Introduction
HCC has been recognized as a hypervascular cancer
(1). The growth of a liver tumor
requires the formation of new blood vessels, which has provided a
strong rationale for anti-angiogenic strategies as therapy
(2,3). Angiogenesis plays an important role
in tumor growth by supplying nutrients and providing a route for
tumor growth, invasion and metastasis (4,5).
Therefore, targeting tumor angiogenesis might be a good option for
HCC treatment. Moreover, a series of signaling pathways
participating in the development of micro-vasculacture have been
identified (5).
Studies have shown that VEGF is strongly involved in
the development of liver tumor vascularization and the infiltration
of cancer cells into the tumor capsule in HCC (2,6).
VEGF can be derived from different sources such as stromal cells,
extracellular matrix, and cancer cells. The VEGF/VEGFR signaling
pathway is essential for drawing endothelial cells from
pre-existing blood vessels and in stimulating their growth. A
number of studies have shown that the vascular endothelial cells
can affect angiogenesis by induction of a number of cytokines such
as VEGF (7,8). Therefore, suppression of VEGF
released from endothelial cells is of vital importance in the
inhibition of tumor angiogenesis, which may achieve enhanced tumor
shrinkage.
Previous studies have shown that PI3K/AKT signaling
regulates angiogenesis through affecting the expression of VEGF
(9–12). It may contribute to tumor
angiogenesis via a paracrine pathway to the surrounding
microvessels by targeting endothelial cells (13,14).
In endothelial cells, the majority of growth factor-induced
responses are mediated by the activation of the PI3K-AKT signaling
cascade (15,16). First, AKT is known to mediate
hypoxia-induced expression of VEGF in vitro and in
vivo (17,18). Studies suggest that the level of
active AKT, as well as its short-term and long-term activation
states, in vascular cells can regulate various signaling pathways
to affect the balance of pro- and anti-angiogenic factors (19–21).
In addition to VEGF, AKT might affect the protein levels and
activities of several key regulators of angiogenesis, including
Anhiopoetin (15,22). Thus, AKT/VEGF pathway may
constitute a therapeutic target for angiogenesis anomaly in
HCC.
Sorafenib, serving as a tyrosine kinase inhibitor,
mainly exerts its anti-angiogenic effect by targeting VEGFR-2, -3,
PDGFR. However, its effect on inhibiting angiogenesis is limited,
as it only improves overall survival of HCC patients by no more
than three months (23,24). Thus, combination with other drugs
which may augment the anti-angiogenic effect of sorafenib should be
encouraged.
Bufalin, extracted from Chinese herbs, has been
proven to induce apoptosis, inhibit proliferation and metastasis
among various cancers (25).
However, little information exists concerning the anti-angiogenic
role of bufalin. An earlier study has demonstrated that the in
vitro angioinhibitory action of bufalin may be induced by the
proliferation inhibition of endothelial cells through the arrest at
the G2/M phase of a cell cycle (26).
Still, it was unknown whether bufalin could enhance
anti-angiogenic effect of sorafenib. Moreover, the underlying
mechanisms involved in this process remain to be clearly defined.
Therefore, in the present study we investigated bufalin-mediated
regulation of angiogenesis and whether it could enhance the
anti-angiogenic effect of sorafenib.
Materials and methods
Reagents and antibodies
Bufalin and dimethyl sulfoxide (DMSO) were obtained
from Sigma Chemical Co. (St. Louis, MO, USA). Sorafenib was
purchased from Bayer Corp. (West Haven, CT, USA). Antibodies
against VEGF were purchased from Bioworld Technology (Minneapolis,
MN, USA). The specific primary antibodies for mTOR,
phosphorylated-mTOR (p-mTOR), AKT, phosphorylated-AKT (p-AKT),
ERK1/2, phosphorylated-ERK1/2 (p-ERK1/2), CD31, and GAPDH were
purchased from Cell Signaling Technology (Danvers, MA, USA); The
PI3K/AKT inhibitor PI103 was purchased from Selleck Chemicals LLC
(Houston, TX, USA). Human Angiogenesis Array Q1 was purchased from
RayBiotech (Norcross, GA, USA).
Cell culture
HUVECs originated from the American Type Culture
Collection (ATCC) and were cultured in DMEM containing 10% fetal
bovine serum (FBS; Biochrom, Berlin, Germany) in 5% CO2
at 37°C. The human HCC cell lines SMMC7721 also originated from the
American Type Culture Collection (ATCC) and were cultured in
RPMI-1640 containing 10% FBS in 5% CO2 at 37°C.
Abdominal tumor model
Six-week-old BALBc nu/nu mice were obtained from the
Shanghai Institute of Material Medica, Chinese Academy of Science.
The mice were bred in laminar flow cabinets under pathogen-free
conditions. We followed internationally recognized guidelines on
animal welfare. The study design was approved by the Animal Ethics
Committee, and the experiments were undertaken in accordance with
the ethical principles of the Animal Experimentation of Fudan
University. The SMMC7721 cells (5×106) were
subcutaneously inoculated into the abdominal intraderma of
6-week-old BALBc nu/nu mice. According to tumor size, the mice were
randomly separated into four groups with three mice per group. The
mice in the experimental group received intra-peritoneal injections
of 1 mg/kg bufalin (5 days/week), oral uptake of 30 mg/kg/day
sorafenib (5 days/week), and the combination of both
intraperitoneal injections of bufalin and oral uptake of sorafenib.
The control mice were injected with the vehicle only. The treatment
was continued for two weeks. Then, the mice were sacrificed, and
the tumors and blood vessel were observed.
Subcutaneous tumor model
To determine the in vivo anti-angiogenic
activity of the combination treatment, viable SMMC7721 cells
(5×106) were subcutaneously inoculated into the right
flank of 6-week-old BALBc nu/nu mice. When the average subcutaneous
tumor volume reached 100–300 mm3, the mice were randomly
divided into four groups as indicated, with six mice per group.
Tumor size was measured every four days after the treatment.
Tumor-bearing mice were sacrificed after 16 days of treatment, and
the tumor weight was evaluated.
Cell viability assay
The cell proliferation analysis was performed as
previously described (27).
Briefly, cells were plated at 5000 cells per well in 96-well
microtiter plates and incubated overnight at 37°C in a humidified
incubator containing 5% CO2. The following day, various
concentrations of drugs were added to the wells, and the cultures
were incubated for an additional 24, 48, or 72 h. Cell viability
was determined using a Cell Counting Kit-8 (Dojindo, Gaithersburg,
MD, USA) according to the manufacturer's instructions.
Cell cycle and apoptosis assay
HUVECs were seeded in 6-well plates at a density of
2×105 cells/well and were subjected to different
treatments for 48 h. Then cells were trypsinized, washed with PBS
and fixed in 70% methanol. Fixed cells were then washed with PBS,
incubated with 100 μg/ml RNAase for 30 min at 37°C, stained with
propidium iodide (50 μg/ml). Cells were then subjected to flow
cytometry (FCM) using Beckman FACScanto. The percentages of cells
in different cell cycle phases were analyzed using ModFit LT
software. Cell apoptosis was determined by Annexin V and PI
staining via FCM. The apoptosis was also observed by PI staining
using Hoechst 33258.
Scratch wound assay
HUVECs were seeded in 6-well plates and grown to
near 100% confluency. The cells were scratched with a pipette tip
to create wounds. Treatment with bufalin (2.5 nM) and sorafenib
(2.5 μM) and their combination was given in serum-free medium after
the scratch was made. Randomly chosen fields were photographed at a
magnification of ×10 with an inverted microscope, and the images
were taken at identical locations at 48 h after treatment.
Percentage of wound was calculated by comparing the final gap width
to the initial gap width using Image pro-plus.
Cell migration assay
For the cell migration assay, the cell migration was
assessed using the Transwell assay (Boyden Chambers, Corning,
Cambridge, MA, USA). Cells (5×104) were seeded in
serum-free medium in the upper chamber and allowed to migrate
towards the lower chamber that contained 10% FBS. After 48 h, the
cells that had traveled through and adhered to the underside of the
membrane were counted at a magnification of ×200.
Tube formation
HUVECs were cultured and subjected to different
treatments as described above. At 48 h after treatment, cell
suspensions were collected as conditioned medium (CM). The CM was
collected after high speed centrifugation and stored at −80°C.
