Introduction
Cancer is a leading cause of death worldwide; 14.1
million new cancer cases were diagnosed in 2012, leading to 8.2
million deaths. Human CRC is the third most common cancer in the
world, with nearly 1.3 million new cases diagnosed in 2012
(1). Despite progress in diagnosis
and treatment, the incidence of CRC is increasing, and there is an
urgent need to identify new agents that suppress the growth of CRC
cells.
The cell cycle is controlled by a regulatory machine
and is tightly regulated in an orderly manner. Three different
transition points during the cell cycle, also known as cell-cycle
checkpoints, namely G1→S, G2→M and
M→G1, as well as the regulatory networks underlying each
transition, are well characterized (2). These cell cycle checkpoints are
regulated by the activities of cyclin-dependent kinases (CDKs),
which cooperate with different classes of cyclins. Specific
cyclin-CDK complexes in turn activate different downstream targets
to promote or prevent cell cycle progression (3). Proper cell cycle transitions are
crucial for the control of cell proliferation, and deregulation or
disruption of cell cycle modulation promotes unrestrained cell
growth, which can cause tumorigenesis. Cancer cells possessing
defective cell cycle checkpoints are defected due to mutations in
the p53 or pRb tumor suppressor genes or through imbalances in
cyclins, cyclin-dependent kinases (CDKs) and their inhibitors
(4). Therefore,
checkpoint-controlling molecules are potential therapeutic targets
for cancer and many checkpoint-modulating agents have been
developed (3,5–7).
The Myb family of transcription factors comprises
three isoforms, a-, b-, and c-Myb. c-Myb was first discovered in
avian myeloblastosis virus, which causes acute myeloblastic
leukemia and can transform hematopoietic cells in culture; a- and
b-Myb were subsequently cloned. Although b-Myb is ubiquitously
expressed, a- and c-Myb show tissue-specific expression patterns.
c-Myb is mainly expressed in hematopoietic stem cells, and a-Myb is
abundant in testes and in B-cells (8). Interestingly, b-Myb is expressed in
embryonic stem cells, and knocking out this gene results in
embryonic lethality in mice (9).
b-Myb is involved in cell growth, differentiation, apoptosis
(10), and senescence (11). In the cell-cycle, the
G2-M transition in cancer cells is regulated through the
activation of the cdc2-cyclin B1 complex, and b-Myb proteins are
involved in the regulation of cyclin B expression (12). b-Myb regulates cyclin B expression
by directly binding its promoter and also upregulates cdc2 and
cyclin B1 expression in conjunction E2F (13).
MicroRNAs (miRNA) are a major class of non-coding
RNAs, which mediate the posttranscriptional regulation of genes
that control multiple cellular processes (14). miRNA biogenesis is tightly
controlled by various regulatory proteins, including DROSHA, DICER
and AGO2 (15). Deregulation of
these regulatory proteins is observed in human cancer cells, and
many oncogenic miRNAs are significantly increased in cancer
(16). The ability of miRNAs to
target oncogenes provides an opportunity to discover antitumor
drugs that interrupt this oncogenic network (17). Cell cycle checkpoints are precisely
controlled by a number of regulatory proteins including cyclins and
CDKs, and recent studies indicated that many miRNAs are also
involved in cell cycle regulation (18). Specifically, miR-34, -29 and -30
family members regulate apoptosis by controlling cell cycle
pathways (19,20).
Ginkgetin is a natural biflavonoid, isolated from
the leaves of Ginkgo biloba, and we previously reported
antitumor activities of ginkgetin (21,22);
however, little information is available on the mechanism of the
anticancer activity of the natural product.
In this study, we examined the effects of ginkgetin
on cell cycle regulation and apoptosis in colorectal cancer cells,
as well as in vivo using human colon tumor xenografts in
nude mice. Our findings show that ginkgetin inhibits tumor growth
through cell cycle arrest and apoptosis via the regulation of b-Myb
and miR-34a expression.
Materials and methods
Reagents and antibodies
Ginkgetin was isolated from Ginkgo biloba
leaves (23,24). Ginkgetin (>95% purity by HPLC
and 1H-NMR) was dissolved in dimethylsulfoxide (DMSO) as
a stock solution, stored at −20°C, and diluted with medium before
each experiment. RPMI-1640, DMEM F-12, Opti-MEM reduced serum
medium, FBS, PBS, trypsin-EDTA, and penicillin-streptomycin
solution were purchased from Invitrogen (Grand Island, NY, USA).
Propidium iodide and the chemicals used in the buffer solutions
were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO,
USA). Primers and small interfering RNAs (siRNAs) were purchased
from Bioneer (Daejeon, Korea). The primary antibodies, anti-PARP,
and anti-β-actin were purchased from Cell Signaling Technology and
anti-b-Myb, anti-cyclin B1, anti-CDC2, anti-pCDC2 (Y15),
anti-pHisone H3 (S10) were purchased from Santa Cruz. The miRNA34a
mimic (hsa-miR-34a, SMI-002), inhibitor (hsa-miR-34a-5p, SMI-001),
miRNA mimic negative control (SMC-2001) and miRNA inhibitor
negative control (SMC-2101) were purchased from Bioneer.
Cell culture and cell viability
assay
HCT116, SW620, and HCA7 were purchased from ATCC and
maintained as monolayer cultures in media recommended by the ATCC.
To investigate cell proliferation, cells were seeded at a density
of 7,000 cells per well in 96-well plates in RPMI-1640 medium
containing 10% FBS. The medium was replaced with fresh complete
medium that contained the test compounds or 0.1% DMSO. After
incubation for 24 or 48 h, the cell proliferation reagent WST-1
(Dojindo Laboratories) was added to each well. WST-1 formazan was
quantitatively measured at 450 nm using an enzyme-linked
immunosorbent assay reader (Bio-Rad Laboratories, Inc., Hercules,
CA, USA).
