Introduction
Malignant rhabdoid tumour (MRT) is an aggressive
paediatric neoplasm, primarily diagnosed in children under three
(1,2). MRTs most frequently arise in the
brain, where they are referred to as atypical/teratoid rhabdoid
tumours (AT/RT), or the kidneys. Common to rhabdoid tumours is an
inactivating mutation in the SWI/SNF related, matrix associated,
actin dependent regulator of chromatin, subfamily b, member 1
(SMARCB1) gene, leading to loss of expression of SMARCB1, a
component of the SWI/SNF chromatin remodelling complex (3,4).
Unlike most cancers, MRTs are genomically stable, with SMARCB1
being the only commonly mutated gene identified (5-7).
Currently, there is no standardised treatment protocol for MRT.
Case reports in the literature, and numerous clinical trials are
testament to this, with treatment regimens differing between
institutions and often dependent upon tumour location. Treatment
primarily consists of surgical resection of the tumour followed by
aggressive chemotherapy, and if appropriate radiation therapy.
Unfortunately, MRT is highly refractory to therapy, with
retrospective reviews placing the 2-year event-free survival rate
at ≤35% (8,9). The most successful trial to date
treated 20 AT/RT patients with surgical resection, intensive
chemotherapy and radiation, resulting in a 2-year overall survival
rate of 70% (10). However, the
dangers of pursuing such an aggressive regimen in children was
evident with several associated toxicities and one toxic death
reported. Given the poor prognosis and lack of treatment options,
there is an urgent need for new therapies for MRT.
TNF-related apoptosis-inducing ligand (TRAIL) is a
proapoptotic cytokine expressed by many tissue types of immune
cells. Unlike other members of the TNF superfamily, TRAIL exhibits
selectivity towards cancer cells whilst sparing healthy cells
(11). While chemotherapeutics and
radiation therapy elicit apoptosis through the intrinsic or
mitochondrial apoptotic pathway, TRAIL, activates the extrinsic
apoptotic pathway (12). Binding
of TRAIL to its cognate death receptors, death receptor 4 (DR4) or
death receptor 5 (DR5), results in receptor oligomerisation and
formation of a membrane associated death inducing signalling
complex (DISC) consisting of the adaptor protein FAS-associated via
death domain (FADD), caspases 8 and 10, and the caspase-8 analogue
FLICE-inhibitory protein (FLIP). The primary result of DISC
formation is caspase-8 activation. Once active, caspase-8 cleaves
and activates caspases 3 and 7, thus inducing apoptosis. In
addition, TRAIL can engage the intrinsic pathway, as caspase-8 may
cleave the pro-apoptotic Bcl-2 protein Bid into its truncated form
tBid, which mediates mitochondrial outer membrane permeabilization
(MOMP). MOMP results in cytochrome c release which in turn
facilitates caspase-9 activation thus potentiating apoptosis. MOMP
and thus the intrinsic apoptotic pathway is regulated by the Bcl-2
family, which consists of pro and antiapoptotic members alongside
regulatory proteins (13). In MRT,
the antiapoptotic Bcl-2 protein myeloid leukaemia cell
differentiation protein 1 (Mcl-1) has been demonstrated to be
overexpressed (14).
Unfortunately, clinical trials with TRAIL have yielded
disappointing outcomes, with drug resistance being a primary issue
(15-19). One approach to combat this, is the
combination of TRAIL treatment with inhibition of negative
regulators of the apoptotic process, such as FLIP-long form
(FLIPL), anti-apoptotic members of the Bcl-2 family and
the inhibitor of apoptosis (IAP) family.
Of the IAP family, X-linked inhibitor of apoptosis
(XIAP), cellular inhibitor of apoptosis protein (cIAP) 1, cIAP2,
livin and survivin have been implicated in cancer progression and
apoptosis inhibition, with their overexpression demonstrated in
various malignancies, including acute myeloid leukaemia,
neuroblastoma, hepatocellular carcinoma and renal cell carcinoma
(20-28). To date, their role in MRT has not
been examined. XIAP is the best characterised of these proteins and
the only IAP which has been demonstrated to inhibit caspase
activity directly (29-34). Specifically, XIAP has been
demonstrated to bind and inhibit caspases 3, 7 and 9 (35,36).
Embelin, the active constituent of the Embelia ribes shrub,
is a small molecule which has been found to inhibit XIAP (37). Its tumour suppressive abilities
have been demonstrated in leukaemia, prostate, breast, gastric and
brain glioma cell lines (38-44).
Its chemotherapeutic and radiation sensitising capacity has also
been demonstrated in various cancers (45,46).
Additionally, there are an increasing number of studies reporting
that embelin can sensitise both cancer cell lines and xenograft
models to TRAIL (47-54). To the best of our knowledge, there
is only one published report on TRAIL treatment in MRT cell lines,
which found 3 of 6 tested cell lines to be sensitive to TRAIL
(55). However, no combination
regimens were tested. Furthermore, there are currently no reports
on the effects of embelin, either alone or in combination, in
MRT.
The results reported in the present study
demonstrated for the first time the expression of a range of IAPs
in MRT cell lines. In addition, embelin was determined to act as a
sensitising agent to TRAIL in otherwise resistant MRT cell lines.
This enhanced apoptosis was mediated in part by XIAP inhibition.
The current findings thus suggest potential novel therapeutic
options for MRT.
Materials and methods
Cells and reagents
BT12 and BT16 cell lines are epithelial AT/RT cells
isolated from a 2 month old female and a 2-year-old male,
respectively. These cell lines were generously donated by Peter
Houghton, St. Jude Children's Research Hospital (Memphis, TN, USA).
G401 cells (American Type Culture Collection, Manassas, VA, USA)
are epithelial kidney MRT cells isolated from a 3 month old male.
BT12 and BT16 cell lines were cultured in Gibco RPMI-1640 GlutaMAX
(Biosciences, Dublin, Ireland). G401 cells were cultured in Gibco
Dulbecco's modified Eagle's medium (DMEM) GlutaMAX (Biosciences).
All media used was supplemented with 10% (v/v) foetal bovine serum
(FBS; Biosciences), 100 U/ml penicillin and 100 μg/ml
streptomycin (Biosciences). Cultured cells were incubated at 37°C
with 5% CO2. Cells were screened for mycoplasma using
PCR and were consistently found to be negative for infection. A 1
mg/ml stock of recombinant human TRAIL (Merck KGaA, Darmstadt,
Germany) was prepared in sterile dH2O and stored at
-80°C. Dilutions were prepared fresh on the day of treatment. A 10
mM stock of embelin (Sigma-aldrich; Merck KGaA) was prepared in
ethanol and stored at -20°C. Stocks (50 mM) of both the general
caspase inhibitor (ZVAD-FMK; Calbiochem; Merck KGaA) and caspase-8
Inhibitor II [Z-IE(OMe) TD(OMe)-FMK; Calbiochem; Merck KGaA] were
prepared in DMSO and ethanol respectively. Dilutions of both
inhibitors were prepared fresh on the day of treatments in sterile
dH2O.
Cell viability assay
BT12, BT16 and G401 cell lines (200 μl) were
seeded at densities of 10x103, 10x103 and
5x103 cells/well, respectively, in 96-well plates. Cells
were left overnight to adhere and then treated with a range of
concentrations of TRAIL or embelin for 72 h. Viability was assessed
via the alamar blue assay. Briefly, 20 μl of AlamarBlue
(Invitrogen; Thermo Fisher Scientific, Inc. Waltham, MA, USA) was
added to each well. The plate was incubated in the dark at 37°C for
5 h. Fluorescence was measured on a SpectraMax plate reader at an
excitation wavelength of 544 nm and an emission wavelength of 590
nm. All alamar blue assays were performed in triplicate.
Cell death analysis
For cell death analysis via propidium iodide (PI)
staining, cells were collected by centrifugation and fixed in 70%
ethanol overnight. The following day, 5 μl of FBS and 1 ml
of PBS were added to each sample. Cells were collected by
centrifugation and resuspended in BD FACSflow sheath fluid (BD
Biosciences, San Jose, CA, USA) supplemented with 10 μg/ml
of RNase A (Sigma-Aldrich; Merck KGaA) and 100 μg/ml of PI
(Sigma-Aldrich; Merck KGaA). The samples were then incubated in the
dark at 37°C for 30 min. Analysis was performed with a BD Accuri C6
flow cytometer using BD Accuri C6 software (BD Biosciences).