After thawed at 4°C overnight, the Matrigel was coated in 96-well
plate then incubated at room temperature for at least 30 min to
gel. HUVECs were seeded at density of 4×104
cells/well/100 μl on the Matrigel. HUVECs were cultured and exposed
to different treatments as indicated. After incubation for
indicated time, the formed networks at 96-well plate were
photographed. Minimum of three fields were analyzed per image.
Chorioallantoic membrane (CAM) of chick
embryos assay
The chicken chorioallantoic membrane (CAM) assay was
performed using eight-day-old fertilized chicken eggs. A 1-cm
diameter window was created in the shell of each egg and the
surface of the dermic sheet was removed to expose the CAM. A 0.5-cm
diameter filter paper was placed on top of the CAM, and a volume of
100 μl drug (control, bufalin, sorafenib, the combination) was
placed on the center of the filter paper. Then the windows in the
shell were closed using sterilized bandages. The eggs were
incubated at 37°C at 90% relative humidity for 48 h. Following
fixation with stationary solution (a mixture of methanol and
acetone with a volume ratio of 1:1) for 15 min, the CAM was excised
and imaged using a digital camera. The morphology of chicken blood
vessels with different treatments was detected.
Rat aortic ring assay
Thoracic aortae were first dissected from 8-week-old
Wistar rats and were then immediately transferred to a culture dish
containing cold DMEM containing 10% fetal bovine serum. The
periaortic fibroadipose tissue was carefully removed with fine
microdissecting forceps and iridectomy scissors, paying special
attention not to damage the aortic wall. Next, rings were embedded
in Matrigel. Once embedded, the rings were fed with DMEM every 2–3
days. Alternatively, different drugs described above were added to
the medium for 48 h to test their effects on angiogenesis. Rings
were photographed under clear field illumination by using an
inverted microscope.
Detection of cytokines related to
angiogenesis using Human Angiogenesis Array
To detect the cytokines related to angiogenesis in
the medium of HUVEC treated with different drugs, we performed
Human Angiogenesis Array. Cytokines closely linked with
angiogenesis were detected. The conditioned media from HUVECs were
placed into wells overlaid with antibody specific for these
cytokines. After binding with specific antibody, the absorbance was
measured at 450 nm using a microplate reader. The concentrations of
different cytokines were evaluated. Specific experimental
procedures were executed according to Human Angiogenesis Array Q1
kit instructions.
Enzyme-linked immunosorbent (ELISA)
assay
Suspensions from HUVECs with different treatments
were collected and the VEGF levels were measured using a sandwich
ELISA kit (R&D Systems, Minneapolis, MN, USA) according to the
manufacturer's instructions and analyzed using a Labsystems
Multiscan reader (Thermo Fisher Scientific, Waltham, MA, USA).
Western blotting
The HUVECs were exposed to the indicated
stimulations and washed with cold PBS and lysed in the culture
dishes using a PhosphoSafe™ Extraction Reagent (Merck, Darmstadt,
Germany) containing 1% Protease Inhibitor Cocktail (EDTA-free,
Thermo, San Jose, CA, USA). The protein concentrations were then
determined using the Bio-Rad detergent compatible protein assays
(Bio-Rad, Hercules, CA, USA). Protein from the control and treated
cell lysates was loaded onto 8–12% gradient NuPAGE gels (Novex, San
Diego, CA, USA), electrophoresed under reducing conditions, and
transferred onto polyvinylidene difluoride membranes (0.22 μm;
Millipore). Western blot analysis was performed as previously
described (27).
Immunohistochemistry
Tumor samples from in vivo studies were
rinsed in PBS and fixed in 10% paraformaldehyde/PBS. Samples were
dehydrated in 70% ethanol, paraffin embedded, and sectioned (4 μm).
Then these sections were stained with CD31 antibody. The mean
positive staining density was analyzed in the randomly selected
areas in each section using image analysis software.
Statistical analysis
The experiments reported herein were repeated in
triplicate. Independent Student's t-test was used to analyze the
variation of two selected groups. A P<0.05 was considered
statistically significant and P<0.01 was considered
highly statistically significant. All statistical analyses were
performed with SPSS software.
Results
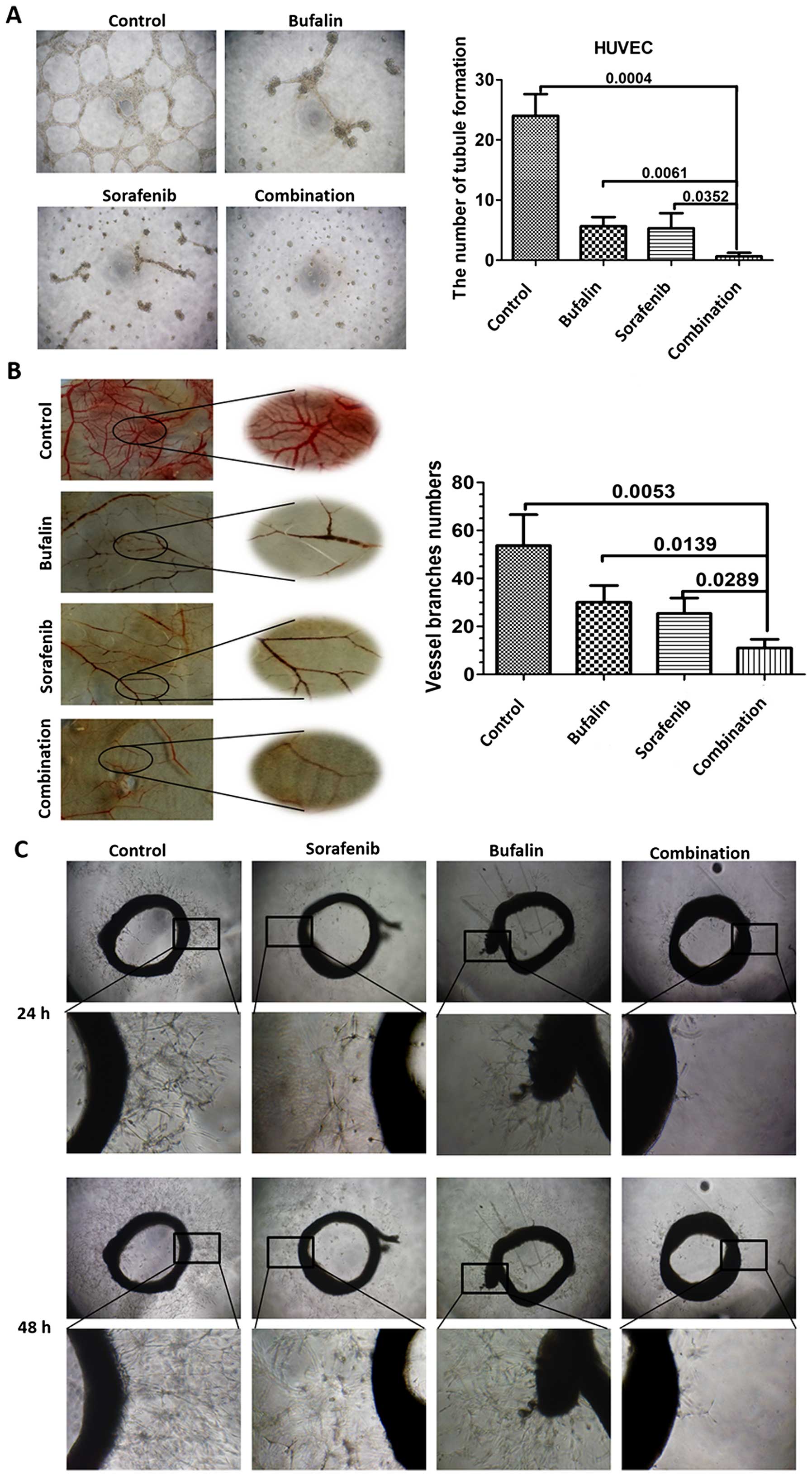
Combination treatment inhibits
intradermal tumor angiogenesis
In order to observe the effect of the combination
treatment on tumor angiogenesis, we used the intradermal tumor
angiogenesis model (28). SMMC7721
cell lines implanted intradermally in nude mice were found to
induce significant angiogenesis within a period of days until the
tumors could be measured. Intraperitoneal injection of bufalin at a
dose of 1 mg/kg/day and oral administration of sorafenib at a dose
of 30 mg/kg/day for consecutive 14 days inhibited blood vessel
formation in the intradermal tumors in nude mice, as manifested by
the vessel numbers and branches. Not surprisingly, the tumor vessel
formation was more inhibited in nude mice with the combination
treatment, as reflected in both vessel numbers and branches
(Fig. 1A and B). We also found
that the tumor weight was significantly attenuated in nude mice
with the combination treatment, as compared with the ones treated
with either bufalin or sorafenib (Fig.
