increasing evidence that epigenetic
alterations, such as histone acetylation and promoter methylation,
play important roles in regulation of gene expression associated
with the cell cycle and apoptosis (2). Chromatin remodeling regulates gene
transcription and is in turn regulated by two enzymes, histone
acetyltransferase (HAT) and histone deacetylase (HDAC), that
control post-translational modifications of histones. Acetylation
of lysine residues on the histones weakens their binding to DNA and
induces a change in DNA conformation and allows transcription
factors to bind to the promoter regions of target genes (3,4).
In mammalian cells, there are 18 different HDACs,
which can be further divided into four types. HDAC 1, 2, 3, and 8
are class I deacetylases that localize almost exclusively to the
nucleus and are ubiquitously expressed in various human cell lines
and tissues. HDAC 4, 5, 6, 7, 9, and 10 are class II deacetylases,
which shuttle between the nucleus and cytoplasm with certain
cellular signals. Class III comprises the conserved nicotinamide
adenine dinucleotide-dependent Sir2 family of deacetylases. HDAC11
is the only member of the class IV HDACs (5). Aberrant levels of HDAC activity have
been found in a variety of human malignancies and result in
repression of tumor-suppressor genes and promotion of tumorigenesis
(6).
Valproic acid (VPA), which has long been used
clinically for treatment of epilepsy and bipolar disorder without
significant toxicity, causes hyperacetylation of the N-terminal
tails of histones H3 and H4 in vitro and in vivo and
inhibits HDAC activity, probably by binding to the catalytic center
and, thereby, blocking substrate access (7,8). VPA
inhibits both class I and II HDACs, with high potency for class I
HDACs (9). Earlier studies
indicated that p21 WAF1, one of the target genes induced by VPA,
affects differentiation and decreases tumor cell growth (10,11).
Another report focused on the apoptotic activity of VPA (12). However, the detailed mechanism of
apoptosis induced by VPA has not been elucidated. In addition,
recent evidence suggests that HDAC inhibitors also enhance the
acetylation of non-histone proteins, such as p53, c-Jun, and
α-tubulin (13–15).
Although VPA has been shown to reduce cancer
proliferation to some extent, there is insufficient amount of data
on its effect in breast cancer cells. Studies on the specificity of
VPA against breast cancer subtypes have often yielded contrasting
results and conflicting conclusions (16–18).
Several studies have found that inhibition of HDAC
increases acetylation levels of the core histones as well as some
non-histone proteins (13,19), raising the possibility that
transcription-independent effects of HDAC inhibitors are also
important for their anticancer activity (5). It has recently been reported that
pan-HDAC inhibitors such as LAQ824, LBH589, and SAHA exert their
antitumor activity by inhibition of HDAC6, a deacetylase of
α-tubulin and heat shock protein (Hsp) 90 (19–21).
The inhibition of HDAC6 results in acetylation of Hsp90 and
disruption of its chaperone function (21–23).
Heat shock proteins (Hsps) are a group of highly
conserved molecular chaperones which were originally identified by
their induction in response to cellular stresses. Hsps are
classified according to their molecular weight and in mammals five
distinct families have been defined: Hsp100, Hsp90, Hsp70, Hsp60
and the small Hsps (24). As Hsp90
controls the intra-cellular trafficking and folding of diverse
cellular proteins, disruption of Hsp90 chaperone function will lead
to the destabilization and eventual degradation of Hsp90 client
proteins and induces apoptosis (25).
HDAC6 is an unusual histone deacetylase, which
harbors two functional catalytic domains and is localized in the
cytoplasm (26). Some recent
reports have demonstrated that HDAC6 is responsible for the
deacetylation of acetyl-α-tubulin and acetyl-Hsp90 (23,27).
It has been reported that the HDAC inhibitor FR901228 disrupts the
chaperone function of Hsp90 and induces apoptosis in human
non-small cell lung cancer cells (13). However, as a class I HDAC
inhibitor, VPA has only a weak effect on inhibition of HDAC6
(14,28).
It is known that Hsp70 is required for the assembly
of the signaling protein-Hsp90 heterocomplex. Hsp90 is involved in
two multi-chaperone complexes and promotes correct folding or
degradation of client proteins, depending on its conformation. When
adenosine triphosphate (ATP) is bound to the amino-terminal
nucleotide-binding pocket, Hsp90 is associated with co-chaperone
proteins p23 and p50Cdc37 and directly binds to the client protein
to stabilize the interactions. When adenosine diphosphate (ADP) is
bound, Hsp90 is assembled into the complex with co-chaperone
proteins Hsp70 and p60Hop. Within the complex, Hsp70 directly
interacts with the client protein to promote ubiquitination and
degradation (25,29). Therefore, the function of Hsp70 may
indirectly affect the chaperone function of Hsp90. However, it is
unknown whether VPA can influence the chaperone function of Hsp70
and the Hsp90 in breast cancer cells. We speculate that VPA could
enhance the acetylation of Hsp70 and Hsp90 through its inhibitory
effect on HDAC6.
In this study, we investigated both inhibitory and
pro-apoptotic effects of VPA on breast cancer cell lines with
various molecular subtypes. In addition, we explored whether VPA
enhanced the acetylation of Hsp70 and Hsp90 and its contribution to
tumor growth inhibition in the VPA-sensitive cell line.
Materials and methods
Materials
VPA and trichostatin A (TSA) was purchased from
Sigma-Aldrich Co. (St. Louis, MO, USA).
Cell lines and cell culture
Two cancer cell lines derived from human breast
cancer, estrogen receptor (ER)-positive and HER2-negative MCF7
cells and ER-negative and HER2-overexpressing SKBR3 cells were
provided by the Cell Resource Center for Biomedical Research,
Institute of Development, Aging and Cancer, Tohoku University. The
other two cell lines derived from human breast cancer, ER-negative
and HER2-negative MDA-MB-231 cells and ER-positive and
HER2-overexpressing BT474 cells, were purchased from the American
Type Culture Collection (Rockville, MD, USA). These breast cancer
cells were seeded in 75-cm2 dishes (Becton-Dickinson,
Franklin Lakes, NJ, USA) and cultured in 10 ml of medium at 37ºC in
a humidified atmosphere of 5% CO2. These cells were
grown in DMEM (Invitrogen, Carlsbad, CA, USA) supplemented with 10%
heat-inactivated fetal bovine serum (Nichirei Bioscience Inc.,
Japan), 100 IU/ml penicillin, 100 mg/ml streptomycin (Invitrogen),
2 mM glutamine (Nissui Pharmaceutical Co., Ltd., Japan), and 0.5 mM
sodium pyruvate. Cells were grown to confluence and harvested by
trypsinization with 0.25 mg/ml trypsin/EDTA solution (Invitrogen)
and suspended in culture medium before use.