Samples were first gated on vehicle controls. A total of
1x105 cells from each sample were counted and results
were visualised on histograms. PI was detected using a 585/40
bandpass filter. The sub G1/G0 population (<2n) was assigned as
apoptotic cells.
For cell death analysis via Annexin V-fluorescein
isothiocyanate (FITC) and PI staining, cells were collected by
centrifugation and washed with 0.5 ml Annexin V binding buffer (5
mM HEPES, 70 mM NaCl, 1.25 mM CaCl2 pH 7.4). The pellet
from this wash was collected and stained with Annexin V-FITC (iQ
Corporation, Groningen, Netherlands) for 30 min in the dark on ice.
Cells were washed with 0.5 ml of Annexin V binding buffer and
collected by centrifugation. The pellet was resuspended in 500
μl PI (0.5 μg/ml). The samples were then analysed
immediately on BD Accuri C6 flow cytometer using BD Accuri C6
software. Samples were first gated on vehicle controls. These gates
were then analysed on a FITC-Annexin V (535 nm; FL1 channel using a
530/30 bandpass filter) vs PI (488 nm; FL2 using a 585/40 bandpass
filter) dot plot.
Gel electrophoresis and western blot
analysis
For whole cell lysates, cell pellets were
resuspended in ice-cold RIPA buffer (Sigma-Aldrich; Merck KGaA)
supplemented with protease inhibitor cocktail II and III
(Sigma-Aldrich; Merck KGaA) and complete ultra protease inhibitor
(Roche Applied Science, Penzberg, Germany) and kept on ice for 30
min. Lysates were centrifuged at 10,000 x g for 10 min and the
pellet was discarded. For the cytosolic lysates, the cell pellet
was resuspended in 75 μl subcellular fractionation buffer
(250 mM sucrose, 20 mM HEPES, 10 mM KCL, 1.5 mM MgCl2, 1
mM EDTA, 1 mM EGTA, 1 mM DTT, protease inhibitor cocktail) and kept
agitated at 4°C for 30 min. The suspension was centrifuged at 720 x
g for 5 min and the resultant supernatant was centrifuged further
at 10,000 x g for 10 min. The final resultant supernatant was the
cytosolic lysate. For all lysates, protein concentration was
determined using the BCA assay (Pierce; Thermo Fisher Scientific,
Inc.) and normalised. Cell lysates were boiled with Laemmli sample
buffer supplemented with 50 μM DTT for 10 min at 90°C. Equal
concentrations of denatured samples were separated using
polyacrylamide gel electrophoresis and transferred to an
immobilion-P PVDF membrane (Merck KGaA). The membrane was blocked
for 1 h in 5% non-fat dried milk and then incubated overnight at
4°C with the relevant primary antibody. The following primary
antibodies were purchased form Cell Signaling Technology, Inc.,
(Danvers, MA, USA): XIAP (1:2,500; cat no. 2045), cIAP1 (1:1,000;
cat no. 7065), cIAP2 (1:1,000; cat no. 3130), livin (1:1,000; cat
no. 5471), survivin (1:1,000; cat no. 2808), caspase-3 (1:1,000;
cat no. 9662), cleaved caspase-3 (1:1,000; cat no. 9664), caspase-8
(1:1,000; cat no. 9746), cleaved caspase-8 (1:1,000; cat no. 9496),
caspase-9 (1:1,000; cat no. 9508) Bid (1:1,000; cat no. 2002) and
Mcl-1 (1:1,000; cat no. 4572). The following primary antibodies
were also used: second mitochondria-derived activator of caspases
(SMAC; 1:1,000; cat no. 612246; BD Biosciences), FLIPL
(1:2,000; cat no. MABC148; Merck KGaA) GAPDH (1:10,000; cat no.
CB1001; Calbiochem), and α tubulin (1:1,000; cat no. CP0;
Calbiochem). The following day the membrane was washed three times
for 10 min each with TBST, probed with the relevant anti-rabbit
(1:2,500; cat no. W402B; Promega Corporation, Madison, WI, U.S.A.)
or anti-mouse (1:2,500; cat no. W401B; Promega Corporation)
horseradish peroxidase (HRP)-conjugated secondary antibody, and
then washed again as aforementioned. The membrane was soaked in
enhanced chemiluminescence detection reagent (Merck KGaA). The
western blot results were visualised using the BioRad Gel Doc™
XR+System with Image Lab software (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA).
XIAP knockdown using small interfering
(si) RNA
On-targetplus human XIAP siRNA SMARTpool (GE
Healthcare Dharmacon, Inc., Lafayette, CO, USA) consisted of four
siRNA sequences targeting XIAP, as follows: GUAGAUAGAUGGCAAUAUG,
GAACUGGGCAGGUUGUAGA, GAAAGAGAUUAGUACUGAA and GGACUCUACUACACAGGUA.
The negative control On-targetplus non-targeting pool (GE
Healthcare Dharmacon, Inc.) consisted of four non-targeting siRNA
sequences: UGGUUUACAUGUCGACUAA, UGGUUUACAUGUUGUGUGA,
UGGUUUACAUGUUUUCUGA, and UGGUUUACAUGUUUUCCUA. BT12 cells were
seeded at 10x104 cells/ml in antibiotic-free media in a
6-well plate. The following day, XIAP siRNA or non-targeting siRNA
was transfected into cells using Lipofectamine (Thermo Fisher
Scientific, Inc.) at a final concentration of 25 nM, as per
manufacturer's instructions. Cell death was assessed 48 h
post-treatment by flow cytometric analysis of PI-stained cells and
subsequent quantification of the sub G1/G0 peak, as described
above. Western blot analysis confirmed XIAP knockdown at 0 and 48 h
of TRAIL treatment.
Determination of synergism
TRAIL and embelin combination treatments were
assessed for synergism based upon the apoptosis they elicited, both
as single drugs and in combination with one another, as determined
by flow cytometric analysis of PI-stained cells, as described
above. The computer software program Compusyn (ComboSyn, Inc., New
Jersey, U.S.A.) was used to study the interactions between the
drugs. This program uses the Chou-Talalay method, based upon the
median effect principle, to determine Combination Index (CI) values
for each drug combination. CI values <1, =1 and >1 indicate
synergistic, additive and antagonistic effects respectively.
Statistical analysis
GraphPad Prism 5 (GraphPad Software, Inc., La Jolla,
CA, USA) was used for statistical analysis of experimental data.
Results are displayed as the mean ± standard error of the mean. To
determine statistical significance, Student's t-test was used for
comparisons between two groups, while a one-way analysis of
variance followed by Tukey's multiple comparison test was performed
for comparisons of more than two groups. P<0.05 was considered
to indicate a statistically significant difference.
Results
MRT cell lines express a range of
antiapoptotic IAP proteins
BT12, BT16 and G401 cell lines were firstly analysed
for expression of XIAP, cIAP1, cIAP2, livin and survivin via
western blotting (Fig. 1A).
Densitometric analysis was then performed (Fig. 1B-G). All three cell lines were
demonstrated to express a range of IAPs, with XIAP, cIAP1 and
survivin universally expressed. Low levels of cIAP2 were detected
in BT12 cells only. Livin exists as two splice variants; livin α
and livin β. Both splice variants were detected in BT12 and G401
cells, while neither was detected in the BT16 cell line.
 | Figure 1MRT cell lines express a range of IAP
family members. (A) Expression levels of cIAP2, cIAP1, XIAP, Livin
α, Livin β and survivin in malignant rhabdoid tumour cell lines,
BT12, BT16 and G401, were detected by western blotting. GAPDH was
used as a loading control. (B-G) Western blots were normalised to
GAPDH and densitometric analysis was performed. Values are mean ±
standard error of the mean of at least three independent
experiments. *P<0.05, **P<0.01 and
***P<0.001 with comparisons indicated by brackets.
MRT, malignant rhabdoid tumour; IAP, inhibitor of apoptosis
protein; cIAP, cellular inhibitor of apoptosis protein; XIAP,
X-linked inhibitor of apoptosis. |
TRAIL and embelin exhibit limited effects
on the viability of MRT cells
Yoshida et al (55) have previously reported a decreased
viability of MRT cell lines upon TRAIL treatment. Given this, and
combined with the XIAP expression demonstrated in Fig. 1, the viability of BT12, BT16 and
G401 cell lines was assessed following treatment with TRAIL or the
XIAP inhibitor, embelin, for 72 h via the alamarBlue assay
(Fig. 2A). Viability dropped to
only 60, 71 and 89% for G401, BT12 and BT16 cells, respectively, at
1,000 ng/ml TRAIL. Embelin reduced the viability of all three cell
lines in a dose dependent manner. However, even at 50 μM
dose, the viability did not decrease below 33% for any cell line.