1C).
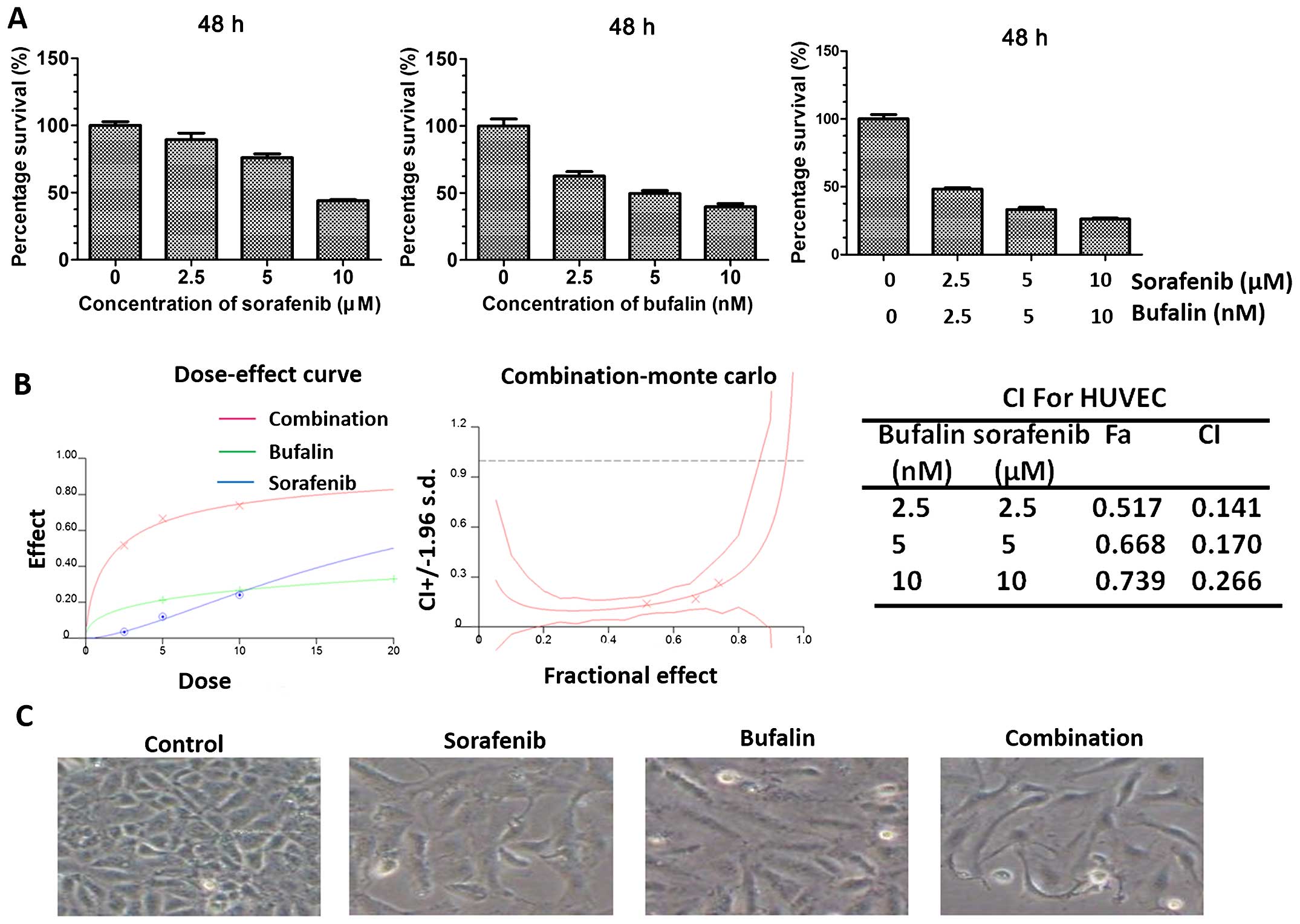
Synergistic inhibitory efficacy of
combination treatment on proliferation of HUVEC in vitro
HUVECs were subjected to the indicated treatments.
They were incubated with either sorafenib or bufalin and the
combination at various concentrations. Sorafenib at concentrations
of 2.5, 5 and 10 μM and bufalin at concentrations of 2.5, 5 and 10
nM were all applied. To test their synergistic effect on the growth
of HUVECs, the combination treatment with sorafenib and bufalin
after 48 h of exposure was also performed using CCK8 assay. The
combination of sorafenib and bufalin resulted in a significant
inhibition in cell growth when compared with either drug alone
(Fig. 2A). The combination index
(CI)/fractional effect curve showed that the synergistic effects
between these two agents became stronger (Fig. 2B). Besides, the morphology of
HUVECs with different treatments was observed. HUVECs with no
treatment were typically pebble-shaped while those with either
sorafenib or bufalin stimulation resembled fibro-blast morphology.
In addition, the combination treatment even gendered stronger
morphological changes (Fig.
2C).
Combination treatment leads to enhanced
inhibition of angiogenesis in blood vessel models
The effect of the combination treatment on
angiogenesis was evaluated. First, we investigated the impact of
different treatments on HUVEC tubule formation. Results revealed
that the combination treatment significantly decreased the
formation of tubule structures compared to those from other groups
(Fig. 3A). To confirm whether the
combination treatment has stronger angiogenic activity in
vivo, we investigated their effect in a chicken chorioallantoic
membrane (CAM) model. As seen from the images, sorafenib or bufalin
exerted a certain inhibitory effect in angiogenesis. However, the
combination treatment successfully disrupted the vasculature and
attenuated the thickness of the vessels (Fig. 3B). Next, arterial ring sprouting
experiment was applied and analyzed. The result showed that under
control conditions the aortic ring was able to generate neo-vessel
sprouting and the density of sprouting decreased when the
microvessels were incubated with either bufalin or sorafenib. The
neo-vessel sprouting almost vanished when incubated with the
combination (Fig. 3C).