Synchronization and
fluorescence-activated cell sorting (FACS) analyses
Cells were synchronized at metaphase by exposure to
nocodazole (500 ng/ml) for 16 h. After treatment, metaphase cells
were collected by a gentle shake-off method, centrifuged for 5 min
at room temperature, and washed twice with fresh medium. To relieve
cells from the M phase arrest, cells were replated in 60-mm cell
culture dishes (4×105/T25 flask) and incubated in fresh
medium for various time periods with or without ginkgetin. To
analyze the DNA content by flow cytometry, cells were trypsinized
from the culture flask. After centrifugation at 300 × g for 5 min
at room temperature, the supernatant was removed. The cells were
then washed twice with PBS solution and fixed overnight with 3 ml
of ice-cold 70% EtOH. Fixed cells were harvested by centrifugation
at 300 × g for 3 min at room temperature and washed twice with PBS.
Collected cells were resuspended in PBS (100
μl/1×105 cells) and treated with 100 μg/ml
of RNase A at 37°C for 30 min. Propidium iodide was then added to a
final concentration of 50 μg/ml for DNA staining and 20,000
fixed cells were analyzed on a FACSCalibur (Becton-Dickinson, San
Jose, CA, USA). Cell cycle distribution was analyzed using ModFit's
program (Becton-Dickinson).
Quantitation of mRNA using quantitative
real-time PCR
RNA was isolated using the RNeasy kit (Qiagen,
Hilden, Germany). Then, 1 μg of total RNA was transcribed
into cDNA with maxime PCR premix (iNTRON, Kyunggi-do, Korea).
Real-time PCR was performed using IQ™ SYBR Green supermix (Bio-Rad)
according to the manufacturer's instructions using an iQ5 real-time
PCR detection system. Triplicate 20 μl PCR reactions were
carried out with polymerase activation at real-time PCR cycles. The
following primers were used for real-time PCR: cyclin B1 forward
primer (5′-ATGGAACTAACTATGTTGGACTATG-3′), reverse primer
(5′-AGATTCTTCAGTATATGACAGGTAATG-3′), CDC2 primer (P149402), b-Myb
(P174647), and GAPDH (P267613). Primers were purchased from
Bioneer. Relative amounts of CDC2, cyclin B1 and b-Myb were
normalized against GAPDH mRNA.
Knockdown of cyclin B1 and B-Myb using
small interfering RNA in HCT116 cells
The sequences of each siRNA are as follows: b-Myb
(5′-GAAACGAGCCUGCCUUACATT-3′), cyclin B1
(5′-GAAUUCUGCACUAGUUCAA-3′) and the negative control
(5′-CCUACGCCACCAAUUUCUU-3′). Cells were plated at a density of
3×105 cells/well in 6-well plates and transfected with
50 nM b-Myb, cyclin B1 and negative control siRNAs after
pre-incubation for 20 min with RNAi max (Invitrogen, Carlsbad, CA,
USA) in serum-free Opti-MEM medium. RPMI-1640 medium containing 10%
FBS was added 5 h after the beginning of the incubation. After
transfection for 48 h, cells were collected and used for
preparation of whole cell lysates.
Western blot analysis
Cell lysates were prepared as reported previously.
Protein concentrations were determined using a Bio-Rad protein
assay kit (Bio-Rad) with BSA as a standard. Then, 30 μg of
protein lysates were subjected to 7.5 or 12% SDS-polyacrylamide gel
electrophoresis (SDS-PAGE) and transferred to polyvinylidene
difluoride membranes (Roche). Membranes were blocked with TBST (50
mM Tris-HCl pH 7.6, 150 mM NaCl, and 0.05% Tween-20) containing
blocking reagents (Roche) for 1 h. Antibodies were used at
dilutions recommended by the manufacturers. Horseradish
peroxidase-conjugated goat anti-rabbit or anti-mouse IgG from
Jackson ImmunoResearch was used as the secondary antibody. Blots
were incubated in primary antibodies for 2 h at room temperature
and washed three times in TBST. The proteins were visualized with
chemiluminescence peroxidase reagents (Roche Applied Science).
Western blot analyses were repeated 2 or 3 times.
Quantitation of microRNA
miR-29a, 29c, 30b, and miR-34a expression levels
were evaluated by using real-time PCR. miRNA expression values were
normalized by using specific primers to U6 as an endogenous
reference RNA. Real-time PCR was performed using IQ SYBR Green
supermix (Bio-Rad, Hercules, CA, USA) according to the
manufacturer's instructions using an iQ5 real-time PCR detection
system. The following primers were used for real-time PCR; miR-29a
(RT, GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACTAACCG; FP,
TCCACACGCATAGCACCATCTG), miR-29c (RT,
GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACTAACCG; FP,
ACACTCCACGCATAGCACCATT), miR-30b (RT,
GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACAGC; FP,
ATCACACTCCACGCATGTAAACATCCTA), miR-34a (RT,
GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACACAAC; FP,
CTCCACGCATGGCAGTGTCTT), U6 (RT, AACGCTTCACGAATTTGCGT; FP,
CTCGCTTCGGCAGCACA), URP (CCAGTGCAGGGTCCGAGGTA).