BT12 and BT16 cells had similar IC50 values of 13.06 and
14.52 μM, respectively, with viability plateauing
approximately 44 and 37%, respectively. The IC50 value
for the G401 cell line was considerably higher at 47.14
μM.
 | Figure 2Embelin enhances TRAIL-mediated cell
death in MRT cell lines. (A) MRT cell lines, BT12, BT16 and G401,
were treated with a range of concentrations of TRAIL or embelin.
Viability was assessed 72 h post-treatment via the alamar blue
assay. (B) MRT cell lines, BT12, BT16 and G401, were treated with a
range of concentrations of TRAIL alone or in combination with 10 μM
embelin. Cell death was assessed at 24, 48 and 72 h post-treatment
via flow cytometric analysis of PI-stained cells and subsequent
quantification of the sub G1/G0 peak. (C) BT12 cells were treated
with 50 ng/ml TRAIL and 10 μM embelin, both alone and in
combination. Cell death was assessed 48 h post-treatment, via flow
cytometric analysis of PI and Annexin-V FITC-stained cells. Cells
which stained negative for PI and Annexin V are considered viable
(Q1), cells which stained positive for Annexin V, but negative for
PI were considered to be undergoing early apoptosis (Q2), and cells
which stained positive for both PI and Annexin V were considered to
be undergoing late apoptosis/necrosis (Q3), as shown in the
representative plots. Values are mean ± standard error of the mean
of at least three independent experiments. *P<0.05,
**P<0.01 and ***P<0.001 with
comparisons indicated by brackets. TRAIL, TNF-related
apoptosis-inducing ligand; MRT, malignant rhabdoid tumour; PI,
propidium iodide; FITC, fluorescein isothiocyanate. |
Embelin synergistically enhances
TRAIL-mediated cell death in MRT cell lines
Having shown the expression of XIAP in a range of
MRT cell lines, and their resistance to TRAIL treatment, the
present study next assessed if embelin could sensitise MRT cell
lines to TRAIL. BT12, BT16 and G401 cell lines were treated with a
range of concentrations of TRAIL, both alone and in combination
with 10 μM embelin, for 24, 48 and 72 h. This concentration of
embelin is at the lowest end of the range (10-50 μM)
previously demonstrated to enhance susceptibility to TRAIL
(47-54). It was selected for the present
study, as lower doses are preferable in cancer therapeutics in
order to minimise potential side effects. Cell death was assessed
via PI staining of permeabilised cells and subsequent
quantification of the sub G1/G0 peak. The computer program Compusyn
was used to determine synergism. This program computes combination
index (CI) values for each drug combination. CI values <1, =1
and >1 indicate synergistic, additive and antagonistic effects
respectively, with lower CI values indicating greater synergism. In
BT12 cells, treatment with TRAIL or embelin alone did not cause
significant cell death compared with vehicle, at any concentration
tested. Combination treatment resulted in a dose and time-dependent
increase in cell death, which was significant and synergistic at
all concentrations and at several timepoints (Fig. 2B; Table I). Treatment with TRAIL at 50 ng/ml
in combination with 10 μM embelin was determined to give the
greatest synergism with a CI value at 0.169 (Table I). Annexin V-PI staining, was then
employed to confirm the increased cell death upon combination
treatment with concurring results obtained (Fig. 2C). Similarly, in BT16 cells,
embelin synergistically enhanced TRAIL-mediated cell death across a
range of timepoints and concentrations, with CI values <1
reported for all concentrations tested at 48 and 72 h (Fig. 2B; Table I). In the G401 cell line,
combination treatment did not result in significantly higher cell
death compared with TRAIL treatment alone, at any concentration
tested (Fig. 2B), possibly owing
to differences between MRTs arising in the kidney and the
brain.
 | Table ISynergism between TRAIL and embelin
in BT12 and BT16 cells. |
Table I
Synergism between TRAIL and embelin
in BT12 and BT16 cells.
| Cell line | Timepoint (h) | TRAIL (ng/ml) | Fa | CI value | Level of
synergism |
|---|
| BT12 | 24 | 25 | 0.144 | 0.35798 | +++ Synergism |
| | 50 | 0.18 | 0.50820 | +++ Synergism |
| | 100 | 0.352 | 0.470532 | +++ Synergism |
| 48 | 25 | 0.264 | 0.28718 | ++++ Strong
Synergism |
| | 50 | 0.434 | 0.16938 | ++++ Strong
Synergism |
| | 100 | 0.479 | 0.14939 | ++++ Strong
Synergism |
| 72 | 25 | 0.39 | 0.43228 | +++ Synergism |
| | 50 | 0.538 | 0.33700 | +++ Synergism |
| | 100 | 0.607 | 0.31940 | +++ Synergism |
| BT16 | 48 | 25 | 0.237 | 0.43825 | +++ Synergism |
| | 50 | 0.275 | 0.36781 | +++ Synergism |
| | 100 | 0.353 | 0.26640 | ++++ Strong
Synergism |
| 72 | 25 | 0.3023 | 0.46910 | +++ Synergism |
| | 50 | 0.3527 | 0.35404 | +++ Synergism |
| | 100 | 0.3399 | 0.38200 | +++ Synergism |
Combination treatment induces apoptosis
through the extrinsic apoptotic pathway
To determine the mode of cell death initiated upon
combination treatment, BT12 cells were pretreated with an inhibitor
of necroptosis, necrostatin 1 or the general caspase inhibitor,
Z-VAD-FMK. Pretreatment with necrostatin 1 had no effect on cell
death (Fig. 3A). By contrast,
pretreatment with Z-VAD-FMK prevented cell death (Fig. 3B), indicating apoptosis was
occurring. In the literature, TRAIL-induced cell death has mostly
been attributed to activation of the extrinsic apoptotic pathway,
resulting firstly in caspase-8 activation, followed by downstream
activation of caspase-3, caspase-7 and in certain cell types
caspase-9 (12). Pretreatment of
BT12 cells with a specific caspase-8 inhibitor, Z-IETD-FMK,
abrogated cell death (Fig. 3C),
suggesting that the extrinsic apoptotic pathway was induced upon
TRAIL and embelin treatment. Caspase inhibition was further
confirmed via western blot analysis (Fig. 3D).
Embelin prevents the recovery of MRT
cells from TRAIL treatment over time
BT12 and BT16 cells were treated with TRAIL and
embelin, both alone and in combination, from 1 to 48 h. In both
cell lines, TRAIL treatment resulted in an initial burst of cell
death from 4 h (Fig. 4A and B).
However, the cells recovered from TRAIL treatment with cell death
decreasing steadily from 16 to 48 h in BT12 cells (Fig. 4A), and from 4 to 48 h in BT16 cells
(Fig. 4B). By contrast, cell death
increased upon combination treatment in a time-dependent manner in
both cell lines. This suggests that embelin prevented the recovery
of MRT cell lines from TRAIL treatment over time. Given the
aforementioned findings that TRAIL and embelin-induced cell death
was dependent on caspase-8 activation, the extrinsic apoptotic
pathway was examined over a range of timepoints. Treatment with
TRAIL alone resulted in a decrease in full length caspase-8 and
generation of active caspase-8 from 4 h (Fig. 4D). However, in agreement with the
cell death results, caspase-8 activation decreased from 4 to 48 h.
By contrast, combination treatment resulted in enhanced caspase-8
cleavage compared with treatment with TRAIL alone from 4 h onwards
(Fig. 4D). Bid, a target of
caspase-8, was determined to be cleaved at 24 h in TRAIL-treated
cells, with cleavage enhanced upon combination treatment (Fig. 4E). To determine if the generation
of tBid was sufficient for MOMP, SMAC release into the cytosol was
assessed from 1 to 24 h. SMAC is a pro-apoptotic protein usually
resident within the mitochondria. However, upon MOMP, SMAC is
released into the cytosol where it is free to carry out its
proapoptotic functions by binding and inhibiting XIAP (33,56,57).