Cell growth assay
The viability of breast cancer cells treated with
VPA was determined by
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2H-tetrazolium
(MTS) assay. Breast cancer cells were seeded at 5×103
cells per well in 96-well plates and incubated overnight at 37ºC.
After incubation, the supernatant was discarded and replaced with
fresh serum-free culture medium. VPA was dissolved in
phosphate-buffered saline (PBS) and added to the cell culture
medium at various concentrations (0–10 mM). At 24, 72, and 120 h
after exposure to VPA, the supernatant was discarded and MTS
solution (CellTiter 96 AQuous One Solution Reagent, Promega,
Fitchburg, WI, USA) was added to each well and incubated at 37ºC
for 2 h. Then, the absorbance of the solution was read at a
wavelength of 490 nm using a microplate reader (Bio-Rad, Fitchburg,
WI, USA). The percentage inhibition was determined by comparing the
cell density of the drug-treated cells with that of untreated
controls. All experiments were repeated at least three times. The
median growth inhibition (GI50) corresponding to the
concentration of the compound that inhibits 50% net cell growth was
calculated for each cell line.
Western blotting
The mechanism of growth inhibition by VPA was
analyzed using SKBR3 cell line, which we found to be the most
sensitive to VPA among the four cell lines studied. The effects of
VPA on acetylation of histone H3 and cell cycle regulatory and
apoptosis-related proteins were analyzed by western blotting.
Breast cancer cells were seeded at a density of 1×106
cells per 75-cm2 dish and cultured in 10 ml of medium
overnight. Lysates were obtained from the cells harvested at 0, 3,
6, 12, 24, and 48 h after incubation with 1 mM VPA, which
corresponded approximately to the maximum level obtained by
administering a clinical dose of VPA. Whole-cell lysates were
prepared in denaturing SDS sample buffer and resolved by
SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred
to a PVDF membrane (ATTO Co. Ltd., Japan). As primary antibodies,
rabbit polyclonal acetyl-histone H3 (Lys9) antibody (1:5,000) (Cell
Signaling Technology, Beverly, MA, USA), rabbit polyclonal histone
H3 antibody (1:1,000) (Cell Signaling Technology), and mouse
monoclonal β-actin antibody (1:5,000) (Sigma-Aidrich) were used. A
mouse monoclonal p21 WAF1 (1:1,000) (Pharmingen, San Diego, CA,
USA) was used against cell cycle regulatory proteins. As antibodies
against apoptosis-related proteins, a rabbit polyclonal cleaved
caspase-3 (Asp175) antibody (1:1,000) (Cell Signaling Technology)
was used. The immunoblots were visualized using an ECL Plus (GE
Healthcare UK, Ltd., Buckinghamshire, UK). Immuno-complex was
detected by an ECL detection system (GE Healthcare).
Chemiluminescent signal was detected by the Light-Capture system
(ATTO), and the intensity of protein bands were quantified using
the CS analyzer program (ATTO).
Immunoprecipitation analyses
Lysates were obtained from the SKBR3 cells harvested
at 0, 6, 12, 24, and 48 h after incubation with 1 mM VPA. Cellular
extracts from ~1×106 cells were prepared in
radio-immunoprecipitation (RIPA) buffer, and ~500 μg of total
proteins was incubated with 2 μg of primary antibody at room
tempreture for 60 min on a rotator. As primary antibodies, rabbit
polyclonal Hsp70 antibody (Upstate Biotechnology, Lake Placid, NY,
USA) and mouse monoclonal Hsp90 (Upstate) antibody were used. Then
20 μl of protein A/G-Plus-Agarose (Santa Cruz Biotechnology, Santa
Cruz, CA, USA) was added to the mixture and incubated at 4ºC for 36
h. Agarose-antibody-protein complexes were washed three times with
lysis buffer. After discarding the supernatant from the final wash,
the immune-complexes were resuspended in 50 μl of 1×
electrophoresis sample buffer and boiled at 95ºC for 3 min. The
immunoprecipitated proteins were separated by SDS-PAGE, and
detected by using an ECL Plus (GE Healthcare). As primary
antibodies, mouse monoclonal acetyl-Lysine antibody (1:1,000)
(Upstate), Hsp70 antibody (1:1,000) and Hsp90 antibody (1:1,000)
were used. The antibody-antigen complex was detected by western
blotting.
Immunohistochemical examination and TUNEL
assay
SKBR3 cells were seeded in 10-cm2 dishes
and incubated overnight at 37ºC. After incubation, the supernatant
was discarded and replaced the cell culture medium including 1 mM
VPA. At 48 h after exposure to VPA, the supernatant was discarded,
and cells were fixed in 10% neutral buffered formalin and embedded
in paraffin. The sections were stained with H&E and
immunostained with a rabbit polyclonal cleaved caspase-3 antibody
(1:200) (Cell Signaling Technology) at 4ºC overnight. The sections
were reacted with EnVision reagent (Dako Co., Japan) for
visualization. For quantitative analysis, the stained cells were
counted under ×400 magnification in 6 randomly chosen fields
representing ≥1,000 cells. The degree of apoptosis was evaluated
using the TdT-mediated dUTP nick-end labeling (TUNEL) method
(Apoptosis In Situ Detection kit; Wako, Osaka, Japan).
Results
Effects of VPA on the growth of breast
cancer cell lines in vitro
To explore whether VPA might be a potential
therapeutic agent against breast cancer, we investigated its
effects of growth inhibition in human breast cancer cell lines
(SKBR3, BT474, MDA-MB-231, and MCF7) that differed in their ER- or
HER2-expression status. Breast cancer cells were treated with
increasing doses of VPA for ≤5 days. The inhibition of cell growth
in breast cancer cell lines was dependent on the dose and
incubation time of VPA (Fig. 1).