Knockdown and overexpression of
miRNA-34a
The miRNA34a mimic (hsa-miR-34a, SMI-002), inhibitor
(hsa-miR-34a-5p, SMI-001), miRNA mimic negative control (SMC-2001)
and miRNA inhibitor negative control (SMC-2101) were purchased from
Bioneer. HCT116 cells were plated at a density of 3×105
cells/well in 6-well plates and transfected with 20, 50 and 100 nM
negative control mimic, miR-34a mimic and 500 nM miRNA inhibitor
negative control, has-miR-34a-5p after pre-incubation for 20 min
with RNAi max (Invitrogen, Carlsbad, CA, USA) in serum-free
Opti-MEM medium. RPMI-1640 medium containing 10% FBS was added 5 h
after the beginning of the incubation. After transfection for 24 h,
cells were collected and used for preparation of RNAs for real-time
PCR and western blotting.
In vivo xenograft model
All animals were maintained in a specific
pathogen-free facility under protocols approved by the
Institutional Animal Care and Use Committee. Five- to six-week-old
female BALB/c nude mice (Oriental Bio., Korea) were used for animal
studies. Xenograft experiments with HCT116 cells were performed in
accordance with protocols approved by the Korea Research Institute
of Bioscience and Biotechnology (KRIBB) Animal Experimentation
Ethics Committee. To establish human colon tumors in mice,
1.2×107 HCT116 cells were subcutaneously implanted in
the right flank region of 6-week-old female S.P.F BALB/c nude mice.
Vehicle control and ginkgetin (10 mg/kg, intraperitoneal injection)
were administered five times per week for 23 days (8 mice/group).
Tumor sizes were calculated using the formula V = length × width ×
height × 0.5 mm3. Tumor volumes and weight at the end of
treatment were compared using Student's t-test.
Statistical analysis
The data are expressed as the means ± standard
deviations (SDs), and the degree of significance was analyzed using
Student's t-test. P-values <0.05 were considered statistically
significant.
Results
Ginkgetin inhibits tumor cell growth and
induces apoptosis
We previously reported that ginkgetin (Fig. 1A) has antitumor activities against
prostate cancer cells via the activation of the caspase pathway or
the inhibition of STAT3 activity (21,22).
To further understand the biological activity of ginkgetin on
cancer cells, we first investigated the inhibitory effect of
ginkgetin of tumor cells by a cell proliferation assay. A variety
of colon cancer cells (HCT116, HCA-7 and SW620) was treated with
different concentrations of ginkgetin for 48 h. As shown in
Fig. 1B, ginkgetin inhibited the
growth of HCT116, SW620, and HCA7 cells with GI50 values
of 4.0, 3.5 and 10 μM, respectively. Because ginkgetin
strongly inhibited the growth of HCT116 (colon tumor cells) in a
dose- and time-dependent manner, with a GI50 value of
4.0 μM for 48 h treatment (Fig.
1C), and the incidence of CRC is sharply increasing, we focused
on the biological activity of ginkgetin on HCT116 cells.
In order to determine whether the growth-inhibitory
effects of ginkgetin were caused by apoptotic death in HCT116
cells, the cleavage of poly(ADP-ribose) polymerase (PARP) were
carried out. Ginkgetin led to the cleavage of PARP (Fig. 1D). These results clearly suggest
that ginkgetin inhibits the growth of HCT116 cells by inducing
apoptosis.
Ginkgetin induces G2/M phase
arrest in HCT116 cells
Because the primary cellular response to
chemotherapy-induced DNA damage includes cell cycle arrest and
apoptosis, we performed fluorescence activated cell sorting (FACS)
analysis, which is a standard tool to analyze the cell cycle
distribution in cell populations. Cells were harvested at different
times (24 and 48 h) after treatment 5 or 10 μM of ginkgetin,
and the cell cycle distribution of treated cells was examined by
flow cytometry. As shown in Fig.
2, the G2/M fraction of ginkgetin-treated cells was
increased and the fraction of cells in G0/G1
phase was decreased in a dose- and time-dependent manner in HCT116
cells (Fig. 2). A 24 h treatment
of HCT116 cells with ginkgetin (5 or 10 μM) resulted in
G2/M distribution rates of 27.05 and 40.85%,
respectively. The G2/M distribution of HCT116 cells was
24.02 and 43.25% at 48 h after treatment with ginkgetin (5 or 10
μM), respectively. In particular, when HCT116 cells were
treated with 10 μM ginkgetin for 48 h, the percentage of
cells in G2/M phase increased by 2.2-fold (43.25%)
versus the untreated control (19.69%).
Previously, it was reported that ginkgetin induced
the accumulation of cells at the G0/G1 phase
through the inhibition of STAT3 activity in DU145 (prostate cancer)
cells, whereas, ginkgetin efficiently induced G2/M phase
arrest in Daoy (medulloblastoma) cells by inhibiting Wnt target
gene expression (21,24). It is interest that the different
types of cell cycle progression were observed in ginkgetin-treated
different tumor cells. Therefore, we examined whether ginkgetin
modulates cell cycle progression in HCT116 cells.
Ginkgetin specifically induces
G2 arrest in HCT116 cells
Cell cycle synchronization was used for observing
the cell cycle progression and many methods have been established
to synchronize cells at specific phases of the cell cycle (25). To identify whether the growth
inhibition by ginkgetin is due to the arrest of cells at
G2 or M phase, synchronize experiment was performed with
nocodazole, because the microtubule-depolymerizing agent
specifically blocks the cell cycle at the metaphase-anaphase
transition and allows for the collection of a cell population that
is in M phase (26).