Both TRAIL alone and combination treatment resulted in SMAC
release, with levels slightly enhanced at 24 h in
combination-treated cells (Fig.
4F). Despite SMAC release into the cytosol, indicative of MOMP,
caspase-9 cleavage fragments, which would be formed upon caspase-9
activation, were not detected using an antibody capable of
detecting both full length and cleaved fragments (Fig. 4G). A further target of active
caspase-8 is caspase-3. Cleavage of caspase-3 results in the
generation of an inactive cleavage product of 19 kDa and active
cleavage products of 17 and 15 kDa. Treatment with TRAIL alone
resulted in generation of both active and inactive cleavage
fragments from 4 h onwards (Fig.
4H). However, at later timepoints it was the inactive fragment
which predominated. By contrast, combination treatment resulted in
enhanced generation of active cleavage fragments over time, with
active caspase-3 levels being higher compared with the TRAIL
alone-treated cells from 4 h onwards (Fig. 4H). Caspase-3 activation was also
assessed in BT16 cells at 24 and 48 h. As per the BT12 cells,
combination treatment resulted in enhanced generation of active
caspase-3 over time (Fig. 4C).
 | Figure 4Embelin prevents the recovery of BT12
cells from TRAIL treatment with enhanced activation of the
extrinsic apoptotic pathway over time. (A) BT12 and (B) BT16 cells
were treated with 50 ng/ml TRAIL and 10 μM embelin, both
alone and in combination. Cell death was assessed at 1, 4, 8, 16,
24 and 48 h post-treatment via flow cytometric analysis of
propidium iodide-stained cells and subsequent quantification of the
sub G1/G0 peak. Values are mean ± standard error of the mean of at
least three independent experiments. *P<0.05,
**P<0.01 and ***P<0.001 vs vehicle;
&P<0.05, &&P<0.01 and
&&&P<0.001 vs. TRAIL; and
#P<0.05, ##P<0.01 and
###P<0.001 vs. embelin. (C) BT16 cells were treated
with 50 ng/mL TRAIL and 10 μM embelin, both alone and in
combination. At 24 and 48 h post-treatment, expression of full
length/cleaved caspase-3 was examined. (D) BT12 cells were treated
with 50 ng/ml TRAIL and 10 μM embelin, both alone and in
combination. At 1, 8, 16, 24 and 48 h post-treatment, expression of
full length/cleaved caspase-8, (G) caspase-9 and (H) full
length/cleaved caspase-3 was assessed via western blotting. (E) Bid
cleavage was examined at 24 h and (F) SMAC release into the cytosol
at 1, 4, 16 and 24 h. Results are representative of at least three
independent experiments, except those for Bid and tBid which are
representative of two independent experiments. TRAIL, TNF-related
apoptosis-inducing ligand; Bid, BH3 interacting domain death
agonist; tBid, truncated Bid; SMAC, second mitochondria-derived
activator of caspases. |
Combination treatment results in
decreased expression of antiapoptotic proteins
BT12 cells were treated with TRAIL and embelin,
either alone or in combination, from 1 to 48 h and protein
expression was analysed via western blotting. The expression levels
of proteins which were implicated in sensitising the cells to
apoptosis in the BT12 cells were then assessed in BT16 cells at 24
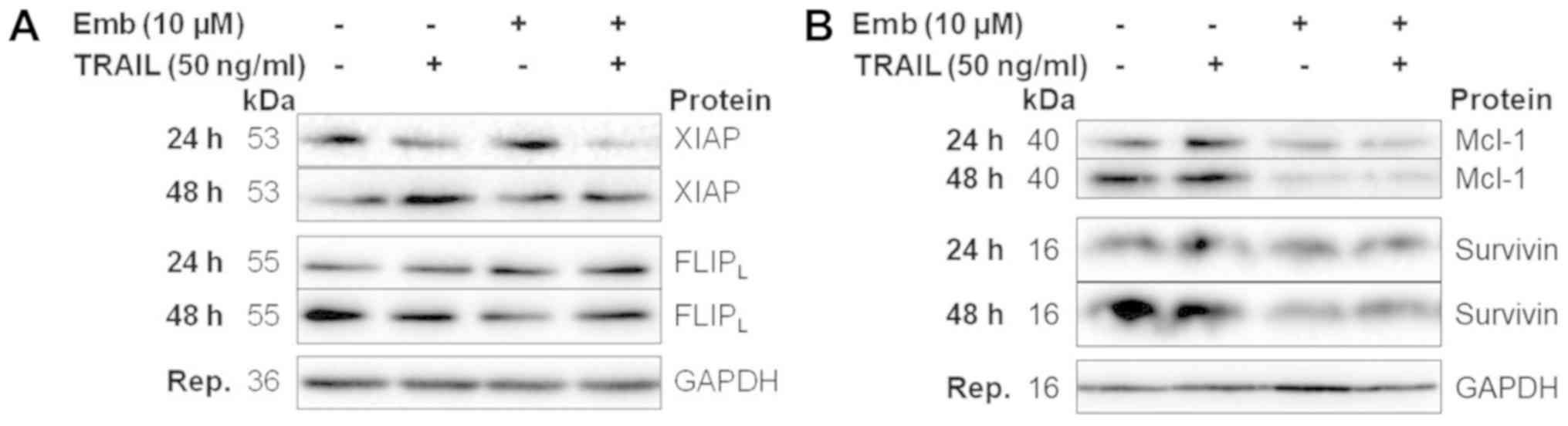
and 48 h. No differences in XIAP expression levels were observed
following TRAIL or embelin treatment at any timepoint (Fig. 5A). Combination treatment resulted
in slightly reduced expression at 24 and 48 h. XIAP expression in
BT16 cells was similar (Fig. 6A).
cIAP1 expression levels in BT12 cells were unaffected by any
treatment at any timepoint (Fig.
5B). cIAP2 levels in BT12 cells were examined at 48 h only, due
to technical limitations with the cIAP2 antibody, and were found to
be slightly decreased following embelin or combination treatment
(Fig. 5C). As aforementioned,
livin exists as two splice variants, both of which can be cleaved
by caspase-3, with the cleaved fragments suggested to be
proapoptotic. In line with the cleaved caspase-3 data, treatment
with TRAIL resulted in livin cleavage from 4 h in BT12 cells
(Fig. 5D). However, by 48 h livin
levels were comparable to vehicle, in accordance with the recovery
of cells from treatment. By contrast, combination treatment
resulted in increased livin cleavage compared with treatment with
TRAIL alone from 4 h, with cleavage increasing in a time-dependent
manner (Fig. 5D). In BT12 cells,
survivin levels were increased upon TRAIL treatment from 24 h
(Fig. 5E). By contrast, treatment
with embelin either alone or in combination with TRAIL resulted in
reduced survivin expression from 16 h (Fig. 5E). Survivin levels in BT16 cells at
24 and 48 h were also decreased upon embelin or combination
treatment (Fig. 6B). Expression
levels of FLIPL, an analog and inhibitor of caspase-8,
were unaffected by TRAIL or embelin single treatment (Fig. 5F). However, combination treatment
resulted in reduced FLIPL expression from 16 h. By
contrast, in BT16 cells, FLIPL levels were unchanged
(Fig. 6A). The anti-apoptotic
Bcl-2 family member Mcl-1, has previously been demonstrated to be
overexpressed in MRT. Mcl-1 levels were unaffected by TRAIL or
embelin single treatment, but were found to be decreased from 4 h
in combination-treated cells (Fig.
5G). Similarly, in BT16 cells, embelin as a single agent or in
combination with TRAIL resulted in reduced Mcl-1 expression at 24
and 48 h (Fig. 6B). Densitometric
analysis of the proteins examined in Figs. 5 and 6 have been included as supplemental
material (Figs. S1 and S2).
 | Figure 5Decreased expression of antiapoptotic
proteins, including early downregulation of survivin,
FLIPL and Mcl-1 accompanies combination treatment in
BT12 cells. BT12 cells were treated with 50 ng/ml TRAIL and 10
μM embelin, both alone and in combination. At 1, 8, 16, 24
and 48 h post-treatment, expression of (A) XIAP, (B) cIAP1, (C)
cIAP2 (examined only at 48 h), (D) livin, (E) survivin, (F)
FLIPL and (G) Mcl-1 was assessed via western blotting.