GI50 values for VPA (mM) in SKBR3, BT474, MDA-MB-231,
and MCF7 at 72 h were 1.8, 3.6, 5.4 and 8.1 mM, respectively
(Table I). SKBR3 cells, which
overexpressed HER2 and were ER-negative, exhibited the most
sensitivity towards VPA in growth inhibition. SKBR3 cell growth was
inhibited by VPA at clinically achievable doses.
 | Table ISubtype and the median growth
inhibition (GI50) for VPA of breast cancer cell
lines. |
Table I
Subtype and the median growth
inhibition (GI50) for VPA of breast cancer cell
lines.
| SKBR3 | BT474 | MDA-MB-231 | MCF7 |
|---|
| ER | − | + | − | + |
| HER2 | ++ | + | − | − |
| GI50
(mM) | 1.8 | 3.6 | 5.4 | 8.1 |
Effects of VPA on histone H3
acetylation
We evaluated the effects of HDAC inhibition by VPA
in SKBR3 cells. The acetylation status of histone H3 in SKBR3 cells
was determined during 48-h incubation with 1 mM VPA, using an
antibody that specifically recognizes hyper-acetylated forms of
histone H3. VPA markedly increased acetyl-histone H3 expression by
2-fold with maximal induction at 12 h of incubation with VPA
(Fig. 2A and B) This time frame of
the maximal acetylation of histone H3 was consistent with that of
the maximal acetylation of histone H3 treated with 100 nM TSA, a
known, strong HDAC inhibitor (Fig. 2C
and D).
Effects of VPA on cell cycle arrest
We studied differentiation effects of VPA by
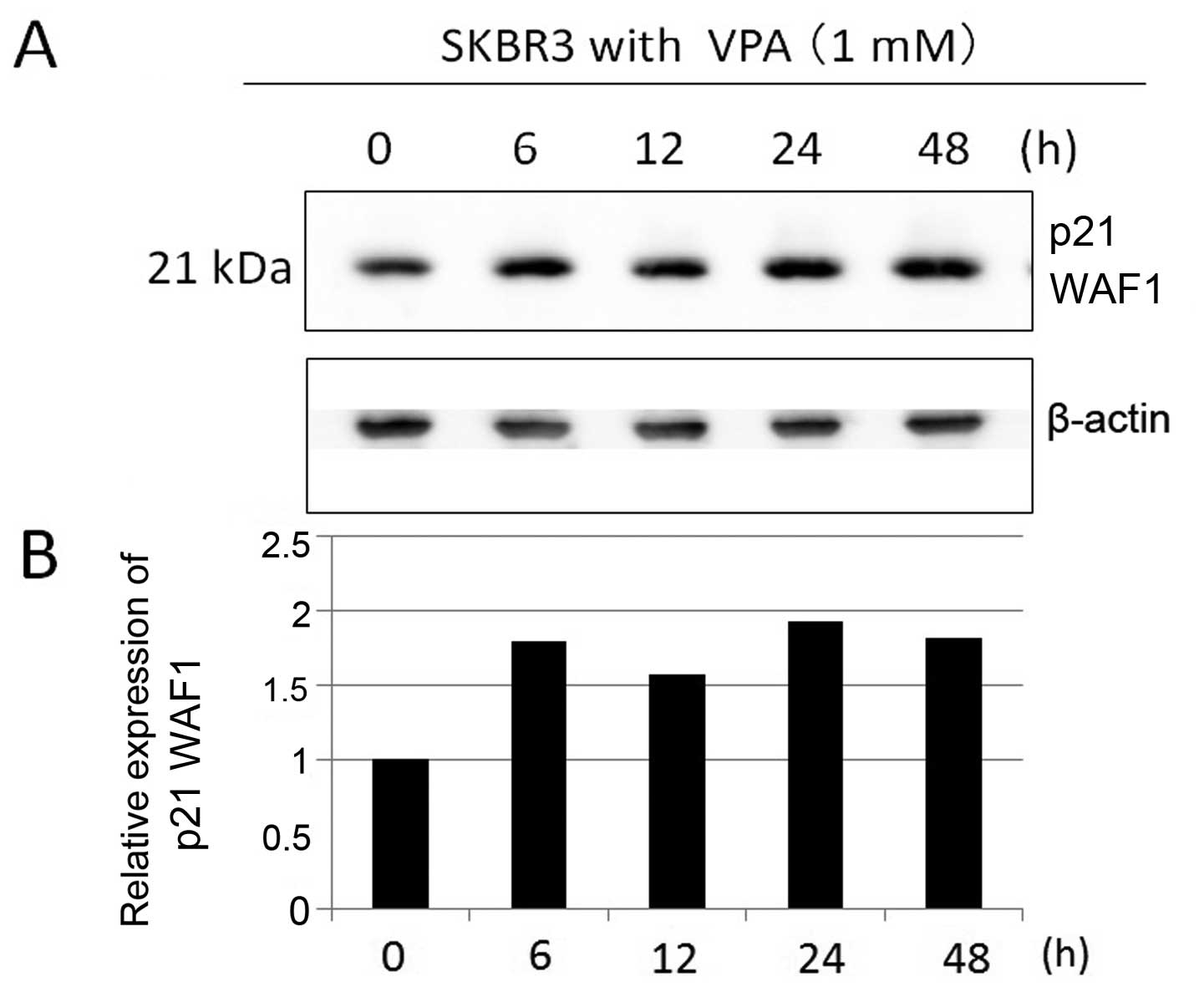
evaluating p21 WAF1 expression in SKBR3 cells by immunoblotting.
Incubation for 48 h in VPA markedly increased p21 WAF1 expression
in SKBR3 cells in a dose-dependent manner (Fig. 3). SKBR3 cells were treated with 1
mM VPA ≤48 h (Fig. 4). The
expression levels of p21 WAF1 increased by ~2–3-fold during the
6–24-h incubation time in the presence of VPA.