To synchronize cells in M phase, HCT116 cells were
treated with nocodazole. M phase-synchronized cells were isolated
by selective detachment, washed to remove the nocodazole, and then
replated in medium with or without ginkgetin (10 μM). The
cells were harvested at the indicated times and the cell cycle
distribution was analyzed by FACS. If ginkgetin induces M phase
arrest, replating cells with ginkgetin should maintain the M phase
arrest. However, the synchronized HCT116 cells were completely
released from M phase arrest 3 h after replating cells both in the
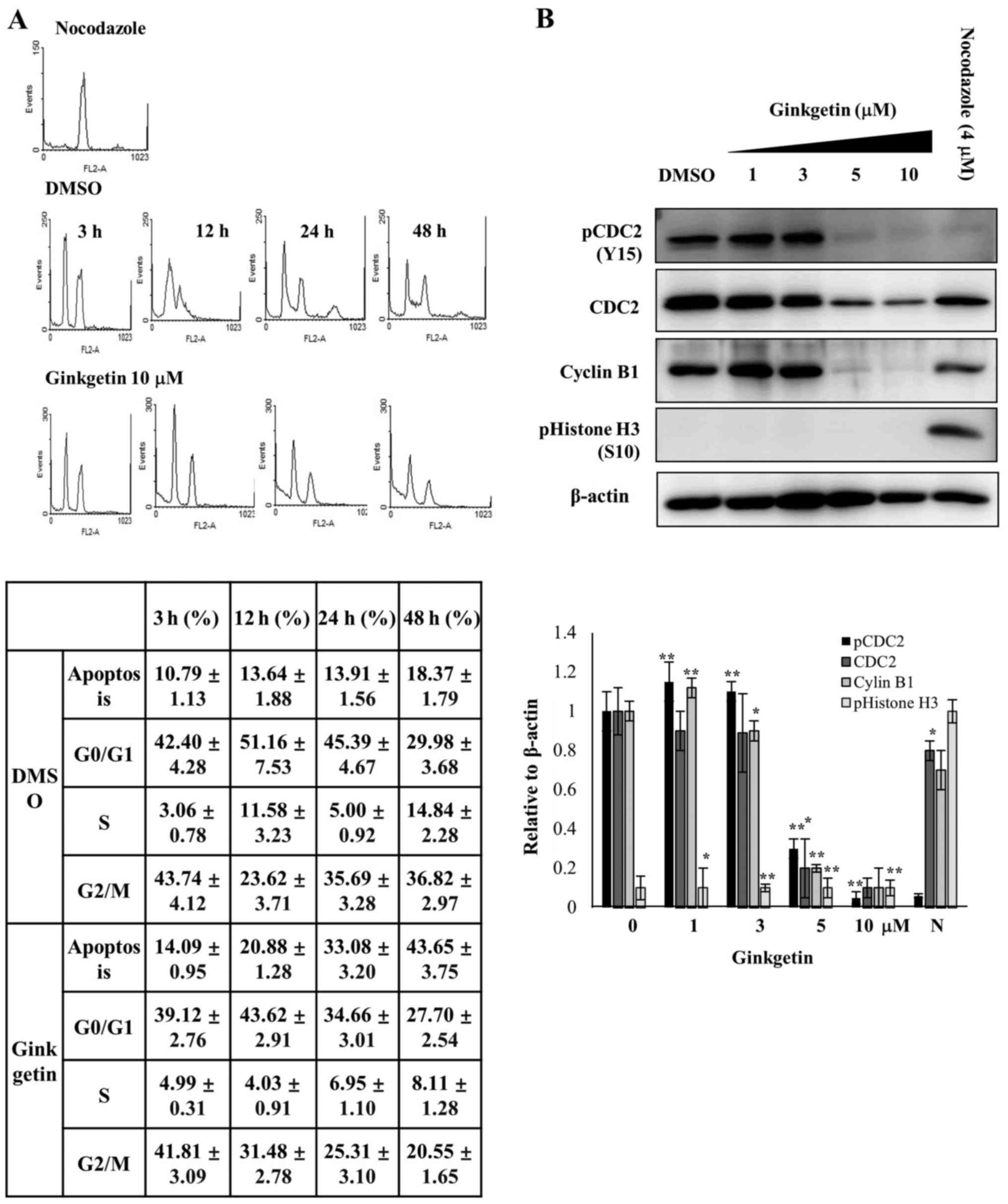
absence and the presence of ginkgetin (Fig. 3A). As shown in Fig. 3A, after 3 h of release, HCT116
cells without and with ginkgetin (10 μM) exhibited
G0/G1 distribution rates of 42.40 and 39.12%,
respectively. The G2/M or G2 distribution of
the HCT116 cells was 43.74 and 41.81% after 3 h of release without
and with ginkgetin, respectively. The apoptotic cell population was
slightly increased, from 10.79 to 18.37%, in HCT116 cells released
from nocodazole-induced arrest in the absence of ginkgetin,
however, cells released with ginkgetin showed an increased
population of apoptotic cells from 14.9 to 43.65% in a
time-dependent manner (Fig.
3A).
To investigate the mode of action of ginkgetin on
G2 cell cycle arrest, the protein levels of pCDC2 (Y15)
(also known as CDK1 kinase), CDC2 and cyclin B, which is important
components for arresting in G2 phase, were measured in
gink-getin-treated HCT116 cells. Immunoblotting demonstrated that
the levels of pCDC2 (Y15), CDC2 and cyclin B1 were decreased at the
concentrations of ginkgetin (5 and 10 μM) that induced
G2 arrest (Fig. 3B).
Phosphorylated histone H3 (S10) (PHH3) correlates closely with M
phase of the cell cycle (27). For
this reason, PHH3 is used as a mitosis-specific marker. After 24 h
of ginkgetin treatment, western blotting with a PHH3 antibody
showed that PHH3 was undetectable in ginkgetin-treated cells;
however, PHH3 was strongly detected in the nocodazole-treated
cells. In addition, CDC2 and cyclin B1 were also detected in
nocodazole-treated cells. These results indicate that ginkgetin
inhibits proliferation and induces apoptosis in HCT116 cells via
G2 phase arrest.