Results are representative of at least three independent
experiments. FLIPL, FLICE-inhibitory protein long form;
Mcl-1, myeloid leukaemia cell differentiation protein 1; TRAIL,
TNF-related apoptosis-inducing ligand; XIAP, X-linked inhibitor of
apoptosis; cIAP, cellular inhibitor of apoptosis protein. |
Knockdown of XIAP enhances TRAIL-mediated
apoptosis
As described above, TRAIL and embelin co-treatment
did not result in downregulation of XIAP until 24 h in MRT cell
lines, despite enhanced caspase activity from 4 h. However, the
initial screening studies which identified embelin as an inhibitor
of XIAP did not suggest degradation was necessary for inhibition
(37). Given this and combined
with previous reports of enhanced susceptibility to TRAIL upon XIAP
knockdown, we next assessed if XIAP inhibition could account for
the sensitisation of MRT cell lines to TRAIL upon embelin
co-treatment. BT12 cells were treated with TRAIL in combination
with XIAP siRNA or non-targeting siRNA and XIAP knockdown was
confirmed via western blotting (Fig.
7A). While significant knockdown was achieved, low levels of
XIAP expression were still detected (Fig. 7A). Despite this, cells treated with
XIAP siRNA exhibited significantly enhanced TRAIL-mediated cell
death, in contrast to cells treated with non-targeting siRNA
(Fig. 7B). It should be noted that
this enhanced cell death was not as pronounced as with the embelin
cotreatment (Figs. 2B and 4A). While the incomplete knockdown of
XIAP may account for this, it is likely that XIAP inhibition via
embelin only partially mediates the enhanced cell death. A complex
multifactorial mechanism of cell death is supported by the reduced
survivin, FLIP and Mcl-1 expression demonstrated upon
cotreatment.
Discussion
MRT is a highly aggressive paediatric tumour, for
which there is no standardised treatment. Its diagnosis carries
with it a poor prognosis, and given its early age of onset,
aggressive treatment is hindered by associated toxicities. As such,
there is an urgent need for the development of safe effective
therapies. TRAIL has demonstrated efficacy in many cancer cell
lines and in in vivo models as a cancer therapeutic with limited
side effects (11). However,
clinical trials with TRAIL have yielded disappointing outcomes,
with resistance being a primary issue (15-19).
In recent years, reports have demonstrated enhanced susceptibility
of cancer cells to TRAIL upon cotreatment with the XIAP inhibitor
embelin (47-54). The present study, therefore,
assessed the efficacy of TRAIL and embelin cotreatment in MRT cell
lines.
The present study is the first to report the
expression levels of antiapoptotic IAP family members in a panel of
MRT cell lines. Further studies are underway into IAP expression in
rhabdoid tumour patient samples (data not shown). XIAP, cIAP1,
cIAP2, livin and survivin have frequently been reported to be
overexpressed in a broad range of tumour types, including acute
myeloid leukaemia, renal cell carcinoma, neuroblastoma and
hepatocellular carcinoma (20-28).
To this end, SMAC mimetics, inhibitors of XIAP, cIAP1 and cIAP2,
have been developed, with several of them currently in clinical
trials. The paediatric preclinical testing program (PPTP) has
previously assessed treatment with the SMAC mimetic L6161 in a
panel of cancer cells, including two MRT cell lines (BT12 and
CHLA-266 cells), with IC50 values >10 μM
obtained for both (58). Despite
the lack of response to LCL161 as a single agent in MRT reported by
the PPTP, it would be interesting to evaluate its effects in
combination with other cancer therapeutics, considering previous
reports on its sensitising ability (59-62).
In addition, YM155 is an agent which suppresses survivin gene
promotion and is currently in clinical trials as a possible
chemotherapeutic sensitising agent (63,64).
Given the demonstrated IAP expression reported in the present
study, there is a rationale for the preclinical evaluation of a
broad range of IAP targeting agents, such as SMAC mimetics, YM155
and the XIAP inhibitor embelin in MRT, both alone and in
combination with various cancer therapeutics.
The current study examined the efficacy of embelin
as a single agent in a panel of MRT cell lines. Embelin was found
to elicit a partial decrease in viability at concentrations up to
50 μM with IC50 values of 13.06, 14.55 and 47.14
μM obtained for BT12, BT16 and G401 cells, respectively,
upon 72 h treatment. While this is the first report on embelin
treatment in MRT cell lines, its anticancer effects have previously
been demonstrated in numerous other malignancies. IC50
values of 65, 28, 56 and 60 μM have been reported for
HCT-116 (colon carcinoma), MCF-7 (breast cancer), MIAPaCa-2
(pancreas ductal carcinoma) and PC-3 (prostate cancer) cells,
respectively, upon 48 h treatment (65). IC50 values <6
μM upon 72 h treatment have been reported in MCF-7 and
MDA-MB-231 breast cancer cells (40). This places MRT within the mid-range
of response to embelin as a single agent. However, the modest
decrease in viability of MRT cells in response to embelin suggests
that its clinical potential may lie in its use as a combinatorial
rather than a single agent.
To date, the only report on the treatment of MRT
with TRAIL was by Yoshida et al (55), which reported sensitivity in 3 out
of 6 tested cell lines upon 18 h treatment. The present study has
reported on a further 3 cell lines, with resistance detected in all
3 upon 72 h treatment. Notably, when tested between 1 and 48 h, MRT
cells initially responded to TRAIL treatment, but recovery was
observed after 16 h. This was confirmed by cell death and caspase
analysis across a broad range of timepoints. The reasons for this
recovery are not entirely obvious. It is possible that the
surviving population increased proliferation upon TRAIL treatment
mediating recovery. In support of this, previous reports have
demonstrated nuclear factor (NF)-κB and extracellular
signal-regulated kinase (ERK) activation upon TRAIL stimulation
(66-70). However, engagement of these
pathways was not detected at a range of early and later timepoints
in the present study (data not shown). Additionally, it is also
possible that the cells became resistant to TRAIL over time. In
line with this, studies by Flusberg et al (71) demonstrated that MCF10A and HCT116
cancer cells treated with TRAIL developed reversible resistance
(71). While their surviving cells
exhibited enhanced NF-κB signalling, this was found to be
dispensable for resistance. The experimental design of the current
study was clearly different to that described by Flusberg et
al, which investigated exclusively the development of
resistance in a survivor population. Yet, both suggest that cancer
cells can develop resistance to TRAIL treatment independently of
enhanced proliferative signalling.
Of note, the recovery of cells from TRAIL was
inhibited by embelin co-treatment which resulted in synergistically
enhanced cell death and caspase activity in BT12 and BT16 cells.
The present results are supported by the literature where embelin
has been reported to enhance TRAIL-mediated cell death in acute
myeloid leukaemia, malignant glioma, pancreatic, inflammatory
breast, and non-small cell lung cancer cell lines (47-54).
Previous reports have differed in their proposed mechanism with
changes in expression of anti-apoptotic proteins FLIP, XIAP,
survivin and Bcl-2 family members, alongside altered regulation of
the NF-κB and ERK pathways among the factors implicated. The
mechanism may thus be concentration or cell type-specific. In MRT,
at the doses used, enhanced cell death was accompanied by reduced
expression of Mcl-1 from 4 h and reduced FLIPL and
survivin levels from 16 h. No changes in XIAP expression were noted
until relatively late timepoints, by which time caspase-3 activity
was already enhanced. While this may suggest that the enhanced cell
death observed did not rely upon embelin-mediated XIAP inhibition,
it should be noted that the original screening studies which
identified embelin as an inhibitor of XIAP did not suggest protein
degradation to be necessary for inhibition (37). In the present study, XIAP
knockdown, via siRNA, significantly enhanced TRAIL-mediated cell
death. This was further supported by analysis of the cleaved
caspase-3 fragments, with the inactive fragment predominating in
TRAIL-treated cells in contrast to further processing evident upon
combination treatment. This is in line with results previously
reported, where enhanced SMAC release resulted in increased XIAP
inhibition and completed caspase-3 processing (72,73).
In addition, livin cleavage, likely mediated by caspase-3 was
observed in correlation with increased caspase activity. Livin has
been demonstrated to bind SMAC and is suggested to inhibit its XIAP
inhibitory activity (74). Livin
cleavage may thus facilitate enhanced SMAC inhibition of XIAP.
