The high mobility group (HMG) proteins, which are
present in only ~3% of the histone content by weight (1), are novel abundant, heterogeneous,
non-histone components of chromatin that were first identified in
1973 (2). The HMG protein family
has been classified into three subfamilies: HMGA, HMGB and HMGN,
previously known as HMGI/Y, HMG1-2 and HMG14/17, respectively
(3). Each of these subfamilies has
a unique protein signature and a characteristic functional sequence
motif: The 'AT-hook' for the HMGA subfamily, the 'HMG-box' for the
HMGB family and the 'nucleosomal binding domain' for the HMGN
family. Through their respective functional motifs, each can bind
to specific structures in DNA or chromatin in a
sequence-independent manner. The HMGA subfamily consists of four
proteins: HMGA1a, HMGA1b, HMGA1c and HMGA2. The first three members
are encoded by the HMGA1 gene through alternative splicing. The
latter is encoded by the separate HMGA2 gene. Although HMGA1 and
HMGA2 have overlapping structures and functions (1,4), a
number of genes are specifically regulated by only one of these,
resulting in different roles in cancer (5,6). The
distinct functions of the HMGA1 and HMGA2 proteins in human
neoplastic diseases have been reviewed previously (7). The human HMGA2 gene is located
at chromosomal band 12q14-15, which contains at least five exons
dispersed over a genomic region of ≥140 kb (Fig. 1). The HMGA2 protein encodes 108
amino acid residues, and although this small, non-histone
chromatin-associated protein has no intrinsic transcriptional
activity, it can modulate gene transcription by altering chromatin
architecture (8). The AT-hook
motif of HMGA2 is a positively charged stretch of nine amino acids
containing the invariant repeat Arg-Gly-Arg-Pro(R-G-R-P) (9), which can bind to B-form DNA and
undergo a disordered-to-ordered conformational change during the
regulation of gene transcription. Depending on the number and
spacing of the AT-rich binding sites in DNA, HMGA2 influences the
conformation of combinative DNA substrates in different ways,
thereby enhancing or suppressing the transcriptional activity of
several human genes, subsequently influencing a variety of
biological processes (10).
Strict regulation of the expression of HMGA2 is
critical for embryonic stem cell development. The dysregulation of
HMGA2 in adult somatic cells renders them prone to tumourigenesis,
and mutation of the HMGA2-encoding gene is widely observed in a
large array of tumours (11). The
basal regulation of the HMGA2 gene promoter is controlled by a
polypyrimidine/polypurine element, which can be positively or
negatively bound by several regulatory elements (12,13).
It is suggested that transforming growth factor (TGF)β induces the
transcription of HMGA2, and it is TGFβ-induced Smad4 that directly
binds to the HMGA2 promoter during-the regulation of HMGA2
(14). In addition, β-catenin
directly binds to the HMGA2 promoter and leads to upregulation of
the expression of HMGA2 (15).
Runt-related transcription factor 1 binds to the HMGA2 promoter and
regulates HMGA2 promoter activity in a cell-type-dependent manner
(16). MicroRNAs (miRNAs) are
small, 21-25 nucleotide lengths of non-coding RNAs, which
post-transcriptionally repress specified messenger RNAs by binding
to the 3' untranslated region (UTR) of their targets (17). Let-7 is one of the founding members
of the miRNA family that can directly bind to the 3'-UTR of the
human HMGA2 gene, resulting in the repressive expression of HMGA2
(18). Inhibition of the
expression of HMGA2 by exogenous Let-7 impairs tumour cell
proliferation, and Let-7 can be packaged and released via exosomes
by tumours cells, thereby inducing a high expression of HMGA2 in
tumour cells (19). By contrast, a
decrease in the expression of Let-7 by oncostatin M treatment has
been shown to cause the expression of HMGA2 to be rapidly elevated,
resulting in enhancement of the invasiveness and metastasis of
breast cancer (20). Therefore,
Let-7 is accepted as an upstream inhibiting factor targeting HMGA2.