Ginkgetin induces G phase arrest by
modulation of b-Myb expression
As shown Fig. 3B,
ginkgetin markedly downregulated the key regulators of the
transition through G2 phase and entry into mitosis such
as cyclin B1 and CDC2. To confirm the G2 arrest by
ginkgetin, the relative mRNA expression levels of G2
arrest-related genes such as b-Myb, CDC2, cyclin G1, and
cyclin B1 were analyzed by quantitative real-time PCR in HCT116
cells treated with 7 μM ginkgetin. Untreated control cells
served as a reference (ratio = 1). Ginkgetin induced the
downregulation of b-Myb, CDC2, cyclin G1, and cyclin B1 mRNA levels
in a time-dependent manner (Fig.
4A). We also confirmed that the levels of G2
arrest-related proteins such as b-Myb, CDC2 and cyclin B1 were
downregulated by ginkgetin (Fig.
4B). As mentioned previously, b-Myb regulates the expression of
S and G2/M genes such as cyclin B and CDC2 (CDK1)
(28), and b-Myb is required for
the transcription of cyclin B1 (29). Furthermore, when b-Myb siRNA was
transfected into HCT116 cells, the cell growth rate was decreased
(Fig. 4C). Therefore, we focused
on the expression of b-Myb, CDC2 and cyclin B1 to investigate the
effects of ginkgetin on cell cycle progression.
We explored the effect of ginkgetin on G2
arrest-related genes, including b-Myb, CDC2 and cyclin B1 in HCT116
cells using siRNA gene knock-down experiments. Because b-Myb
regulates the transcription of cyclin B1 and CDC2, we tested
whether repression of b-Myb has reverse effect on the expression of
CDC2 and cyclin B1 by transfecting HCT116 cells with siRNA against
b-Myb. As shown in Fig. 4D, b-Myb
protein expression was completely abolished by b-Myb siRNA, and
cyclin B1 protein levels were also strongly decreased in repressed
cells. We also observed an approximately 40% reduction in the
amount of CDC2 in b-Myb-depleted cells. These results are
consistent with those in ginkgetin-treated cells (Fig. 4B). Next we repeated the experiments
using siRNA-mediated repression of cyclin B1 and detected no
effects on CDC2 protein levels, however, b-Myb protein levels were
decreased by ~50% compared with the negative-control cells
(Fig. 4D). Thus, b-Myb is pivotal
for ginkgetin-induced G2 arrest in HCT116 cells.
Ginkgetin regulates b-Myb expression by
modulating miR-34a expression
MicroRNAs can act as potent oncogenes or tumor
suppressors and can regulate the progression of cancer via cell
cycle control (18). Martinez
et al reported that overexpression of miR-29 and miR-30
inhibits b-Myb expression and reduces DNA synthesis (30) and an inverse correlation between
overexpression and downregulation of b-Myb has also been reported
(31). Therefore, we hypothesized
that ginkgetin downregulates b-Myb expression through the induction
of microRNAs and set out to characterize the mechanism of b-Myb
transcriptional repression by ginkgetin. Based on our hypothesis,
we measured the expression of several miRNAs, including miR-29a,
-29c, -30b and-34a, which are reported to regulate b-Myb
expression. As shown in Fig. 5A,
ginkgetin increased the expression levels of miR-29a, -29c, -30b
and-34a in HCT116 cells. In particular, we observed a significant
induction of miR-29c and -34a in response to ginkgetin treatment in
a dose- and time-dependent manner (Fig. 5A and B). To test the effect of the
miRNA on b-Myb expression in HCT116 cells, we transfected HCT116
cells with the precursor to miR-29c and-34a (designated miR-29c and
-34a mimic), and as control, the cells were transfected with
scrambled miRNA. We found that only the miR-34a mimic downregulated
b-Myb protein expression (data not shown). In addition, miR-34a was
upregulated in ginkgetin-treated cells a time-dependent manner.
Therefore, we selected miR-34a as a modulator of b-Myb in
ginkgetin-treated HCT116 cells. miR-34a mimic significantly
decreased b-Myb expression in a dose-dependent manner and b-Myb was
downregulated by >80% at 100 nM miR-34a mimic. An ~40% reduction
in cyclin B1 expression was also observed in 100 nM miR-34a
mimic-treated cells (Fig. 5C),
consistent with a previous report in which miR-34a overexpression
downregulated b-Myb expression (31). To confirm these results, we treated
HCT116 cells with a specific miR-34a inhibitor and measured the
expression levels of b-Myb and cyclin B1. miR-34a inhibitor
strongly induced the expression of endogenous b-Myb and also
markedly rescued the reduced b-Myb expression caused by ginkgetin
(Fig. 5D). miR-34a inhibitor also
increased the expression of endogenous cyclin B1 and rescued the
reduced cyclin B1 expression caused by ginkgetin. These results
suggest that ginkgetin exerts its anticancer activity in part
through a newly defined mechanism, i.e., the miR-34a/b-Myb
cascade.