Furthermore, the cleaved livin fragments generated have previously
been demonstrated to be proapoptotic (75,76).
The present results suggest that the enhanced cell death observed
upon embelin cotreatment is likely multifactorial, with reduced
expression of the antiapoptotic proteins survivin, FLIPL
and Mcl-1 being combined with livin cleavage and XIAP inhibition,
to most likely account for the synergism observed.
In conclusion, the current study has highlighted the
antiapoptotic IAP family members as possible targets for the
treatment of MRT and suggests that further preclinical evaluation
of their inhibition is warranted. Furthermore, the present results
suggest that the XIAP inhibitor embelin may be combined with TRAIL
as a potential novel treatment strategy for MRT, an aggressive
malignancy desperately in need of new therapeutic options.
Supplementary Materials
Funding
This study was supported by a Trinity College Dublin
postgraduate Ussher fellowship (Grant no. 1826 9050332).
Availability of data and materials
The datasets used and/or analysed during the present
study are available from the corresponding author on reasonable
request.
Author's contributions
DMZ and MJO'S decided upon this line of study.
Experiments were planned and designed by DMZ and RC. Experiments
were executed by RC, with KS and LE performing initial optimisation
experiments and repeating a number of experiments. RC prepared and
labelled all figures, executed experiments for Figs. 2B-7 independently and performed repeats for
Figs. 1 and 2A. KS performed a number of replicates
for Fig. 2A and optimisation
experiments for Fig. 2B. LE
performed optimisation experiments for Figs. 1 and 2, and a number of replicates for Fig. 1A. The manuscript was written by RC
and reviewed by DMZ and MJO'S. DMZ and MJO'S reviewed all figures
for publication. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Brennan B, Stiller C and Bourdeaut F:
Extracranial rhabdoid tumours: What we have learned so far and
future directions. Lancet Oncol. 14:e329–e336. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Geller JI, Roth JJ and Biegel JA: Biology
and Treatment of Rhabdoid Tumor. Crit Rev Oncog. 20:199–216. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Biegel JA, Kalpana G, Knudsen ES, Packer
RJ, Roberts CW, Thiele CJ, Weissman B and Smith M: The role of INI1
and the SWI/SNF complex in the development of rhabdoid tumors:
Meeting summary from the workshop on childhood atypical
teratoid/rhabdoid tumors. Cancer Res. 62:323–328. 2002.PubMed/NCBI
|
|
4
|
Versteege I, Sévenet N, Lange J,
Rousseau-Merck MF, Ambros P, Handgretinger R, Aurias A and Delattre
O: Truncating mutations of hSNF5/INI1 in aggressive paediatric
cancer. Nature. 394:203–206. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hasselblatt M, Isken S, Linge A, Eikmeier
K, Jeibmann A, Oyen F, Nagel I, Richter J, Bartelheim K, Kordes U,
et al: High-resolution genomic analysis suggests the absence of
recurrent genomic alterations other than SMARCB1 aberrations in
atypical teratoid/rhabdoid tumors. Genes Chromosomes Cancer.
52:185–190. 2013. View Article : Google Scholar
|
|
6
|
Kieran MW, Roberts CW, Chi SN, Ligon KL,
Rich BE, Macconaill LE, Garraway LA and Biegel JA: Absence of
oncogenic canonical pathway mutations in aggressive pediatric
rhabdoid tumors. Pediatric Blood Cancer. 59:1155–1157. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
McKenna ES, Sansam CG, Cho YJ, Greulich H,
Evans JA, Thom CS, Moreau LA, Biegel JA, Pomeroy SL and Roberts CW:
Loss of the epigenetic tumor suppressor SNF5 leads to cancer
without genomic instability. Mol Cell Biol. 28:6223–6233. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lafay-Cousin L, Hawkins C, Carret AS,
Johnston D, Zelcer S, Wilson B, Jabado N, Scheinemann K, Eisenstat
D, Fryer C, et al: Central nervous system atypical teratoid
rhabdoid tumours: The Canadian Paediatric Brain Tumour Consortium
experience. Eur J Cancer. 48:353–359. 2012. View Article : Google Scholar
|
|
9
|
Tekautz TM, Fuller CE, Blaney S, Fouladi
M, Broniscer A, Merchant TE, Krasin M, Dalton J, Hale G, Kun LE, et
al: Atypical teratoid/rhabdoid tumors (ATRT): Improved survival in
children 3 years of age and older with radiation therapy and
high-dose alkylator-based chemotherapy. J Clin Oncol. 23:1491–1499.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chi SN, Zimmerman MA, Yao X, Cohen KJ,
Burger P, Biegel JA, Rorke-Adams LB, Fisher MJ, Janss A, Mazewski
C, et al: Intensive multimodality treatment for children with newly
diagnosed CNS atypical teratoid rhabdoid tumor. J Clin Oncol.
27:385–389. 2009. View Article : Google Scholar :
|
|
11
|
Walczak H, Miller RE, Ariail K, Gliniak B,
Griffith TS, Kubin M, Chin W, Jones J, Woodward A, Le T, et al:
Tumoricidal activity of tumor necrosis factor-related
apoptosis-inducing ligand in vivo. Nat Med. 5:157–163. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Walczak H: Death receptor-ligand systems
in cancer, cell death, and inflammation. Cold Spring Harb Perspect
Biol. 5:a0086982013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Czabotar PE, Lessene G, Strasser A and
Adams JM: Control of apoptosis by the BCL-2 protein family:
Implications for physiology and therapy. Nat Rev Mol Cell Biol.
15:49–63. 2014. View
Article : Google Scholar
|
|
14
|
Ouchi K, Kuwahara Y, Iehara T, Miyachi M,
Katsumi Y, Tsuchiya K, Konishi E, Yanagisawa A and Hosoi H: A
NOXA/MCL-1 Imbalance Underlies Chemoresistance of Malignant
Rhabdoid Tumor Cells. J Cell Physiol. 231:1932–1940. 2016.
View Article : Google Scholar
|
|
15
|
Cai JH, Fu SM, Tu ZH, Deng LQ, Liang Z,
Chen XP, Gong XJ and Wan LH: Survivin gene functions and
relationships between expression and prognosis in patients with
nasopharyngeal carcinoma. Asian Pac J Cancer Prev. 16:2341–2345.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen W, Qiu L, Hou J, Zhang X, Ke X, Wang
Z, Zhou F, Yang S, Zhao Y, Leng Y, et al: Phase Ib Study of
Recombinant Circularly Permuted TRAIL (CPT) in Relapsed or
Refractory multiple Myeloma Patients. Blood. 120:18572012.
|
|
17
|
Hotte SJ, Hirte HW, Chen EX, Siu LL, Le
LH, Corey A, Iacobucci A, MacLean M, Lo L, Fox NL and Oza AM: A
phase 1 study of mapatumumab (fully human monoclonal antibody to
TRAIL-R1) in patients with advanced solid malignancies. Clin Cancer
Res. 14:3450–3455. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pan Y, Xu R, Peach M, Huang CP,
Branstetter D, Novotny W, Herbst RS, Eckhardt SG and Holland PM:
Evaluation of pharmacodynamic biomarkers in a Phase 1a trial of
dulanermin (rhApo2L/TRAIL) in patients with advanced tumours. Br J
Cancer. 105:1830–1838. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Soria JC, Márk Z, Zatloukal P, Szima B,
Albert I, Juhász E, Pujol JL, Kozielski J, Baker N, Smethurst D, et
al: Randomized phase II study of dulanermin in combination with
paclitaxel, carboplatin, and bevacizumab in advanced non-small-cell
lung cancer. J Clin Oncol. 29:4442–4451. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cho SB, Lee WS, Park YL, Kim N, Oh HH, Kim
MY, Oak CY, Chung CY, Park HC, Kim JS, et al: Livin is associated
with the invasive and oncogenic phenotypes of human hepatocellular
carcinoma cells. Hepatol Res. 45:448–457. 2015. View Article : Google Scholar
|
|
21
|
Dasgupta A, Alvarado CS, Xu Z and Findley
HW: Expression and functional role of inhibitor-of-apoptosis
protein livin (BIRC7) in neuroblastoma. Biochem Biophys Res Commun.