Effects of VPA on apoptosis
induction
To examine the apoptosis induction effects of VPA,
we evaluated cleaved caspase-3 expression in SKBR3 cells by
immunoblotting. SKBR3 cells were treated with 1 mM VPA for ≤48 h
(Fig. 5). The expression levels of
cleaved caspase-3 increased by ~2-fold during the incubation times
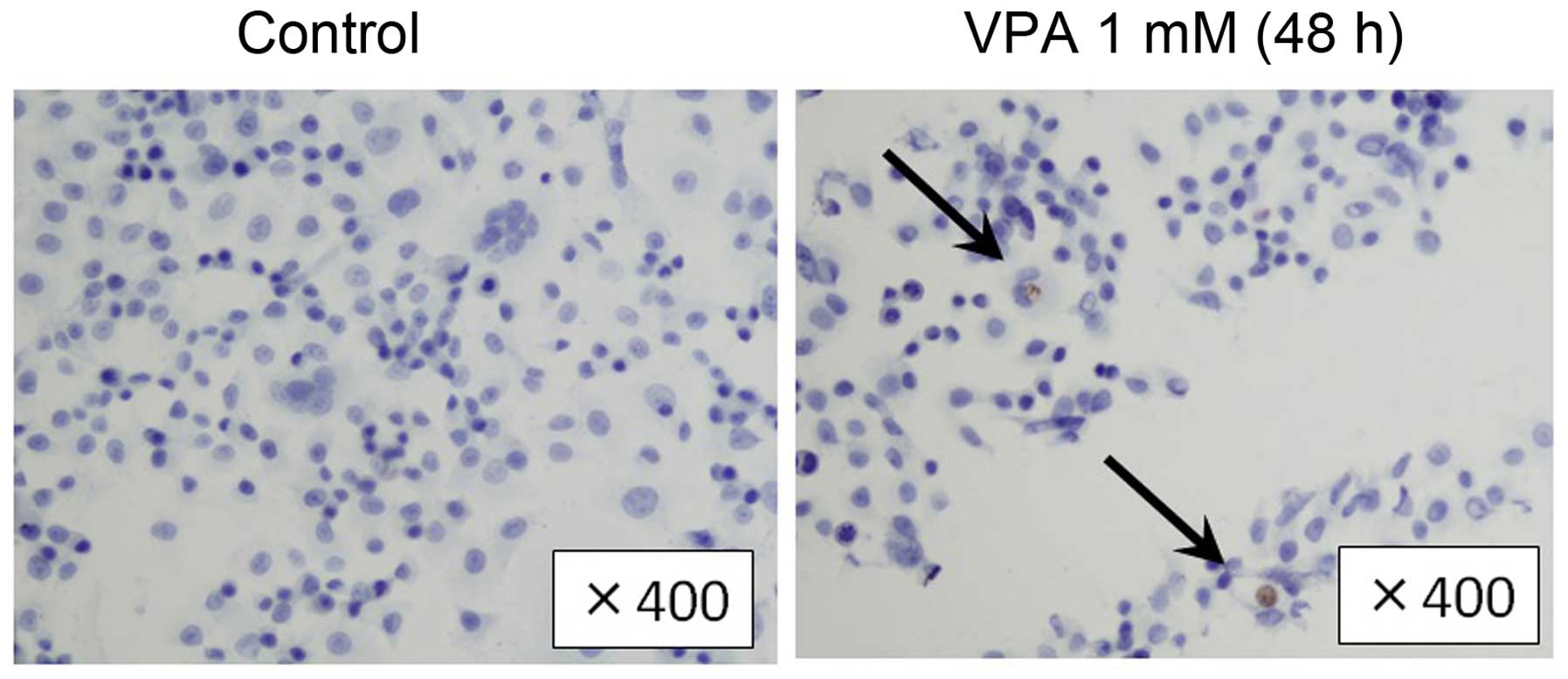
of 6–24 h in VPA. Immunohistochemical examination showed that the
population of cleaved caspase-3-positive SKBR3 cells was higher in
VPA-treated group in comparison to the control group (16.6 vs.
7.4%, Fig. 6). Consistently, in
TUNEL assay, the population of TUNEL-positive cells was higher in
VPA-treated SKBR3 cells than in the control group (Fig. 7).
Effects of VPA on acetylation of Hsp70
and Hsp90
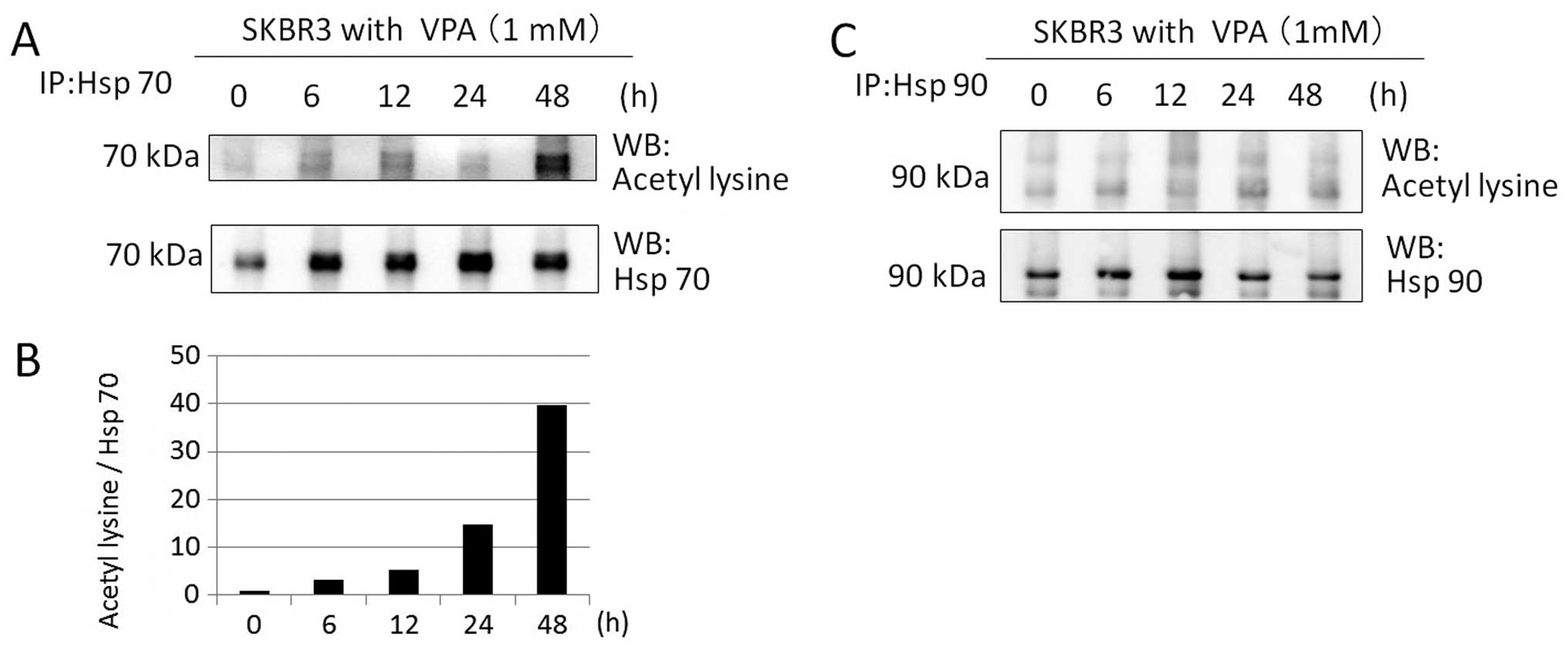
We investigated whether VPA acetylates Hsp70 and
Hsp90 in SKBR3 cells by immunoprecipitation and immunoblotting.