Ginkgetin inhibits tumor growth in a
mouse xenograft model of HCT116 cells
To measure the ginkgetin efficacy in vivo, a
HCT116 tumor xenograft model of nude mice was used. HCT116 cells
were subcutaneously implanted into the right flank of a nude mouse
on day 0, and ginkgetin feeding was started at day 1 after HCT116
xenograft implantation. Vehicle (0.5% Tween-80) and ginkgetin in
0.5% Tween-80 were intraperitoneally administered five times per
week to tumor-bearing nude mice at a concentration of 10 mg/kg/day
for 20 days (eight mice/group). Compared with control (vehicle),
ginkgetin treatment suppressed tumor xenograft growth throughout
the study. On day 21, the mice were sacrificed, and tumor volume
and weight were measured. At the end of the experiment, tumor
volume per mouse was 456.7±54 mm3 in the
ginkgetin-treatment group compared with 716.4±63 mm3 in
the control group, reflecting a 36.5% decrease in tumor volume
(Fig. 6A). Consistent with this
observation, ginkgetin caused a 37.6% decrease in tumor weight
(P=0.01) compared with control (Fig.
6B). Ginkgetin did not affect body weight gain and diet
consumption profiles, which were almost identical to those of the
control group.
Discussion
We consume flavonoid-rich foods, which are widely
associated with the reduced risk of chronic diseases based on
evidence from epidemiological and human intervention studies
(32). However, whether isolated
flavonoids can confer the same benefits in preventing and
controlling chronic diseases is unclear; therefore, extensive
pre-clinical studies are needed to understand the mechanisms of
natural flavonoids against different types of human diseases
(33). Ginkgetin is a natural
biflavonoid isolated from the leaves of Ginkgo biloba, and
we previously reported on the antitumor activities of ginkgetin
(21,22); however, less is known about the
mechanisms underlying the anticancer activities of this
biflavonoid. In this study, we examined the effects of ginkgetin on
various tumor cells and found that ginkgetin inhibited the growth
of human tumor cells (Fig. 1A).
Based on these results, we investigated the effect of ginkgetin on
cell cycle regulation (Fig. 2),
inducing apoptosis (Fig. 1C and
D), as well as its in vivo anti-tumor activities against
human colon tumor (HCT116 cells) xenografts in nude mice (Fig. 6), because colon cancer is the
third-most prevalent cancer worldwide.
Many natural products including flavonoids, act on
the G2 checkpoint and induced G2/M cell cycle
arrest in human cancer cells (34), however, a few natural product
selectively induced G2 arrest in human cancer cells.
Recently, Liberio et al reported that eusynstyelamide B,
marine natural product, induced G2 cell cycle arrest and
increased the G2/M cell population without elevating the
levels of the mitotic marker PHH3 in MDA-MB-231 (breast cancer) and
LNCap (prostate cancer) cells (35). Although ginkgetin induced
G0/G1 phase arrest in DU145 cells
(STAT3-dependent prostate cancer) by inhibiting STAT3 activity and
downregulating cyclin D1 expression (21), we found that ginkgetin selectively
induced G2 cell cycle arrest, as confirmed by M
phase-synchronized experiments and using the mitotic marker PHH3 in
HCT116 colon cancer cells (Fig. 3A and
B). As shown Fig. 3, the
microtubule depolymerizing agent nocodazole, which arrests cells in
mitosis, increased the level of a mitotic marker PHH3; however, the
protein was not detected in ginkgetin-treated HCT116 cells by
western blotting. The mitosis-promoting activity of the cyclin
B/CDC2 kinase is a critical target for inducing G phase arrest, and
this activity is regulated by multiple G2 checkpoint
mediators, including the CDK inhibitor p21CIP1/WAF1,
CHK1, ATM, ATR, Polo-like kinases, and the CDC25 family (4). However, ginkgetin did not inhibit
kinase activity and has no effect on the expression of p21 (data
not shown). Interestingly, ginkgetin downregulated the expression
of cyclin B1 and CDC2 instead of inhibiting the activity of cyclin
B/CDC2 complexes (Fig. 4B). Cyclin
B1 expression levels began to decrease after 3 h of treatment,
followed by the loss of CDC2 and cyclin B1 protein levels that were
dramatically reduced after 24 h of treatment.
Therefore, we postulated the involvement of a
transcriptional regulator of cyclin B1 in the G phase arrest by
ginkgetin. At early stages of the cell cycle, a very small amount
of b-Myb binds to the cyclin B1 promoter; however, binding is
significantly increased at the end of S phase (28). b-Myb shRNA significantly decreased
the activity of the cyclin B1 promoter compared with control
shRNA-transfected cells (29). To
gain insight into the mechanism by which ginkgetin regulates b-Myb
expression, real-time PCR, siRNA, and western blotting were
performed to examine whether ginkgetin directly regulates the
expression of cyclin B1. Our results showed that ginkgetin
downregulated b-Myb expression both at the mRNA and protein level
in a time-dependent manner; b-Myb expression levels were
significantly decreased after 12 h of ginkgetin treatment (Fig. 4A and B). When b-Myb was depleted by
siRNA, HCT116 cell growth was inhibited, and cyclin B1 expression
was dramatically decreased. We did not detect any changes in b-Myb
and CDC2 expression in cyclin B1 siRNA-transfected HCT116 cells.
Our results suggest that ginkgetin regulates cell cycle progression
through b-Myb-mediated cyclin B1 and CDC2 expression. This finding
is consistent with previous studies, which suggest that Myb is
important for regulating the G2/M transition and is a
key modulator for cyclin B1 expression in certain human tumor cells
(28,29,36).
In ginkgetin-treated HCT116 cells, b-Myb and cyclin
B1 expression was downregulated. In cells in which b-Myb was
silenced by siRNA, cyclin B1 expression was completely inhibited,
and cell growth was also inhibited; therefore, identifying the
transcriptional regulator of b-Myb is central to elucidating the
mechanism of ginkgetin-induced G2 arrest in HCT116
cells. To characterize the mechanism of b-Myb transcription
repression by ginkgetin, we have searched published reports and
found that miR-29a, miR-29c, miR-30b, and miR-34a are associated
with b-Myb expression in human cancer cells (30,31).