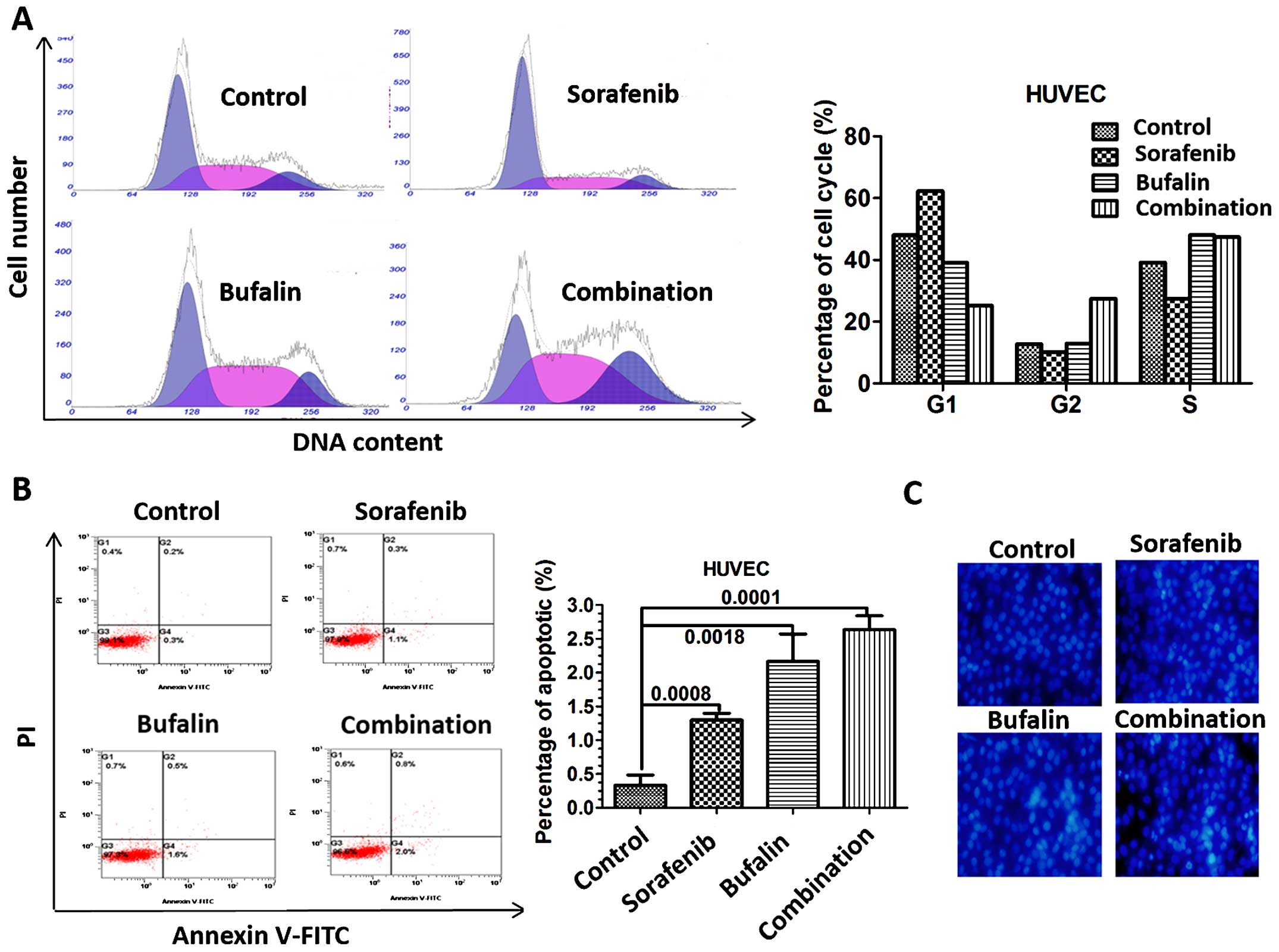
Combination treatment leads to cell cycle
arrest and apoptosis in HUVECs
As we have demonstrated the inhibitory effect of the
combination treatment on angiogenesis, specific mechanisms need to
be further analyzed. The percentage of cells in G2 phase increased
in HUVECs treated with the combination compared to HUVECs treated
with either agent alone (Fig. 4A).
We then determined the effect of sorafenib, bufalin and the
combination on apoptosis of HUVECs in vitro. To clarify the
effect of different treatments on apoptosis in HUVECs, flow
cytometry was employed. HUVECs treated with sorafenib and bufalin
experienced more apoptosis than the untreated ones, as quantified
by Annexin V and PI staining. Not surprisingly, the result showed
that apoptotic rate was significantly higher in HUVECs treated with
the combination of sorafenib and bufalin, suggesting that bufalin
cooperates with sorafenib to induce apoptosis of HUVECs (Fig. 4B). A drastic increase in the
apoptosis by the combination treatment was also confirmed, as
determined by the quantification of fluorescent intensity of
Hoechst 33258 via fluorescent microscope (Fig. 4C).
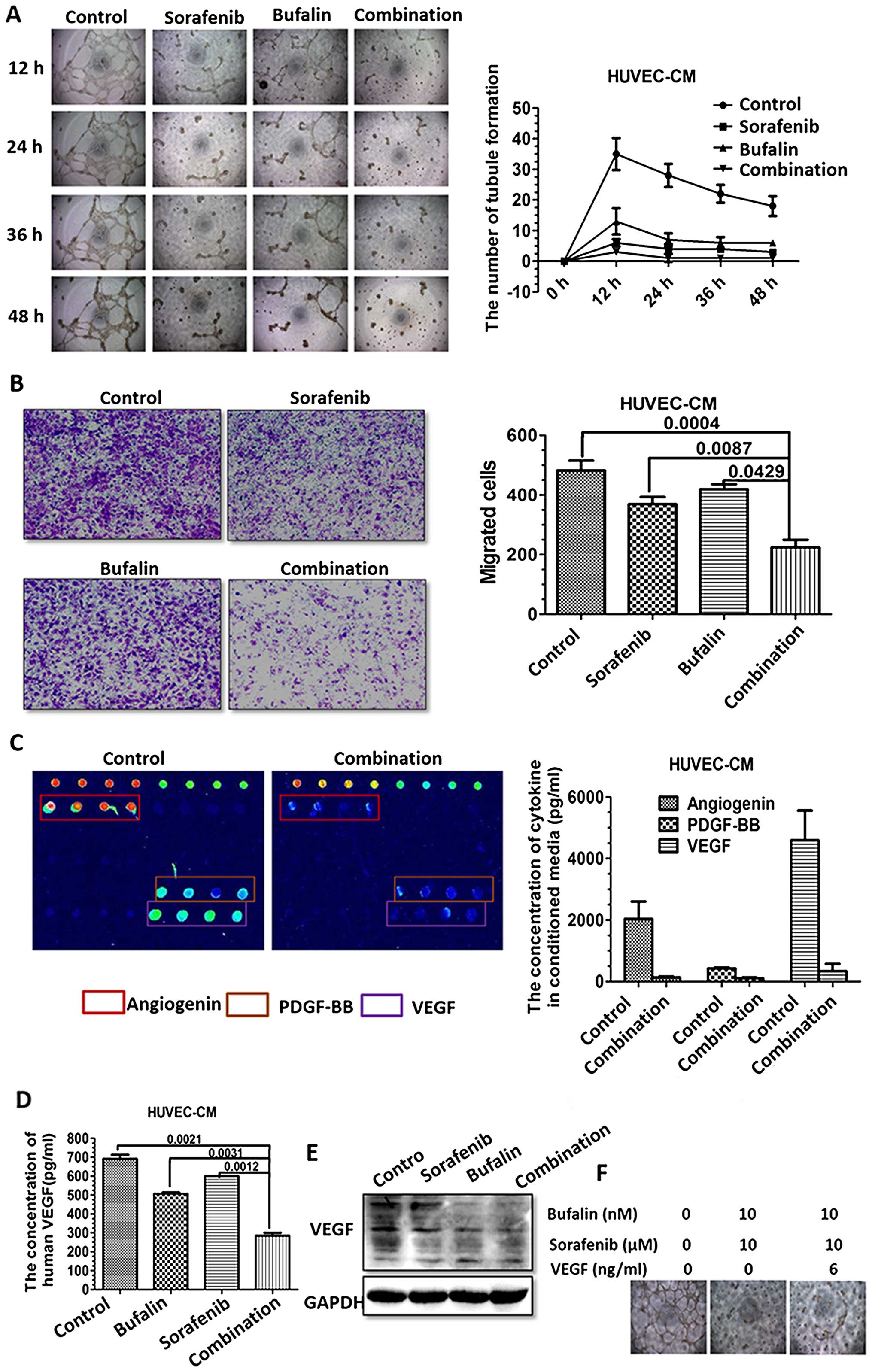
Combination treatment inhibits migration
in HUVECs
Since endothelial migration plays a critical role in
tumor angiogenesis, we next observed the migration ability of
HUVECs with the combination treatment. Wound healing was used to
study whether different treatments would impact migration of
HUVECs. The combination-treated HUVECs showed decreased motility
compared with the controls or single drug treatment group (Fig. 5A). Transwell assay was further used
to detect the migration in HUVEC after different treatments. The
combination treatment also showed a significant decrease in
migration compared with the controls or single drug treatment group
(Fig. 5B).
Combination-treated CM impairs HUVEC
angiogenesis and the expression of angiogenesis-related cytokines
in vitro
As we found that HUVECs showed increased apoptosis
with the combination treatment, we assume the anti-angiogenic
capacity of the combination treatment may, at least in part, be
attributed to the induced apoptosis acceleration. Vessel formation
is a process regulated by a network of cytokines, released from
endothelial cells in an autocrine manner. These cytokines may lead
to the maturation of the adjacent cells and migration, thus
affecting angiogenesis. Firstly, the effect of different treatments
on angiogenesis in HUVECs was investigated using the HUVEC tubule
formation assay. CM derived from HUVECs with different treatments
for 48 h were harvested. The effect of CM on tubule formation and
migration of HUVECs were observed at 12, 24, 36, 48 h after
incubation. CM derived from the combination treatment significantly
decreased the formation of tubule structures compared to CM from
other groups (Fig. 6A).