SKBR3 cells were treated with 1 mM VPA for ≤48 h. Western blotting
with anti-aceyl-lysine, anti-Hsp70, and anti-Hsp90 antibodies after
immunoprecipitation revealed that VPA markedly increased Hsp70
acetylation of SKBR3 cells at 1 mM for ≤48 h in a time-dependent
manner (40-fold increase at 48 h) (Fig. 8A and B). However, VPA did not
induce Hsp90 acetylation at 1 mM within the time frame of the study
(Fig. 8C).
Effects of VPA on acetylation of
α-tubulin
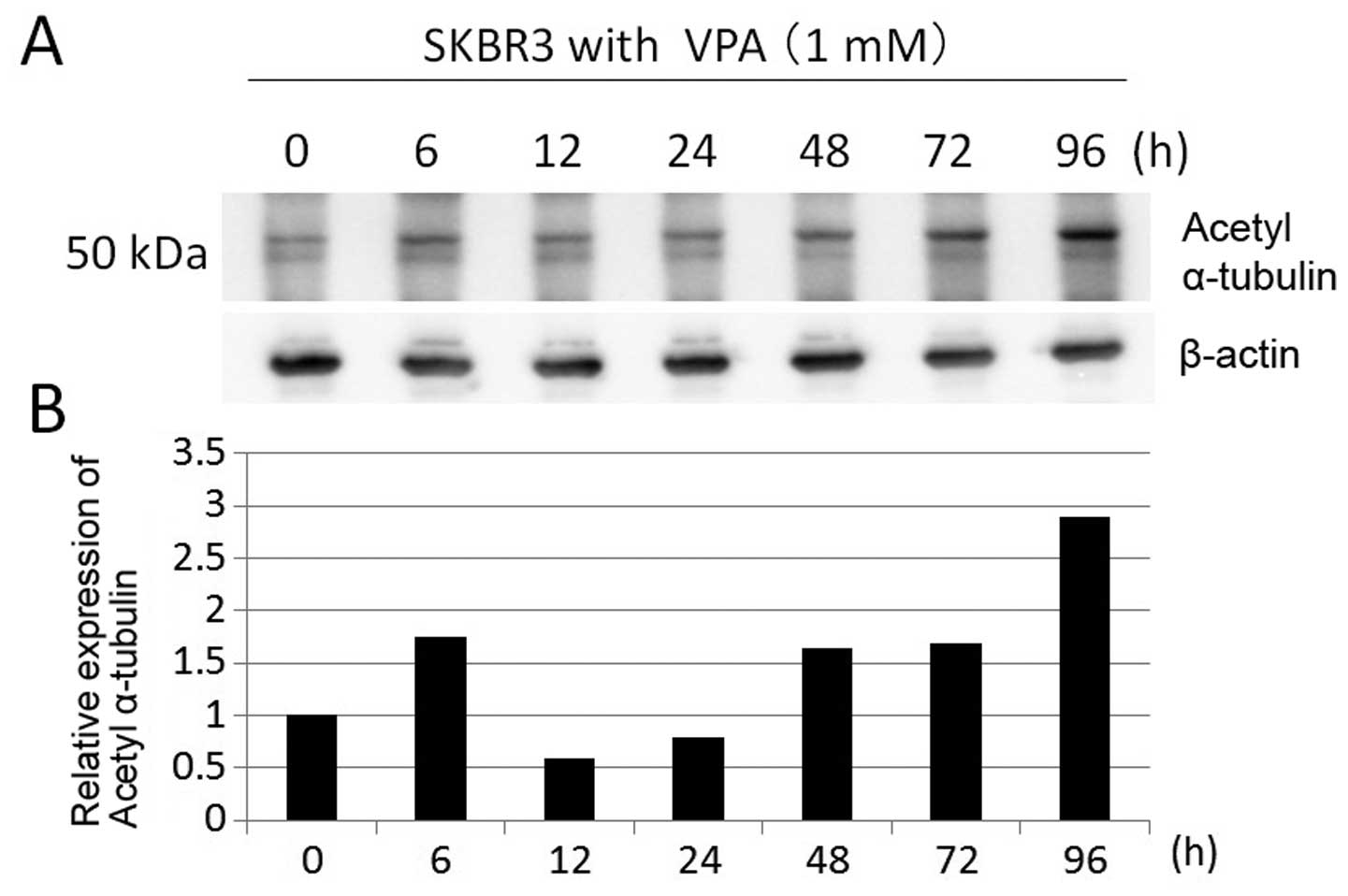
To confirm whether VPA has inhibitory effect on
HDAC6 activity and leads to Hsp90 acetylation, we investigated
acetylation of α-tubulin, which is deacetylated by HDAC6, in SKBR3
cells by immunoblotting. SKBR3 cells were treated with 1 mM VPA for
≤96 h. The expression levels of acetyl-α-tubulin were increased at
later time (3-fold increase at 96 h) (Fig. 9). These data suggest that VPA has
weak HDAC6 inhibitory activity.
Discussion
The present study reports that VPA has
anti-proliferative activity in HER2-overexpressing breast cancer
cells through cell cycle arrest and apoptosis induction at a
clinically achievable dose of 1 mM. Proliferation-inhibiting effect
of VPA in HER2-overexpressing breast cancer cells might be
attributable to dysfunction of Hsp90, which affects its client
protein HER2, indirectly through hyper-acetylation of Hsp70.