MicroRNAs regulate various cellular processes, including cancer
cell proliferation and tumor progression (37). In particular, miR-34a has been
identified as a therapeutic agent against human cancer, because
miR-34a is an important tumor suppressor whose expression is
epigenetically silenced in various human cancers; miR-34a induces
cell cycle arrest, apoptosis, senescence, suppresses the
epithelial-mesenchymal transition and inhibits the proliferation of
cancer stem cells (38). Our study
showed that ginkgetin markedly increased the expression of miR-29a,
miR-29c, miR-30b, and miR-34a, but only miR-34a affected b-Myb
expression in HCT116 cells. Upregulation of miR-34a by ginkgetin
contributed to the downregulation of b-Myb, leading to
G2 arrest and apoptosis in HCT116 cells (Figs. 2, 4A and 5B). These results were confirmed by
overexpression using a miR-34a mimic and miR-34a inhibitor
(Fig. 5C and D). Recent progress
in cancer research has demonstrated that changes in DNA
methylation, histone modifications, and miRNAs are intricately
linked to the initiation, promotion and progression of cancer. The
epigenetic silencing of miR-34a by aberrant CpG methylation
frequently occurs in various types of human cancer (39). Dihydroxyflavone has been reported
to demethylate miR-34a and increases its expression in breast
cancer cells (40). Therefore, we
hypothesize that ginkgetin controls the expression of miR-34a
through its epigenetic modulation; however, ginkgetin did not
affect the demethylation of miR-34a (data not shown). A deeper
investigation of the underlying mechanism is therefore needed and
we are investigating the epigenetic modification by ginkgetin.
In this study, we focused on investigating the
mechanism of ginkgetin-inducing G2 cell cycle arrest and
found that ginkgetin induces G2 arrest and leads to
apoptosis in HCT116 cells (CRC) through a newly defined mechanism,
i.e., the miR-34a/b-Myb/cyclin B1 pathway. To our knowledge, this
report is the first systematic study to demonstrate selective
G2 cell cycle arrest in CRC through the
miR-34a/b-Myb/cyclin B1 cascade using a natural flavonoid. These
findings suggest the usefulness of therapeutic approaches aimed at
modulating the levels of miR-34a by a natural product and are
useful to understand the mechanisms of natural flavonoids against
human cancer cells (41).
Abbreviations:
|
CRC
|
colorectal cancer
|
|
CDK
|
cyclin-dependent kinase
|
|
PARP
|
poly(ADP-ribose) polymerase
|
|
FACS
|
fluorescence activated cell
sorting
|
|
PHH3
|
phosphorylated histone H3 (S10)
|
Acknowledgments
This study was supported by the KRIBB Research
Initiative Program, the Bio-Synergy Research Project
(2012M3A9C4048777), the Bio and Medical Technology Development
Program (2015M3A9B5030311), funded by the Ministry of Science, ICT
and Future Planning.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cooper GM: The eukaryotic cell cycle. The
cell: A Molecular Approach. 2nd edition. Cooper GM: ASM Press;
Washington, DC: 2000
|
|
3
|
Lapenna S and Giordano A: Cell cycle
kinases as therapeutic targets for cancer. Nat Rev Drug Discov.
8:547–566. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kastan MB and Bartek J: Cell-cycle
checkpoints and cancer. Nature. 432:316–323. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen T, Stephens PA, Middleton FK and
Curtin NJ: Targeting the S and G2 checkpoint to treat cancer. Drug
Discov Today. 17:194–202. 2012. View Article : Google Scholar
|
|
6
|
Marzo I and Naval J: Antimitotic drugs in
cancer chemotherapy: Promises and pitfalls. Biochem Pharmacol.
86:703–710. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dickson MA: Molecular pathways: CDK4
inhibitors for cancer therapy. Clin Cancer Res. 20:3379–3383. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lyon J, Robinson C and Watson R: The role
of Myb proteins in normal and neoplastic cell proliferation. Crit
Rev Oncog. 5:373–388. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tanaka Y, Patestos NP, Maekawa T and Ishii
S: B-myb is required for inner cell mass formation at an early
stage of development. J Biol Chem. 274:28067–28070. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sala A: B-MYB, a transcription factor
implicated in regulating cell cycle, apoptosis and cancer. Eur J
Cancer. 41:2479–2484. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mowla SN, Lam EWF and Jat PS: Cellular
senescence and aging: The role of B-MYB. Aging Cell. 13:773–779.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Down CF, Millour J, Lam EW and Watson RJ:
Binding of FoxM1 to G2/M gene promoters is dependent upon B-Myb.
Biochim Biophys Acta. 1819:855–862. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu N, Lucibello FC, Zwicker J, Engeland K
and Müller R: Cell cycle-regulated repression of B-myb
transcription: Cooperation of an E2F site with a contiguous
corepressor element. Nucleic Acids Res. 24:2905–2910. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Esteller M: Non-coding RNAs in human
disease. Nat Rev Genet. 12:861–874. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang JS and Lai EC: Alternative miRNA
biogenesis pathways and the interpretation of core miRNA pathway
mutants. Mol Cell. 43:892–903. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Garzon R, Calin GA and Croce CM: MicroRNAs
in cancer. Annu Rev Med. 60:167–179. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rupaimoole R, Calin GA, Lopez-Berestein G
and Sood AK: miRNA Deregulation in cancer cells and the tumor
microenvironment. Cancer Discov. 6:235–246. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liang LH and He XH: Macro-management of
microRNAs in cell cycle progression of tumor cells and its
implications in anti-cancer therapy. Acta Pharmacol Sin.