Moreover, CM from the combination treatment
significantly decreased the migration capacity of HUVECs, as
demonstrated in Transwell assay (Fig.
6B). Such phenomenon is a good indication of cytokine changes
in HUVECs with different treatments. Thus, the conditioned media
were collected and detected. Cytokines crucial for angiogenesis
were detected using the Human Angiogenesis Array. The array has
shown that angiogenic cytokines such as angiogenin, PDGF-BB and
VEGF vary significantly between the untreated and the
combination-treated group (Fig.
6C). As VEGF plays a central role in the regulation of
angiogenesis, we examined the expression of VEGF in untreated and
the combination-treated HUVECs. The concentration of VEGF was
significantly downregulated in the medium from HUVEC with the
combination treatment, as detected by ELISA (Fig. 6D). Additionally, the expression of
VEGFs proteins were detected in HUVECs. The expressions of VEGFs in
HUVECs as detected through western blotting were consistent with
the VEGFs released from CM (Fig.
6E). Noteworthy, the addition of VEGF reversed the inhibitory
effect on tube formation by the combination treatment, which was
indicative of the role of VEGF in vessel formation (Fig. 6F).
Combination treatment regulates
VEGF-mediated HUVEC via PI3K/AKT pathway
We have already proved that the combination
treatment-induced VEGF reduction may be one of the reasons leading
to attenuated angiogenesis. Therefore, the effects of different
treatments on pathways regulating VEGF were analyzed. Thus, we
investigated whether the AKT, mTOR and ERK pathways could be
affected by the combination treatment. For this purpose, we
performed western blotting to detect the activation of AKT, mTOR
and ERK in HUVECs. We found that HUVECs treated with bufalin showed
a significant decrease in AKT and mTOR phosphorylation and a
significant increase in ERK phosphorylation compared with the
control group. HUVECs treated with sorafenib showed a significant
decrease in ERK phosphorylation compared with the control group.
However, HUVECs treated with combination treatment showed a
significant decrease in AKT phosphorylation compared with the
control group or monotherapy group, implying that AKT may be
involved in the collaborative medication process (Fig. 7A).
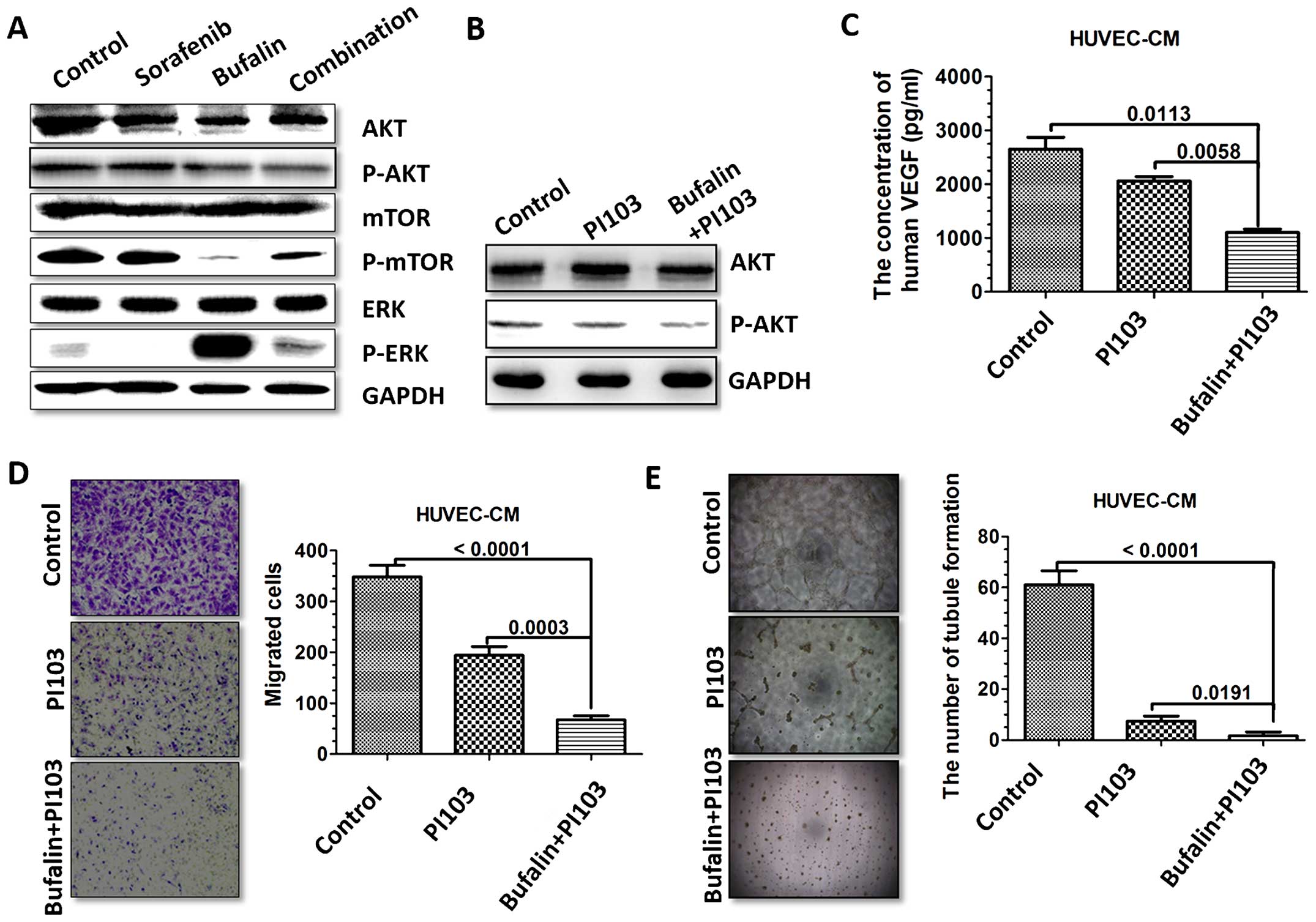 | Figure 7The combination treatment regulates
VEGF-mediated HUVEC via PI3K/AKT pathway. (A) The expression of
p-AKT, p-mTOR in PI3K/AKT/mTOR signaling pathway and p-ERK was
detected in HUVECs with different treatments via western blotting.
(B) HUVECs were treated with PI3K/AKT inhibitor, PI103, and
stimulated with bufalin for 48 h. HUVECs were harvested, and
western blotting was performed for detection of p-AKT. (C) HUVECs
were treated with PI3K/AKT inhibitor, PI103 (2 μM), and stimulated
with bufalin for 48 h. Secretions of VEGF of different treatments
were detected by ELISA. (D) HUVECs were treated with PI3K/AKT
inhibitor, PI103, and stimulated with bufalin for 48 h.
Representative images of the number of migrated HUVECs after
incubation in conditioned medium (CM) derived from different
treatments in the Transwell migration assay are shown. (E) HUVECs
were treated with PI3K/AKT inhibitor, PI103, and stimulated with
bufalin for 48 h. Representative images of the tubule formation
after incubation in CM derived from different treatment and
analysis of the number of tubule formation are shown. |
To further determine whether the AKT signaling
pathway mediated VEGF expression induced by combination treatment,
we treated HUVECs with the PI3K/AKT inhibitor PI103 (2 μM)
(Fig. 7B). We observed that VEGF
expression was significantly downregulated by PI103 treatment,
suggesting a role of the AKT pathway in regulating VEGF production
induced by combination treatment (Fig.
7C). We next evaluated the effect of the CM treated by PI103 on
HUVEC migration and tubule formation. Compared with untreated CM,
HUVEC migration and tubule formation were obviously suppressed by
the CM treated with PI103. Such phenomenon was even stronger in the
CM treated with PI103 combined with bufalin (Fig. 7D and E).