We suggested that the diverse cellular responses to
VPA treatment largely depend on the intrinsic characteristics of
breast cancer cells. It is controversial whether ER-expression
status affects the growth-inhibitory effect of HDAC inhibitors.
Fortunati et al (16)
showed that growth of ER-positive breast cancer cells were
suppressed by VPA in lower dose than ER-negative cells, while
Travaglini et al (17)
showed estrogen sensitivity had no relation with the extent of cell
growth suppression by VPA. On the other hand, Giacinti et al
(30) showed that ER-negative
cells were more sensitive to HDAC inhibitor Scriptaid than
ER-positive cells. Consistent with the results of Giacinti et
al, ER-negative cell lines seem to be more sensitive to VPA in
this study.
In our study, VPA exhibits a greater efficacy
against HER2-overexpressing SKBR3 cells than HER2-negative cells,
regardless of their ER-expression status. We focused on
HER2-overexpressing SKBR3 cell line to investigate the mechanism of
anti-proliferative activity of VPA.
Anti-proliferative activity of VPA has previously
been reported in HER2-negative breast cancer cell lines (16–18,31–34)
(Table II). Huang et al
(35) showed HDAC inhibitor
SNDX-275 induced strong apoptosis in HER2-overexpressing breast
cancer cells compared with HER2-negative cells. Giacinti et
al (30) also showed that
HER2-overexpressing breast cancer cells were more sensitive to HDAC
inhibitor Scriptaid than HER2-negative cells. To explore the
differences among intrinsic subtypes of breast cancer cell lines,
we studied the inhibition of cell growth in four different human
breast cancer cell lines: ER-positive and HER2-negative MCF7 cells,
ER-negative and HER2-overexpressing SKBR3 cells, ER-negative and
HER2-negative MDA-MB-231 cells, and ER-positive and HER2-negative
BT474 cells. We confirmed that HER2-overexpressing breast cancer
cells were more sensitive to VPA treatment than HER2-negative ones.
The results suggested that anti-proliferative mechanism of breast
cancer cells by VPA is related to their HER2-expression status.
| Table IIThe characteristics of breast cancer
cell lines treated with VPA in past reports. |
Table II
The characteristics of breast cancer
cell lines treated with VPA in past reports.
| Authors (Ref.) | Year | |
|---|
| Olsen et al
(31) | 2004 | MCF7
(ER+, HER2−) |
| Chavez-Blanco et
al (32) | 2006 | MCF7
(ER+, HER2−) |
| Hodges-Gallagher
et al (33) | 2007 | MCF7
(ER+, HER2−) |
| Fortunati et
al (16) | 2008 | MCF7
(ER+, HER2−) > ZR-75-1 (ER+,
HER2+) > MDA-MB-231 (ER−,
HER2−) > MDA-MB435 (ER−, HER2) |
| Travaglini et
al (17) | 2009 | MCF7
(ER+, HER2−) = MDA-MB-231 (ER−,
HER2−) |
| Li et al
(34) | 2012 | MDA-MB-231
(ER−, HER2−) |
| Zhang et al
(18) | 2012 | MDA-MB-231
(ER−, HER2−) |
| This study | 2015 | SKBR3
(ER+, HER2++) > BT474 (ER−,
HER2+) > MDA-MB-231 (ER−,
HER2−) > MCF7 (ER+, HER2−) |
The cyclin-dependent kinase inhibitor p21 WAF1,
which is involved in both the G1-S and the G2-M transition,
regulates cell cycle progression. Fortunati et al (16) showed the anti-proliferative effects
of VPA were associated with upregulation of p21. In this
investigation, we confirmed that VPA induced cell cycle arrest with
upregulation of p21 at 1 mM, in concurrence with the times of
histone H3 acetylation.
The present study further showed that VPA induces
apoptosis in SKBR3 cells in immunohistochemical examination and
TUNEL assay. Caspase-3 is a critical executioner of apoptosis in
its activated form of cleaved caspase-3. We showed that caspase-3
is activated in SKBR3 by VPA simultaneously as histone H3
acetylation. Thus VPA seemed to affect proliferation of breast
cancer cells by cell cycle arrest and apoptosis induction.
Hsp90 is involved in two multi-chaperone complexes
and promotes correct folding or degradation of client proteins,
depending on its conformation (25). As class II deacetylase, HDAC6 is
known as the deacetylase of Hsp90, and inhibiting the activity of
HDAC6 leads to hyper-acetylation of Hsp90, disruption of its
chaperone function, and cell apoptosis (21–23).
VPA is known to have a weak inhibitory effect on HDAC6, and we
could not directly observe VPA-induced acetylation of Hsp90 in
SKBR3 cells. However, we showed that VPA had acetylation effect
against α-tubulin. Considering that HDAC6 is the only known
deacetylase of α-tubulin, the increased acetylation of α-tubulin
indicates that VPA can partially inhibit HDAC6 activity. HDAC
inhibition by VPA might be insufficient to acetylate Hsp90. On the
other hand, we showed that VPA could acetylate Hsp70. It is known
that Hsp70 is required for the assembly of the signaling
protein-Hsp90 heterocomplex.
We thought VPA may disrupt the function of Hsp90
indirectly through hyper-acetylation of Hsp70. Wang et al
(36) showed that FK228, a class I
HDAC inhibitor, blocked Hsp90 chaperone function in K562 CML cells
via hyper-acetylation of Hsp70. Fuino et al (37) showed that LAQ824, a hydroxamic acid
analogue HDAC inhibitor, induced acetylation of Hsp90, and reduced
its binding to ATP. In SKBR3 and BT474 cells, treatment with LAQ824
shifted the chaperone association of HER2 from Hsp90 to Hsp70,
which efficiently ubiquitinates and downregulates HER2. Scott et
al (38) showed that treatment
with TSA, a strong HDAC inhibitor, in SKBR3 and BT474 cells
produced the expected marked decline in their endogenous HER2
protein levels. Meng et al (39) showed that treatment with
carbamazepine (CBZ), an anti-epileptic drug that acts as an HDAC
inhibitor in SKBR3 cells, produced a marked decline in their HER2
protein levels. In the same study, CBZ was shown to synergize with
trastuzumab inhibiting breast cancer cell proliferation more
strongly than CBZ or trastuzumab alone. Therefore, VPA may
synergize with trastuzumab to inhibit HER2-overexpressing breast
cancer cell proliferation more strongly. The fact that
HER2-overexpressing breast cancer cells were more sensitive to VPA
than HER2-negative cells seems to be associated with the role of
HER2 as a client protein of Hsp90. We speculate VPA may disrupt the
function of Hsp90 indirectly through hyper-acetylation of Hsp70 and
downregulation of HER2 expression, thus suppressing cell
growth.