400:53–59. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kempkensteffen C, Hinz S, Christoph F,
Köllermann J, Krause H, Schrader M, Schostak M, Miller K and
Weikert S: Expression parameters of the inhibitors of apoptosis
cIAP1 and cIAP2 in renal cell carcinomas and their prognostic
relevance. Int J Cancer. 120:1081–1086. 2007. View Article : Google Scholar
|
|
23
|
Kim DK, Alvarado CS, Abramowsky CR, Gu L,
Zhou M, Soe MM, Sullivan K, George B, Schemankewitz E and Findley
HW: Expression of inhibitor-of-apoptosis protein (IAP) livin by
neuroblastoma cells: correlation with prognostic factors and
outcome. Pediatr Dev Pathol. 8:621–629. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mizutani Y, Nakanishi H, Li YN, Matsubara
H, Yamamoto K, Sato N, Shiraishi T, Nakamura T, Mikami K, Okihara
K, et al: Overexpression of XIAP expression in renal cell carcinoma
predicts a worse prognosis. Int J Oncol. 30:919–925.
2007.PubMed/NCBI
|
|
25
|
Ramp U, Krieg T, Caliskan E, Mahotka C,
Ebert T, Willers R, Gabbert HE and Gerharz CD: XIAP expression is
an independent prognostic marker in clear-cell renal carcinomas.
Hum Pathol. 35:1022–1028. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shi YH, Ding WX, Zhou J, He JY, Xu Y,
Gambotto AA, Rabinowich H, Fan J and Yin XM: Expression of X-linked
inhibitor-of-apoptosis protein in hepatocellular carcinoma promotes
metastasis and tumor recurrence. Hepatology. 48:497–507. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tamm I, Kornblau SM, Segall H, Krajewski
S, Welsh K, Kitada S, Welsh K, Kitada S, Scudiero DA, Tudor G, Qui
YH, Monks A, et al: Expression and prognostic significance of
IAP-family genes in human cancers and myeloid leukemias. Clin
Cancer Res. 6:1796–1803. 2000.PubMed/NCBI
|
|
28
|
Tamm I, Richter S, Oltersdorf D, Creutzig
U, Harbott J, Scholz F, Karawajew L, Ludwig WD and Wuchter C: High
expression levels of x-linked inhibitor of apoptosis protein and
survivin correlate with poor overall survival in childhood de novo
acute myeloid leukemia. Clin Cancer Res. 10:3737–3744. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Huang Y, Park YC, Rich RL, Segal D, Myszka
DG and Wu H: Structural basis of caspase inhibition by XIAP:
Differential roles of the linker versus the BIR domain. Cell.
104:781–790. 2001.PubMed/NCBI
|
|
30
|
Riedl SJ, Renatus M, Schwarzenbacher R,
Zhou Q, Sun C, Fesik SW, Liddington RC and Salvesen GS: Structural
basis for the inhibition of caspase-3 by XIAP. Cell. 104:791–800.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Scott FL, Denault JB, Riedl SJ, Shin H,
Renatus M and Salvesen GS: XIAP inhibits caspase-3 and -7 using two
binding sites: evolutionarily conserved mechanism of IAPs. EMBO J.
24:645–655. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shiozaki EN, Chai J, Rigotti DJ, Riedl SJ,
Li P, Srinivasula SM, Alnemri ES, Fairman R and Shi Y: Mechanism of
XIAP-mediated inhibition of caspase-9. Mol Cell. 11:519–527. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Srinivasula SM, Hegde R, Saleh A, Datta P,
Shiozaki E, Chai J, Lee RA, Robbins PD, Fernandes-Alnemri T, Shi Y,
et al: A conserved XIAP-interaction motif in caspase-9 and
Smac/DIABLO regulates caspase activity and apoptosis. Nature.
410:112–116. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Suzuki Y, Nakabayashi Y, Nakata K, Reed JC
and Takahashi R: X-linked inhibitor of apoptosis protein (XIAP)
inhibits caspase-3 and -7 in distinct modes. J Biol Chem.
276:27058–27063. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Deveraux QL, Roy N, Stennicke HR, Van
Arsdale T, Zhou Q, Srinivasula SM, Srinivasula SM, Alnemri ES,
Salvesen GS and Reed JC: IAPs block apoptotic events induced by
caspase-8 and cytochrome c by direct inhibition of distinct
caspases. EMBO J. 17:2215–2223. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Deveraux QL, Takahashi R, Salvesen GS and
Reed JC: X-linked IAP is a direct inhibitor of cell-death
proteases. Nature. 388:300–304. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nikolovska-Coleska Z, Xu L, Hu Z, Tomita
Y, Li P, Roller PP, Wang R, Fang X, Guo R, Zhang M, et al:
Discovery of embelin as a cell-permeable, small-molecular weight
inhibitor of XIAP through structure-based computational screening
of a traditional herbal medicine three-dimensional structure
database. J Med Chem. 47:2430–2440. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hu R, Zhu K, Li Y, Yao K, Zhang R, Wang H,
Yang W and Liu Z: Embelin induces apoptosis through down-regulation
of XIAP in human leukemia cells. Med Oncol. 28:1584–1588. 2011.
View Article : Google Scholar
|
|
39
|
Park N, Baek HS and Chun YJ:
Embelin-Induced Apoptosis of Human Prostate Cancer Cells Is
Mediated through Modulation of Akt and β-Catenin Signaling. PloS
One. 10:e01347602015. View Article : Google Scholar
|
|
40
|
Shah P, Djisam R, Damulira H, Aganze A and
Danquah M: Embelin inhibits proliferation, induces apoptosis and
alters gene expression profiles in breast cancer cells. Pharmacol
Rep. 68:638–644. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sumalatha KR, Abiramasundari G, Chetan GK,
Divya T, Sudhandiran G and Sreepriya M: XIAP inhibitor and
antiestrogen embelin abrogates metastasis and augments apoptosis in
estrogen receptor positive human breast adenocarcinoma cell line
MCF-7. Mol Biol Rep. 41:935–946. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang A, Zhang B, Zhang J and Wu W and Wu
W: Embelin-induced brain glioma cell apoptosis and cell cycle
arrest via the mitochondrial pathway. Oncol Rep. 29:2473–2478.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wang DG, Sun YB, Ye F, Li W, Kharbuja P,
Gao L, Zhang DY and Suo J: Anti-tumor activity of the X-linked
inhibitor of apoptosis (XIAP) inhibitor embelin in gastric cancer
cells. Mol Cell Biochem. 386:143–152. 2014. View Article : Google Scholar
|
|
44
|
Xu CL, Zheng B, Pei JH, Shen SJ and Wang
JZ: Embelin induces apoptosis of human gastric carcinoma through
inhibition of p38 MAPK and NF-κB signaling pathways. Mol Med Rep.
14:307–312. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Cheng YJ, Jiang HS, Hsu SL, Lin LC, Wu CL,
Ghanta VK and Hsueh CM: XIAP-mediated protection of H460 lung
cancer cells against cisplatin. Eur J Pharmacol. 627:75–84. 2010.
View Article : Google Scholar
|
|
46
|
Dai Y, Desano J, Qu Y, Tang W, Meng Y,
Lawrence TS and Xu L: Natural IAP inhibitor Embelin enhances
therapeutic efficacy of ionizing radiation in prostate cancer. Am J
Cancer Res. 1:128–143. 2011.PubMed/NCBI
|
|
47
|
Allensworth JL, Aird KM, Aldrich AJ,
Batinic-Haberle I and Devi GR: XIAP inhibition and generation of
reactive oxygen species enhances TRAIL sensitivity in inflammatory
breast cancer cells. Mol Cancer Ther. 11:1518–1527. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Hu R, Yang Y, Liu Z, Jiang H, Zhu K, Li J
and Xu W: The XIAP inhibitor Embelin enhances TRAIL-induced
apoptosis in human leukemia cells by DR4 and DR5 upregulation.