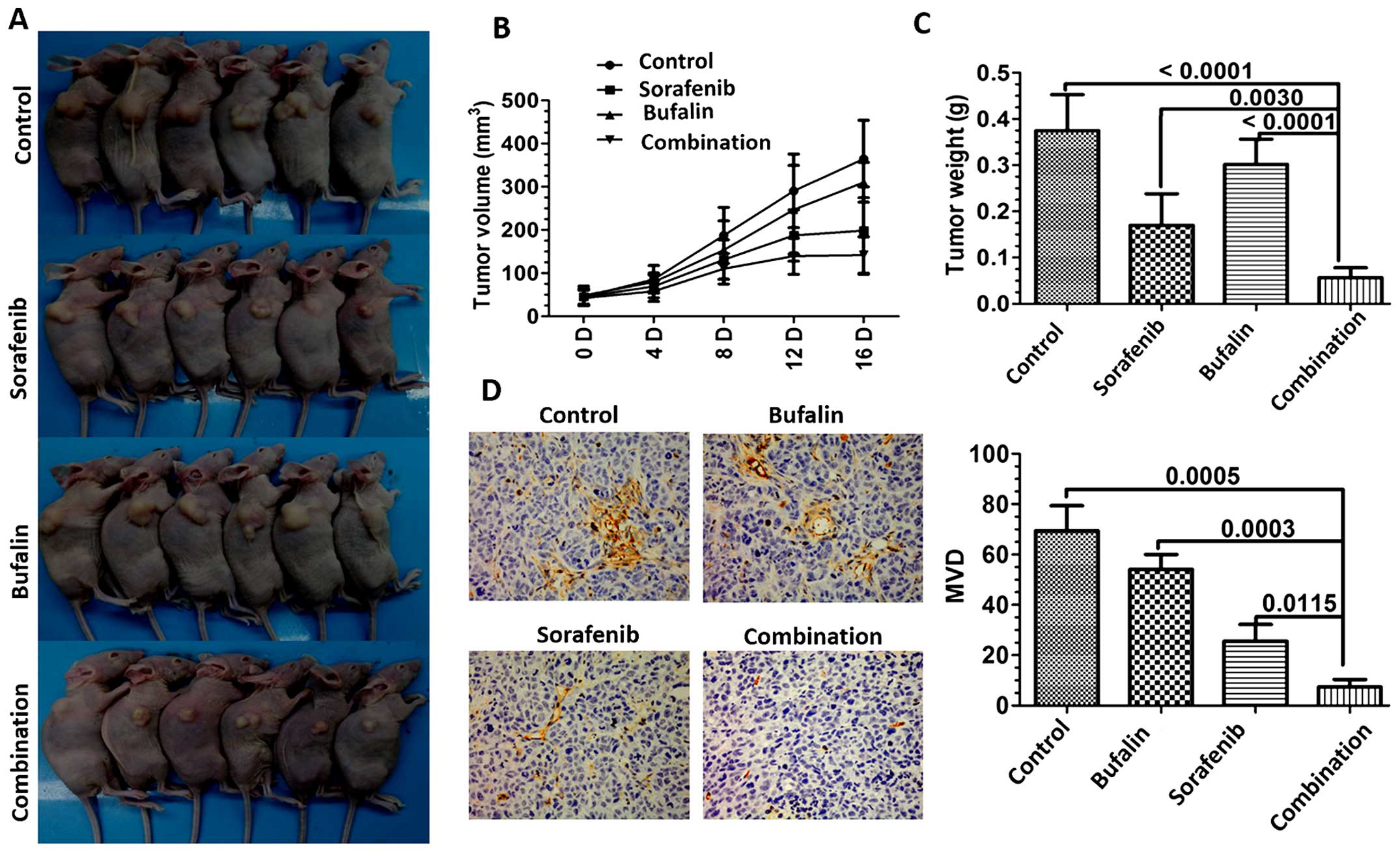
Combination treatment inhibits tumor
growth and tumor angiogenesis in vivo
To further investigate whether bufalin facilitates
the sorafenib antitumor activity in vivo, HCC tumor
xenografts were established in BALBc nu/nu mice. As shown in
Fig. 8A, sorafenib combined with
bufalin inhibited the growth of HCC-derived tumors more potently
than the individual group (Fig.
8A). Tumor volumes were smaller in the combination-treated
group than the individual group (Fig.
8B). Tumor weights of mice of all the groups were analyzed. As
shown in Fig. 8C, mice with the
combination treatment showed lower weights of tumor, as compared to
the other groups (Fig. 8C). Since
CD31 is the prominent endothelial marker which binds specifically
to blood microvessels, the tumors were then evaluated for
expression of CD31 by immunohistochemical analysis. The combination
treatment group showed reduced microvessel density (MVD) more than
any other group (Fig. 8D).
Discussion
At present, HCC remains a considerable challenge in
clinical practice, and current treatments are inadequate (29). Abundant blood supply has been noted
in HCC, which leads to the application of many anti-angiogenic
drugs in HCC treatments.
Sorafenib, a commonly known anti-angiogenic tyrosine
kinase inhibitor, has been approved for liver cancer. Approved by
the FDA to treat unresectable HCC, sorafenib is currently the main
drug used in the treatment of patients with advanced HCC (23). It is a small molecule that inhibits
tumor angiogenesis. It acts by inhibiting the serine-threonine
kinases Raf-1 and B-Raf and the receptor tyrosine kinase activity
of vascular endothelial growth factor receptors (VEGFRs) 1, 2 and 3
and platelet-derived growth factor receptor β. Sorafenib targets
VEGF receptors, and is now thought to exert its effect primarily by
blocking VEGF signaling, as its efficacy against B-Raf is
questionable. Although sorafenib has been shown to improve overall
survival from a median of 7.9 to 10.7 months in patients with
advanced HCC, its effect remains to be improved (24). Therefore, recently attention has
been focused on the finding and development of potent angiogenesis
inhibitors.
Bufalin, one of the prominent components of
bufadienolides, was reported to treat various tumors by inducing
apoptosis, inhibiting proliferation and matastasis (25). However, there are few
investigations concerning the effect of bufalin on angiogenesis. An
earlier study demonstrated that the in vitro angioinhibitory
action of bufalin may be induced by the proliferation inhibition of
endothelial cells through the arrest at the G2/M phase of a cell
cycle (26). Thus, we hypothesized
that they may exert more potent efficacy against angiogenesis than
either alone.
In the present study, we first examined the effect
of bufalin and sorafenib on angiogenesis of human HCC intradermal
tumor in nude mice. Results confirmed that the combination
treatment significantly inhibited tumor angiogenesis compared with
mice administered the vehicle or the individual treatment. Next, we
demonstrated that the combination application significantly
suppressed vessel formation as demonstrated in the tube formation,
chick chorioallantoic membrane and rat aortic rings. As tumor
angiogenesis can be modulated through apoptosis and migration of
endothelial cells, we anticipated that apoptosis and migration
alteration could be discovered in HUVECs with the combination
treatment. The assumption was validated, as tested in the apoptosis
and migration assay.
Considering the pro-angiogenic role of cytokines
released from endothelial cells, suspensions from HUVECs with
different treatments were collected as CM. The combination-treated
CM significantly inhibited the migration of HUVEC cells and blood
vessels formation in vitro. Of note, multiple cytokines
associated with angiogenesis were downregulated in the
combination-treated CM, as detected by Human Angiogenesis Array,
among which VEGF was the most saliently downregulated. The
inhibition of PI3K/AKT pathway upon the combination treatment was
observed, as evidenced by the evaluation of p-AKT. Finally, we
revealed that bufalin-induced VEGF reduction may be attributed to
the inhibition of the PI3K/AKT pathway, using PI3K inhibitor.
Together, our study may provide better insight of the application
of sorafenib in combination with bufalin.
Over the years, researchers have developed a wide
range of experimental and integrative approaches, including
conventional migration and proliferation assays, tubule formation
assays, the chick chorioallantoic membrane (CAM) assay, the aortic
ring and many other methods, to investigate the process of
angiogenesis (30). Among them,
the model of intradermal tumor angiogenesis was adopted according
to previous methods, as it may provide a more direct and vivid
result (28). Our results showed
that the combination treatment achieved enhanced effect against
tumor angiogenesis, as distinctly observed in this model.