Our results showed that the class I and II HDAC
inhibitor VPA preferably inhibited cell proliferation and induced
cell cycle arrest and apoptosis of HER2-overexpressing breast
cancer cells. This effect might be a direct function of VPA as an
HDAC inhibitor. Moreover, we propose another mechanism of
anti-proliferation that might be related to its non-histone
acetylation effect by indirectly disrupting Hsp90 function via
acetylation of Hsp70 and leading to downregulation of its client
proteins, including HER2. We showed that, in addition to the direct
function of VPA in histone acetylation that results in cell cycle
arrest or apoptosis, its indirect function of acetylation of
non-histone proteins could result in the anti-proliferative
activity of VPA. To confirm this hypothesis, further study is
required.
Acknowledgements
We thank Lynn Kimlicka for editing the
manuscript.
Abbreviations:
|
VPA
|
valproic acid
|
|
HDAC
|
histone deacetylase
|
|
Hsp
|
heat shock protein
|
|
HER2
|
human epidermal receptor 2
|
|
HAT
|
histone acetyltransferase
|
|
ATP
|
adenosine triphosphate
|
|
ADP
|
adenosine diphosphate
|
|
TSA
|
trichostatin A
|
|
ER
|
estrogen receptor
|
|
RIPA
|
radio-immunoprecipitation
|
|
TUNEL
|
TdT-mediated dUTP nick-end
labeling
|
|
CBZ
|
carbamazepine
|
References
|
1
|
von Minckwitz G, Untch M, Blohmer JU,
Costa SD, Eidtmann H, Fasching PA, Gerber B, Eiermann W, Hilfrich
J, Huober J, et al: Definition and impact of pathologic complete
response on prognosis after neoadjuvant chemotherapy in various
intrinsic breast cancer subtypes. J Clin Oncol. 30:1796–1804. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fang JY and Lu YY: Effects of histone
acetylation and DNA methylation on p21(WAF1) regulation. World J
Gastroenterol. 8:400–405. 2002.PubMed/NCBI
|
|
3
|
Jenuwein T and Allis CD: Translating the
histone code. Science. 293:1074–1080. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Davie JR and Moniwa M: Control of
chromatin remodeling. Crit Rev Eukaryot Gene Expr. 10:303–325.
2000. View Article : Google Scholar
|
|
5
|
Bolden JE, Peart MJ and Johnstone RW:
Anticancer activities of histone deacetylase inhibitors. Nat Rev
Drug Discov. 5:769–784. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Monneret C: Histone deacetylase
inhibitors. Eur J Med Chem. 40:1–13. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sami S, Höti N, Xu HM, Shen Z and Huang X:
Valproic acid inhibits the growth of cervical cancer both in vitro
and in vivo. J Biochem. 144:357–362. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Krämer OH, Zhu P, Ostendorff HP,
Golebiewski M, Tiefenbach J, Peters MA, Brill B, Groner B, Bach I,
Heinzel T, et al: The histone deacetylase inhibitor valproic acid
selectively induces proteasomal degradation of HDAC2. EMBO J.
22:3411–3420. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Göttlicher M, Minucci S, Zhu P, Krämer OH,
Schimpf A, Giavara S, Sleeman JP, Lo Coco F, Nervi C, Pelicci PG,
et al: Valproic acid defines a novel class of HDAC inhibitors
inducing differentiation of transformed cells. EMBO J.
20:6969–6978. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hrzenjak A, Moinfar F, Kremser ML,
Strohmeier B, Staber PB, Zatloukal K and Denk H: Valproate
inhibition of histone deacetylase 2 affects differentiation and
decreases proliferation of endometrial stromal sarcoma cells. Mol
Cancer Ther. 5:2203–2210. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rocchi P, Tonelli R, Camerin C, Purgato S,
Fronza R, Bianucci F, Guerra F, Pession A and Ferreri AM:
p21Waf1/Cip1 is a common target induced by short-chain fatty acid
HDAC inhibitors (valproic acid, tributyrin and sodium butyrate) in
neuroblastoma cells. Oncol Rep. 13:1139–1144. 2005.PubMed/NCBI
|
|
12
|
Takai N and Narahara H, Takai N and
Narahara H: Human endometrial and ovarian cancer cells: Histone
deacetylase inhibitors exhibit antiproliferative activity, potently
induce cell cycle arrest, and stimulate apoptosis. Curr Med Chem.
14:2548–2553. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yu X, Guo ZS, Marcu MG, Neckers L, Nguyen
DM, Chen GA and Schrump DS: Modulation of p53, ErbB1, ErbB2, and
Raf-1 expression in lung cancer cells by depsipeptide FR901228. J
Natl Cancer Inst. 94:504–513. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Blagosklonny MV, Robey R, Sackett DL, Du
L, Traganos F, Darzynkiewicz Z, Fojo T and Bates SE: Histone
deacetylase inhibitors all induce p21 but differentially cause
tubulin acetylation, mitotic arrest, and cytotoxicity. Mol Cancer
Ther. 1:937–941. 2002.PubMed/NCBI
|
|
15
|
Catalano MG, Poli R, Pugliese M, Fortunati
N and Boccuzzi G: Valproic acid enhances tubulin acetylation and
apoptotic activity of paclitaxel on anaplastic thyroid cancer cell
lines. Endocr Relat Cancer. 14:839–845. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fortunati N, Bertino S, Costantino L,
Bosco O, Vercellinatto I, Catalano MG and Boccuzzi G: Valproic acid
is a selective antiproliferative agent in estrogen-sensitive breast
cancer cells. Cancer Lett. 259:156–164. 2008. View Article : Google Scholar
|
|
17
|
Travaglini L, Vian L, Billi M, Grignani F
and Nervi C: Epigenetic reprogramming of breast cancer cells by
valproic acid occurs regardless of estrogen receptor status. Int J
Biochem Cell Biol. 41:225–234. 2009. View Article : Google Scholar
|
|
18
|
Zhang L, Wang G, Wang L, Song C, Leng Y,
Wang X and Kang J: VPA inhibits breast cancer cell migration by
specifically targeting HDAC2 and down-regulating Survivin. Mol Cell
Biochem. 361:39–45. 2012. View Article : Google Scholar
|
|
19
|
Nimmanapalli R, Fuino L, Bali P,
Gasparetto M, Glozak M, Tao J, Moscinski L, Smith C, Wu J, Jove R,
et al: Histone deacetylase inhibitor LAQ824 both lowers expression
and promotes proteasomal degradation of Bcr-Abl and induces
apoptosis of imatinib mesylate-sensitive or -refractory chronic
myelogenous leukemia-blast crisis cells. Cancer Res. 63:5126–5135.