Tumour Biol. 36:769–777. 2015. View Article : Google Scholar
|
|
49
|
Jiang L, Hao JL, Jin ML, Zhang YG and Wei
P: Effect of Embelin on TRAIL receptor 2 mAb-induced apoptosis of
TRAIL-resistant A549 non-small cell lung cancer cells. Asian Pac J
Cancer Prev. 14:6115–6120. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Mori T, Doi R, Kida A, Nagai K, Kami K,
Ito D, Toyoda E, Kawaguchi Y and Uemoto S: Effect of the XIAP
inhibitor Embelin on TRAIL-induced apoptosis of pancreatic cancer
cells. J Surg Res. 142:281–286. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Qian H, Chen Y, Huang T, Liu T, Li X,
Jiang G, Zhang W, Cheng S and Li P: Combined application of Embelin
and tumor necrosis factor-related apoptosis-inducing ligand
inhibits proliferation and invasion in osteosarcoma cells via
caspase-induced apoptosis. Oncol Lett. 15:6931–6940.
2018.PubMed/NCBI
|
|
52
|
Siegelin MD, Gaiser T and Siegelin Y: The
XIAP inhibitor Embelin enhances TRAIL-mediated apoptosis in
malignant glioma cells by down-regulation of the short isoform of
FLIP. Neurochem Int. 55:423–430. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Yang S, Li SS, Yang XM, Yin DH and Wang L:
Embelin prevents LMP1-induced TRAIL resistance via inhibition of
XIAP in nasopharyngeal carcinoma cells. Oncol Lett. 11:4167–4176.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Yang T, Lan J, Huang Q, Chen X, Sun X, Liu
X, Yang P, Jin T, Wang S and Mou X: Embelin sensitizes acute
myeloid leukemia cells to TRAIL through XIAP inhibition and NF-κB
inactivation. Cell Biochem Biophys. 71:291–297. 2015. View Article : Google Scholar
|
|
55
|
Yoshida S, Narita T, Koshida S, Ohta S and
Takeuchi Y: TRAIL/Apo2L ligands induce apoptosis in malignant
rhabdoid tumor cell lines. Pediatr Res. 54:709–717. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Du C, Fang M, Li Y, Li L and Wang X: Smac,
a mitochondrial protein that promotes cytochrome c-dependent
caspase activation by eliminating IAP inhibition. Cell. 102:33–42.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Ekert PG, Silke J, Hawkins CJ, Verhagen AM
and Vaux DL: DIABLO promotes apoptosis by removing MIHA/XIAP from
processed caspase 9. J Cell Biol. 152:483–490. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Houghton PJ, Kang MH, Reynolds CP, Morton
CL, Kolb EA, Gorlick R, Keir ST, Carol H, Lock R, Maris JM, et al:
Initial testing (stage 1) of LCL161, a SMAC mimetic, by the
Pediatric Preclinical Testing Program. Pediatr Blood Cancer.
58:636–639. 2012. View Article : Google Scholar :
|
|
59
|
Langemann D, Trochimiuk M, Appl B,
Hundsdoerfer P, Reinshagen K and Eschenburg G: Sensitization of
neuroblastoma for vincristine-induced apoptosis by Smac mimetic
LCL161 is attended by G2 cell cycle arrest but is independent of
NFκB, RIP1 and TNF-α. Oncotarget. 8:87763–87772. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Najem S, Langemann D, Appl B, Trochimiuk
M, Hundsdoerfer P, Reinshagen K and Eschenburg G: Smac mimetic
LCL161 supports neuroblastoma chemotherapy in a drug
class-dependent manner and synergistically interacts with ALK
inhibitor TAE684 in cells with ALK mutation F1174L. Oncotarget.
7:72634–72653. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Qin Q, Zuo Y, Yang X, Lu J, Zhan L, Xu L,
Zhang C, Zhu H, Liu J, Liu Z, et al: Smac mimetic compound LCL161
sensitizes esophageal carcinoma cells to radiotherapy by inhibiting
the expression of inhibitor of apoptosis protein. Tumour Biol.
35:2565–2574. 2014. View Article : Google Scholar
|
|
62
|
Yang C, Wang H, Zhang B, Chen Y, Zhang Y,
Sun X, Xiao G, Nan K, Ren H and Qin S: LCL161 increases
paclitaxel-induced apoptosis by degrading cIAP1 and cIAP2 in NSCLC.
J Exp Clin Cancer Res. 35:1582016. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Clemens MR, Gladkov OA, Gartner E,
Vladimirov V, Crown J, Steinberg J, Jie F and Keating A: Phase II,
multicenter, open-label, randomized study of YM155 plus docetaxel
as first-line treatment in patients with HER2-negative metastatic
breast cancer. Breast Cancer Res Treat. 149:171–179. 2015.
View Article : Google Scholar :
|
|
64
|
Kelly RJ, Thomas A, Rajan A, Chun G,
Lopez-Chavez A, Szabo E, Spencer S, Carter CA, Guha U, Khozin S, et
al: A phase I/II study of sepantronium bromide (YM155, survivin
suppressor) with paclitaxel and carboplatin in patients with
advanced non-small-cell lung cancer. Ann Oncol. 24:2601–2606. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Singh B, Guru SK, Sharma R, Bharate SS,
Khan IA, Bhushan S, Bharate SB and Vishwakarma RA: Synthesis and
anti-proliferative activities of new derivatives of embelin. Bioorg
Med Chem Lett. 24:4865–4870. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Hartwig T, Montinaro A, von Karstedt S,
Sevko A, Surinova S, Chakravarthy A, Taraborrelli L, Draber P,
Lafont E, Arce Vargas F, et al: The TRAIL-Induced Cancer Secretome
Promotes a Tumor-Supportive Immune Microenvironment via CCR2. Mol
Cell. 65:730–742.e5. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Henry CM and Martin SJ: Caspase-8 Acts in
a Non-enzymatic Role as a Scaffold for Assembly of a
Pro-inflammatory 'FADDosome' Complex upon TRAIL Stimulation. Mol
Cell. 65:715–729.e5. 2017. View Article : Google Scholar
|
|
68
|
Lafont E, Kantari-Mimoun C, Draber P, De
Miguel D, Hartwig T, Reichert M, Kupka S, Shimizu Y, Taraborrelli
L, Spit M, et al: The linear ubiquitin chain assembly complex
regulates TRAIL-induced gene activation and cell death. EMBO J.
36:1147–1166. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Varfolomeev E, Maecker H, Sharp D,
Lawrence D, Renz M, Vucic D and Ashkenazi A: Molecular determinants
of kinase pathway activation by Apo2 ligand/tumor necrosis
factor-related apoptosis-inducing ligand. J Biol Chem.
280:40599–40608. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
von Karstedt S, Conti A, Nobis M,
Montinaro A, Hartwig T, Lemke J, Legler K, Annewanter F, Campbell
AD, Taraborrelli L, et al: Cancer cell-autonomous TRAIL-R signaling
promotes KRAS-driven cancer progression, invasion, and metastasis.
Cancer Cell. 27:561–573. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Flusberg DA, Roux J, Spencer SL and Sorger
PK: Cells surviving fractional killing by TRAIL exhibit transient
but sustainable resistance and inflammatory phenotypes. Mol Biol
Cell. 24:2186–2200. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Hörnle M, Peters N, Thayaparasingham B,
Vörsmann H, Kashkar H and Kulms D: Caspase-3 cleaves XIAP in a
positive feedback loop to sensitize melanoma cells to TRAIL-induced
apoptosis. Oncogene. 30:575–587. 2011. View Article : Google Scholar
|
|
73
|
Thayaparasingham B, Kunz A, Peters N and
Kulms D: Sensitization of melanoma cells to TRAIL by UVB-induced
and NF-kappaB-mediated downregulation of xIAP. Oncogene.
28:345–362. 2009. View Article : Google Scholar
|
|
74
|
Ma L, Huang Y, Song Z, Feng S, Tian X, Du
W, Qiu X, Heese K and Wu M: Livin promotes Smac/DIABLO degradation
by ubiquitin-proteasome pathway. Cell Death Differ. 13:2079–2088.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Abd-Elrahman I, Hershko K, Neuman T,
Nachmias B, Perlman R and Ben-Yehuda D: The inhibitor of apoptosis
protein Livin (ML-IAP) plays a dual role in tumorigenicity. Cancer
Res. 69:5475–5480. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Nachmias B, Ashhab Y, Bucholtz V, Drize O,
Kadouri L, Lotem M, Peretz T, Mandelboim O and Ben-Yehuda D:
Caspase-mediated cleavage converts Livin from an antiapoptotic to a
proapoptotic factor: Implications for drug-resistant melanoma.
Cancer Res. 63:6340–6349. 2003.PubMed/NCBI
|