It has been demonstrated that VEGF secreted by
stromal cells such as endothelial cells has multiple functions
(31). VEGF functions as a primary
stimulus for angiogenesis, which is a process that involves the
ability of VEGF receptors to stimulate signaling pathways that
induce the proliferation and the migration of endothelial cells
(32). The prevailing idea is that
therapies target angiogenesis and endothelial cell functions, and
this aspect of VEGF-targeted therapy has been extensively studied
(14,33,34).
VEGF binds to VEGF receptor, which leads to the activation of
phosphatidylinositol 3-kinase (PI3K)/AKT signaling pathway.
PI3K/AKT signaling regulates angiogenesis through affecting the
expression of VEGF (9–12,14).
In addition, forced expression of PI3K alone is sufficient to
increase angiogenesis via increased VEGF expression. The
phospholipid second messengers generated by PI3K provide a common
mechanism for multiple steps during angiogenesis (35,36).
Serine-threonine protein kinase AKT is a major downstream target of
PI3K for regulating tumor growth and angiogenesis (37,38).
For instance, PI3K/AKT may regulate angiogenesis by several
downstream targets such as mTOR/p70S6K1, FOXO, NOS, and GSK-3β
(39–41). A study has reported that Arnebin-1
promotes angiogenesis by inducing eNOS, VEGF and HIF-1 expression
through the PI3K-dependent pathway (42).
In this study, we found that sorafenib was able to
slightly decrease the level of VEGF expression in HUVECs. As
expected, such phenomenon was more salient when combined with
bufalin, as evidenced by VEGF expression in the HUVEC and HUVEC-CM
with different treatments. However, the exact mechanism of the
bufalin regulation of VEGF expression remains to be elucidated.
Previous studies have reported that PI3K/AKT stimulation of
angiogenesis is mediated, in part, by mTOR and HIF-1 (43–45).
Sustained endothelial activation of AKT has been shown to induce
the formation of structurally and functionally abnormal blood
vessels that recapitulate the aberrations of tumor vessels
(46). Besides, inhibition of AKT
signaling was able to inhibit the vascularization (47).
In search of mechanisms underlying the process, we
tested the effect of different treatments on p-AKT and p-mTOR in
PI3K/AKT/mTOR pathway and p-ERK expression. We found that p-AKT was
decreased while p-ERK was increased by the combination treatment.
It has been reported that p-AKT and ERK enhance the expression of
pro-angiogenesis target genes, such as VEGF, and then promote
endothelial cell migration and proliferation, thus contributing to
tumor angiogenesis (48–50). The seemingly paradoxical phenomenon
that the combination treatment enhances ERK phosphorylation can be
explained by the following: i) The combination-evoked increase in
p-ERK may be due to ER stress. ii) Its inhibition on other pathways
contributing to angiogenesis is more potent than its effect on
p-ERK activation, thus may offset this pro-angiogenesis process.
iii) Although the phosporylation of ERK may upregulate VEGF
expression, it may downregulate other angiogenic factors potently,
thus resulting in inhibition of angiogenesis.
The PI3K inhibition with PI103 was used to determine
whether the PI3K pathway has a role in AKT and VEGF expression and
whether combination treatment can decrease VEGF expression via
inhibiting the PI3K/AKT pathway. VEGF expression was almost
completely abolished by PI103, which suggest that the PI3K pathway
is a positive regulator of VEGF expression. The suppression of
HUVEC migration and tube formation by PI103, the PI3K inhibitor, as
well as bufalin implies that combination treatment may inhibit VEGF
expression via the PI3K/AKT pathway.
Collectively, the present study aids the
understanding of the antitumor effect of bufalin in HCC in terms of
its anti-angiogenic effect on HUVECs, which may support a
breakthrough in the use of TCM for the treatment of HCC. It also
uncovered an important functional role of bufalin in enhancing the
anti-angiogenic effect of sorafenib, suggesting that bufalin
combined with sorafenib can serve as an effective treatment for
anti-angiogenesis therapy in HCC.
Acknowledgements
We thank Huiying Chi (Shanghai Geriatric Institute
of Chinese Medicine, Longhua Hospital, Shanghai University of
Traditional Chinese Medicine, Shanghai) for the technical help in
the experiments. This study was supported jointly by the National
Natural Science Foundation of China (no. 81273954) and the Hundred
Talents Program of the Health System in Shanghai (no.
JGRC1302).
References
|
1
|
Sun HC and Tang ZY: Angiogenesis in
hepatocellular carcinoma: The retrospectives and perspectives. J
Cancer Res Clin Oncol. 130:307–319. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Folkman J: Angiogenesis: An organizing
principle for drug discovery? Nat Rev Drug Discov. 6:273–286. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Van de Veire S, Stalmans I, Heindryckx F,
Oura H, Tijeras-Raballand A, Schmidt T, Loges S, Albrecht I, Jonckx
B, Vinckier S, et al: Further pharmacological and genetic evidence
for the efficacy of PlGF inhibition in cancer and eye disease.
Cell. 141:178–190. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Weis SM and Cheresh DA: Tumor
angiogenesis: Molecular pathways and therapeutic targets. Nat Med.
17:1359–1370. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sharma PS, Sharma R and Tyagi T:
VEGF/VEGFR pathway inhibitors as anti-angiogenic agents: Present
and future. Curr Cancer Drug Targets. 11:624–653. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Carmeliet P and Jain RK: Angiogenesis in
cancer and other diseases. Nature. 407:249–257. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ribatti D, Ranieri G, Basile A, Azzariti
A, Paradiso A and Vacca A: Tumor endothelial markers as a target in
cancer. Expert Opin Ther Targets. 16:1215–1225. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Avramis IA, Kwock R and Avramis VI:
Taxotere and vincristine inhibit the secretion of the angiogenesis
inducing vascular endothelial growth factor (VEGF) by wild-type and
drug-resistant human leukemia T-cell lines. Anticancer Res.
21:2281–2286. 2001.PubMed/NCBI
|
|
9
|
Lee JH, Choi S, Lee Y, Lee HJ, Kim KH, Ahn
KS, Bae H, Lee HJ, Lee EO, Ahn KS, et al: Herbal compound
farnesiferol C exerts antiangiogenic and antitumor activity and
targets multiple aspects of VEGFR1 (Flt1) or VEGFR2 (Flk1)
signaling cascades. Mol Cancer Ther. 9:389–399. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yue GG, Fan JT, Lee JK, Zeng GZ, Ho TW,
Fung KP, Leung PC, Tan NH and Lau CB: Cyclopeptide RA-V inhibits
angiogenesis by down-regulating ERK1/2 phosphorylation in HUVEC and
HMEC-1 endothelial cells. Br J Pharmacol. 164:1883–1898. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li R, Zhao H and Lin Y: Anti-tumor effect
and protective effect on chemotherapeutic damage of water soluble
extracts from Hedyotis diffusa. J Chin Pharmaceut Sci. 11:pp.
54–58. 2002, http://118.145.16.238/Jwk_zgyxen/EN/abstract/abstract509.shtml.
|
|
12
|
Gupta S, Zhang D, Yi J and Shao J:
Anticancer activities of Oldenlandia diffusa. J Herb Pharmacother.
4:21–33. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Italiano JE Jr, Richardson JL, Patel-Hett
S, Battinelli E, Zaslavsky A, Short S, Ryeom S, Folkman J and
Klement GL: Angiogenesis is regulated by a novel mechanism: Pro-
and anti-angiogenic proteins are organized into separate platelet
alpha granules and differentially released. Blood. 111:1227–1233.
2008. View Article : Google Scholar
|
|
14
|
Kerbel RS: Tumor angiogenesis. N Engl J
Med. 358:2039–2049. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shiojima I and Walsh K: Role of Akt
signaling in vascular homeostasis and angiogenesis. Circ Res.
90:1243–1250. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kandel ES and Hay N: The regulation and
activities of the multi-functional serine/threonine kinase Akt/PKB.
Exp Cell Res. 253:210–229. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mazure NM, Chen EY, Laderoute KR and
Giaccia AJ: Induction of vascular endothelial growth factor by
hypoxia is modulated by a phosphatidylinositol 3-kinase/Akt
signaling pathway in Ha-ras-transformed cells through a hypoxia
inducible factor-1 transcriptional element. Blood. 90:3322–3331.