2003.PubMed/NCBI
|
|
20
|
Hubbert C, Guardiola A, Shao R, Kawaguchi
Y, Ito A, Nixon A, Yoshida M, Wang XF and Yao TP: HDAC6 is a
microtubule-associated deacetylase. Nature. 417:455–458. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bali P, Pranpat M, Bradner J, Balasis M,
Fiskus W, Guo F, Rocha K, Kumaraswamy S, Boyapalle S, Atadja P, et
al: Inhibition of histone deacetylase 6 acetylates and disrupts the
chaperone function of heat shock protein 90: A novel basis for
antileukemia activity of histone deacetylase inhibitors. J Biol
Chem. 280:26729–26734. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bali P, Pranpat M, Swaby R, et al:
Activity of suberoylanilide hydroxamic Acid against human breast
cancer cells with amplification of her-2. Clin Cancer Res.
11:6382–6389. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kovacs JJ, Murphy PJ, Gaillard S, Zhao X,
Wu JT, Nicchitta CV, Yoshida M, Toft DO, Pratt WB and Yao TP: HDAC6
regulates Hsp90 acetylation and chaperone-dependent activation of
gluco-corticoid receptor. Mol Cell. 18:601–607. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Powers MV, Clarke PA and Workman P: Death
by chaperone: HSP90, HSP70 or both? Cell Cycle. 8:518–526. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pratt WB and Toft DO: Regulation of
signaling protein function and trafficking by the hsp90/hsp70-based
chaperone machinery. Exp Biol Med (Maywood). 228:111–133. 2003.
|
|
26
|
Boyault C, Zhang Y, Fritah S, Caron C,
Gilquin B, Kwon SH, Garrido C, Yao TP, Vourc'h C, Matthias P, et
al: HDAC6 controls major cell response pathways to cytotoxic
accumulation of protein aggregates. Genes Dev. 21:2172–2181. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang Y, Li N, Caron C, Matthias G, Hess
D, Khochbin S and Matthias P: HDAC-6 interacts with and
deacetylates tubulin and microtubules in vivo. EMBO J.
22:1168–1179. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Furumai R, Matsuyama A, Kobashi N, Lee KH,
Nishiyama M, Nakajima H, Tanaka A, Komatsu Y, Nishino N, Yoshida M,
et al: FK228 (depsipeptide) as a natural prodrug that inhibits
class I histone deacetylases. Cancer Res. 62:4916–4921.
2002.PubMed/NCBI
|
|
29
|
Neckers L: Hsp90 inhibitors as novel
cancer chemotherapeutic agents. Trends Mol Med. 8(Suppl): S55–S61.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Giacinti L, Giacinti C, Gabellini C,
Rizzuto E, Lopez M and Giordano A: Scriptaid effects on breast
cancer cell lines. J Cell Physiol. 227:3426–3433. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Olsen CM, Meussen-Elholm ET, Røste LS and
Taubøll E: Antiepileptic drugs inhibit cell growth in the human
breast cancer cell line MCF7. Mol Cell Endocrinol. 213:173–179.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chavez-Blanco A, Perez-Plasencia C,
Perez-Cardenas E, Carrasco-Legleu C, Rangel-Lopez E, Segura-Pacheco
B, Taja-Chayeb L, Trejo-Becerril C, Gonzalez-Fierro A, Candelaria
M, et al: Antineoplastic effects of the DNA methylation inhibitor
hydralazine and the histone deacetylase inhibitor valproic acid in
cancer cell lines. Cancer Cell Int. 6:22006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hodges-Gallagher L, Valentine CD, Bader SE
and Kushner PJ: Inhibition of histone deacetylase enhances the
anti-proliferative action of antiestrogens on breast cancer cells
and blocks tamoxifen-induced proliferation of uterine cells. Breast
Cancer Res Treat. 105:297–309. 2007. View Article : Google Scholar
|
|
34
|
Li GF, Qian TL, Li GS, Yang CX, Qin M,
Huang J, Sun M and Han YQ: Sodium valproate inhibits MDA-MB-231
breast cancer cell migration by upregulating NM23H1 expression.
Genet Mol Res. 11:77–86. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Huang X, Gao L, Wang S, Lee CK, Ordentlich
P and Liu B: HDAC inhibitor SNDX-275 induces apoptosis in
erbB2-overexpressing breast cancer cells via down-regulation of
erbB3 expression. Cancer Res. 69:8403–8411. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang Y, Wang SY, Zhang XH, Zhao M, Hou CM,
Xu YJ, Du ZY and Yu XD: FK228 inhibits Hsp90 chaperone function in
K562 cells via hyperacetylation of Hsp70. Biochem Biophys Res
Commun. 356:998–1003. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Fuino L, Bali P, Wittmann S, Donapaty S,
Guo F, Yamaguchi H, Wang HG, Atadja P and Bhalla K: Histone
deacetylase inhibitor LAQ824 down-regulates Her-2 and sensitizes
human breast cancer cells to trastuzumab, taxotere, gemcitabine,
and epothilone B. Mol Cancer Ther. 2:971–984. 2003.PubMed/NCBI
|
|
38
|
Scott GK, Marden C, Xu F, Kirk L and Benz
CC: Transcriptional repression of ErbB2 by histone deacetylase
inhibitors detected by a genomically integrated ErbB2
promoter-reporting cell screen. Mol Cancer Ther. 1:385–392.
2002.PubMed/NCBI
|
|
39
|
Meng Q, Chen X, Sun L, Zhao C, Sui G and
Cai L: Carbamazepine promotes Her-2 protein degradation in breast
cancer cells by modulating HDAC6 activity and acetylation of Hsp90.
Mol Cell Biochem. 348:165–171. 2011. View Article : Google Scholar
|