32:1311–1320. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Martinez I, Cazalla D, Almstead LL, Steitz
JA and DiMaio D: miR-29 and miR-30 regulate B-Myb expression during
cellular senescence. Proc Natl Acad Sci USA. 108:522–527. 2011.
View Article : Google Scholar :
|
|
20
|
Tazawa H, Tsuchiya N, Izumiya M and
Nakagama H: Tumor-suppressive miR-34a induces senescence-like
growth arrest through modulation of the E2F pathway in human colon
cancer cells. Proc Natl Acad Sci USA. 104:15472–15477. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jeon YJ, Jung SN, Yun J, Lee CW, Choi J,
Lee YJ, Han DC and Kwon BM: Ginkgetin inhibits the growth of DU-145
prostate cancer cells through inhibition of signal transducer and
activator of transcription 3 activity. Cancer Sci. 106:413–420.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
You OH and Kim SH, Kim B, Sohn EJ, Lee HJ,
Shim BS, Yun M, Kwon BM and Kim SH: Ginkgetin induces apoptosis via
activation of caspase and inhibition of survival genes in PC-3
prostate cancer cells. Bioorg Med Chem Lett. 23:2692–2695. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kang SS, Kim JS, Kwak WJ and Kim KH:
Flavonoids from the leaves of Ginkgo biloba. Korean J Pharmacogn.
21:111–120. 1990.
|
|
24
|
Ye ZN, Yu MY, Kong LM, Wang WH, Yang YF,
Liu JQ, Qiu MH and Li Y: Biflavone ginkgetin, a novel Wnt
inhibitor, suppresses the growth of medulloblastoma. Nat Prod
Bioprospect. 5:91–97. 2015. View Article : Google Scholar :
|
|
25
|
Merrill GF: Cell synchronization. Methods
Cell Biol. 57:229–249. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Schorl C and Sedivy JM: Analysis of cell
cycle phases and progression in cultured mammalian cells. Methods.
41:143–150. 2007. View Article : Google Scholar :
|
|
27
|
Wei Y, Mizzen CA, Cook RG, Gorovsky MA and
Allis CD: Phosphorylation of histone H3 at serine 10 is correlated
with chromosome condensation during mitosis and meiosis in
Tetrahymena. Proc Natl Acad Sci USA. 95:7480–7484. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pilkinton M, Sandoval R, Song J, Ness SA
and Colamonici OR: Mip/LIN-9 regulates the expression of B-Myb and
the induction of cyclin A, cyclin B, and CDK1. J Biol Chem.
282:168–175. 2007. View Article : Google Scholar
|
|
29
|
Knight AS, Notaridou M and Watson RJ: A
Lin-9 complex is recruited by B-Myb to activate transcription of
G2/M genes in undifferentiated embryonal carcinoma cells. Oncogene.
28:1737–1747. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Martinez I, Cazalla D, Almstead LL, Steitz
JA and DiMaio D: MiR-29 and miR-30 regulate B-Myb expression uring
cellular senescence. Proc Natl Acad Sci USA. 92:9363–9367.
2011.
|
|
31
|
Zauli G, Voltan R, di Iasio MG, Bosco R,
Melloni E, Sana ME and Secchiero P: miR-34a induces the
downregulation of both E2F1 and B-Myb oncogenes in leukemic cells.
Clin Cancer Res. 17:2712–2724. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Del Rio D, Rodriguez-Mateos A, Spencer JP,
Tognolini M, Borges G and Crozier A: Dietary (poly)phenolics in
human health: Structures, bioavailability, and evidence of
protective effects against chronic diseases. Antioxid Redox Signal.
18:1818–1892. 2013. View Article : Google Scholar :
|
|
33
|
Lu MF, Xiao ZT and Zhang HY: Where do
health benefits of flavonoids come from? Insights from flavonoid
targets and their evolutionary history. Biochem Biophys Res Commun.
434:701–704. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Singh RP and Agarwal R: Natural flavonoids
targeting deregulated cell cycle progression in cancer cells. Curr
Drug Targets. 7:345–354. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liberio MS, Sadowski MC, Davis RA,
Rockstroh A, Vasireddy R, Lehman ML and Nelson CC: The ascidian
natural product eusynstyelamide B is a novel topoisomerase II
poison that induces DNA damage and growth arrest in prostate and
breast cancer cells. Oncotarget. 6:43944–43963. 2015. View Article : Google Scholar
|
|
36
|
Nakata Y, Shetzline S, Sakashita C, Kalota
A, Rallapalli R, Rudnick SI, Zhang Y, Emerson SG and Gewirtz AM:
c-Myb contributes to G2/M cell cycle transition in human
hematopoietic cells by direct regulation of cyclin B1 expression.
Mol Cell Biol. 27:2048–2058. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Calin GA and Croce CM: MicroRNA signatures
in human cancers. Nat Rev Cancer. 6:857–866. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Agostini M and Knight RA: miR-34: From
bench to bedside. Oncotarget. 5:872–881. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wong KY, Yu L and Chim CS: DNA methylation
of tumor suppressor miRNA genes: A lesson from the miR-34 family.
Epigenomics. 3:83–92. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Peng X, Chang H, Gu Y, Chen J, Yi L, Xie
Q, Zhu J, Zhang Q and Mi M: 3,6-Dihydroxyflavone suppresses breast
carcinogenesis by epigenetically regulating miR-34a and miR-21.
Cancer Prev Res (Phila). 8:509–517. 2015. View Article : Google Scholar
|
|
41
|
Li XJ, Ren ZJ and Tang JH: MicroRNA-34a: A
potential therapeutic target in human cancer. Cell Death Dis.
5:e13272014. View Article : Google Scholar : PubMed/NCBI
|