1997.PubMed/NCBI
|
|
18
|
Arsham AM, Plas DR, Thompson CB and Simon
MC: Akt and hypoxia-inducible factor-1 independently enhance tumor
growth and angiogenesis. Cancer Res. 64:3500–3507. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sun JF, Phung T, Shiojima I, Felske T,
Upalakalin JN, Feng D, Kornaga T, Dor T, Dvorak AM, Walsh K, et al:
Microvascular patterning is controlled by fine-tuning the Akt
signal. Proc Natl Acad Sci USA. 102:128–133. 2005. View Article : Google Scholar :
|
|
20
|
Shiojima I, Sato K, Izumiya Y, Schiekofer
S, Ito M, Liao R, Colucci WS and Walsh K: Disruption of coordinated
cardiac hypertrophy and angiogenesis contributes to the transition
to heart failure. J Clin Invest. 115:2108–2118. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nagoshi T, Matsui T, Aoyama T, Leri A,
Anversa P, Li L, Ogawa W, del Monte F, Gwathmey JK, Grazette L, et
al: PI3K rescues the detrimental effects of chronic Akt activation
in the heart during ischemia/reperfusion injury. J Clin Invest.
115:2128–2138. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang Q, Oh CK, Messadi DV, Duong HS,
Kelly AP, Soo C, Wang L and Le AD: Hypoxia-induced HIF-1 alpha
accumulation is augmented in a co-culture of keloid fibroblasts and
human mast cells: Involvement of ERK1/2 and PI-3K/Akt. Exp Cell
Res. 312:145–155. 2006. View Article : Google Scholar
|
|
23
|
Llovet JM, Ricci S, Mazzaferro V, Hilgard
P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A,
et al; SHARP Investigators Study Group. Sorafenib in advanced
hepatocellular carcinoma. N Engl J Med. 359:378–390. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wilhelm S, Carter C, Lynch M, Lowinger T,
Dumas J, Smith RA, Schwartz B, Simantov R and Kelley S: Discovery
and development of sorafenib: A multikinase inhibitor for treating
cancer. Nat Rev Drug Discov. 5:835–844. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Meng Z, Yang P, Shen Y, Bei W, Zhang Y, Ge
Y, Newman RA, Cohen L, Liu L, Thornton B, et al: Pilot study of
huachansu in patients with hepatocellular carcinoma, non-small-cell
lung cancer, or pancreatic cancer. Cancer. 115:5309–5318. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lee DY, Yasuda M, Yamamoto T, Yoshida T
and Kuroiwa Y: Bufalin inhibits endothelial cell proliferation and
angiogenesis in vitro. Life Sci. 60:127–134. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gao Y, Li HX, Xu LT, Wang P, Xu LY, Cohen
L, Yang PY, Gu K and Meng ZQ: Bufalin enhances the
anti-proliferative effect of sorafenib on human hepatocellular
carcinoma cells through downregulation of ERK. Mol Biol Rep.
39:1683–1689. 2012. View Article : Google Scholar
|
|
28
|
Wedge SR, Ogilvie DJ, Dukes M, Kendrew J,
Chester R, Jackson JA, Boffey SJ, Valentine PJ, Curwen JO, Musgrove
HL, et al: ZD6474 inhibits vascular endothelial growth factor
signaling, angiogenesis, and tumor growth following oral
administration. Cancer Res. 62:4645–4655. 2002.PubMed/NCBI
|
|
29
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar
|
|
30
|
Auerbach R, Lewis R, Shinners B, Kubai L
and Akhtar N: Angiogenesis assays: A critical overview. Clin Chem.
49:32–40. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Koch S, Tugues S, Li X, Gualandi L and
Claesson-Welsh L: Signal transduction by vascular endothelial
growth factor receptors. Biochem J. 437:169–183. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ferrara N: VEGF as a therapeutic target in
cancer. Oncology. 69(Suppl 3): 11–16. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Carmeliet P and Jain RK: Molecular
mechanisms and clinical applications of angiogenesis. Nature.
473:298–307. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Olsson AK, Dimberg A, Kreuger J and
Claesson-Welsh L: VEGF receptor signalling - in control of vascular
function. Nat Rev Mol Cell Biol. 7:359–371. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Skinner HD, Zheng JZ, Fang J, Agani F and
Jiang BH: Vascular endothelial growth factor transcriptional
activation is mediated by hypoxia-inducible factor 1alpha, HDM2,
and p70S6K1 in response to phosphatidylinositol 3-kinase/AKT
signaling. J Biol Chem. 279:45643–45651. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Graupera M, Guillermet-Guibert J, Foukas
LC, Phng LK, Cain RJ, Salpekar A, Pearce W, Meek S, Millan J,
Cutillas PR, et al: Angiogenesis selectively requires the p110alpha
isoform of PI3K to control endothelial cell migration. Nature.
453:662–666. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Staal SP: Molecular cloning of the akt
oncogene and its human homologues AKT1 and AKT2: Amplification of
AKT1 in a primary human gastric adenocarcinoma. Proc Natl Acad Sci
USA. 84:5034–5037. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tanaka H, Fujita N and Tsuruo T:
3-Phosphoinositide-dependent protein kinase-1-mediated IkappaB
kinase beta (IkkB) phosphorylation activates NF-kappaB signaling. J
Biol Chem. 280:40965–40973. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Gómez-Raposo C, Mendiola M, Barriuso J,
Casado E, Hardisson D and Redondo A: Angiogenesis and ovarian
cancer. Clin Transl Oncol. 11:564–571. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ellis L, Hammers H and Pili R: Targeting
tumor angiogenesis with histone deacetylase inhibitors. Cancer
Lett. 280:145–153. 2009. View Article : Google Scholar
|
|
41
|
Li Q, Michaud M, Canosa S, Kuo A and Madri
JA: GSK-3β: A signaling pathway node modulating neural stem cell
and endothelial cell interactions. Angiogenesis. 14:173–185. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zeng Z, Huang WD, Gao Q, Su ML, Yang YF,
Liu ZC and Zhu BH: Arnebin-1 promotes angiogenesis by inducing
eNOS, VEGF and HIF-1α expression through the PI3K-dependent
pathway. Int J Mol Med. 36:685–697. 2015.PubMed/NCBI
|
|
43
|
Marimpietri D, Nico B, Vacca A, Mangieri
D, Catarsi P, Ponzoni M and Ribatti D: Synergistic inhibition of
human neuroblastoma-related angiogenesis by vinblastine and
rapamycin. Oncogene. 24:6785–6795. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Nakamura K, Martin KC, Jackson JK, Beppu
K, Woo CW and Thiele CJ: Brain-derived neurotrophic factor
activation of TrkB induces vascular endothelial growth factor
expression via hypoxia-inducible factor-1alpha in neuroblastoma
cells. Cancer Res. 66:4249–4255. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Jiang BH, Jiang G, Zheng JZ, Lu Z, Hunter
T and Vogt PK: Phosphatidylinositol 3-kinase signaling controls
levels of hypoxia-inducible factor 1. Cell Growth Differ.
12:363–369. 2001.PubMed/NCBI
|
|
46
|
Bajaj A, Zheng Q, Adam A, Vincent P and
Pumiglia K: Activation of endothelial ras signaling bypasses
senescence and causes abnormal vascular morphogenesis. Cancer Res.
70:3803–3812. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Phung TL, Ziv K, Dabydeen D, Eyiah-Mensah
G, Riveros M, Perruzzi C, Sun J, Monahan-Earley RA, Shiojima I,
Nagy JA, et al: Pathological angiogenesis is induced by sustained
Akt signaling and inhibited by rapamycin. Cancer Cell. 10:159–170.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Takahashi T, Yamaguchi S, Chida K and
Shibuya M: A single autophosphorylation site on KDR/Flk-1 is
essential for VEGF-A-dependent activation of PLC-gamma and DNA
synthesis in vascular endothelial cells. EMBO J. 20:2768–2778.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Jiang BH and Liu LZ: PI3K/PTEN signaling
in angiogenesis and tumorigenesis. Adv Cancer Res. 102:19–65. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Claesson-Welsh L and Welsh M: VEGFA and
tumour angiogenesis. J Intern Med. 273:114–127. 2013. View Article : Google Scholar
|