In addition, Lin-28, an embryonic stem cell-specific protein,
serves as a competitor RNA that can mimic the binding site of Let-7
and prevent the Let-7 precursor from being processed to mature
miRNAs by inducing terminal uridylation and degradation of Let-7
precursors. Therefore, the overexpression of Lin-28 impairs Let-7
function and derepresses the expression of HMGA2 (21). The Lin28-Let-7-HMGA2 axis is a
critical regulatory system for maintaining an undifferentiated
state in cancer cells (18,22,23).
Raf-1 kinase inhibitory protein (RKIP) is a member of the
evolutionarily conserved phosphatidylethanolamine-binding protein
family that is poorly expressed in tumour cells. It has been shown
that RKIP negatively modulated Raf-1/MEK/ERK1/2 cascade activity
and subsequently impaired the Lin28/Let-7/HMGA2 axis, thereby
inhibiting the transcription of HMGA2 (24,25).
Several other miRNAs, including miRNA (miR)-33b, miR-145, miR-9,
miR-93 and miR539, have also been reported to be involved in the
regulation of HMGA2 (26-30). Furthermore, long non-coding RNAs
(IncRNAs) are also likely to affect the expression of HMGA2
(31). RPSAP52 is an antisense
lncRNA transcribed from the HMGA2 locus, and it can form an R-loop
at the promoter of HMGA2, thereby improving accessibility to the
transcription machinery (31,32).
Notably, HMGA2 is expressed in pluripotent embryonic
stem cells during embryogenesis, but is absent or present only at
low levels in adult tissue cells (33). However, HMGA2 is re-expressed in
human malignancies, indicating that HMGA2 may be essential in
development and carcinogenesis. Heterozygous HMGA2−/+
mice and homozygous HMGA2−/− mice exhibit a pygmy
phenotype, with a body size of 80 and 40% of wild-type littermates,
respectively, due to a reduction in cell growth (33). A common variant of HMGA2 is
associated with human growth height (34), and HMGA2 disruption may lead to
foetal growth restriction (35).
Several experimental models have shown the potent neoplastic
transforming ability of HMGA2. Full-length HMGA2 transgenic mice
and truncated HMGA2 transgenic mice produce a similar benign
mesenchymal neoplastic phenotype, including fibroadenomas of the
breast and salivary gland adenomas (36). Transgenic mice carrying wild-type
HMGA2 genes develop pituitary adenomas (8). When fibroblast cells with ectopic
overexpression of HMGA2 are injected into athymic nude mice,
fibrosarcomas are formed and develop distant metastases (4). Transgenic mice bearing the human
HMGA2 gene under the control of the VH promoter/Eµ enhancer suffer
from precursor T-cell lymphoblastic leukaemia (37). Therefore, the dysregulation of
HMGA2 may be an important step in the pathogenesis of malignancies.
Furthermore, the dysregulation of HMGA2 in different human tumour
tissues suggests the role of carcinogenesis. Non-random chromosomal
translocations (38) lead to the
overexpression of HMGA2 in several types of mesenchymal tumour,
including conventional and intramuscular lipomas,
well-differentiated and dedifferentiated liposarcoma, benign
fibrous histiocytomas, nodular fasciitis and aggressive angiomyxoma
(39). In addition, it has been
suggested that the overexpression of HMGA2 in human epithelial
malignancies is correlated with a highly malignant phenotype and
poorer survival rates (Table I).
The same phenomenon is seen in acute myeloid leukaemia (40). To illustrate, HMGA2
immunoreactivity is observed in primary colorectal cancer cells and
metastatic colon cancer to liver, but not in the adjacent normal
colorectal epithelium. HMGA2 also correlates positively and
significantly with distant metastasis and poor survival rates,
which support the use of HMGA2 as a potential diagnostic and
prognostic tumour marker (41).
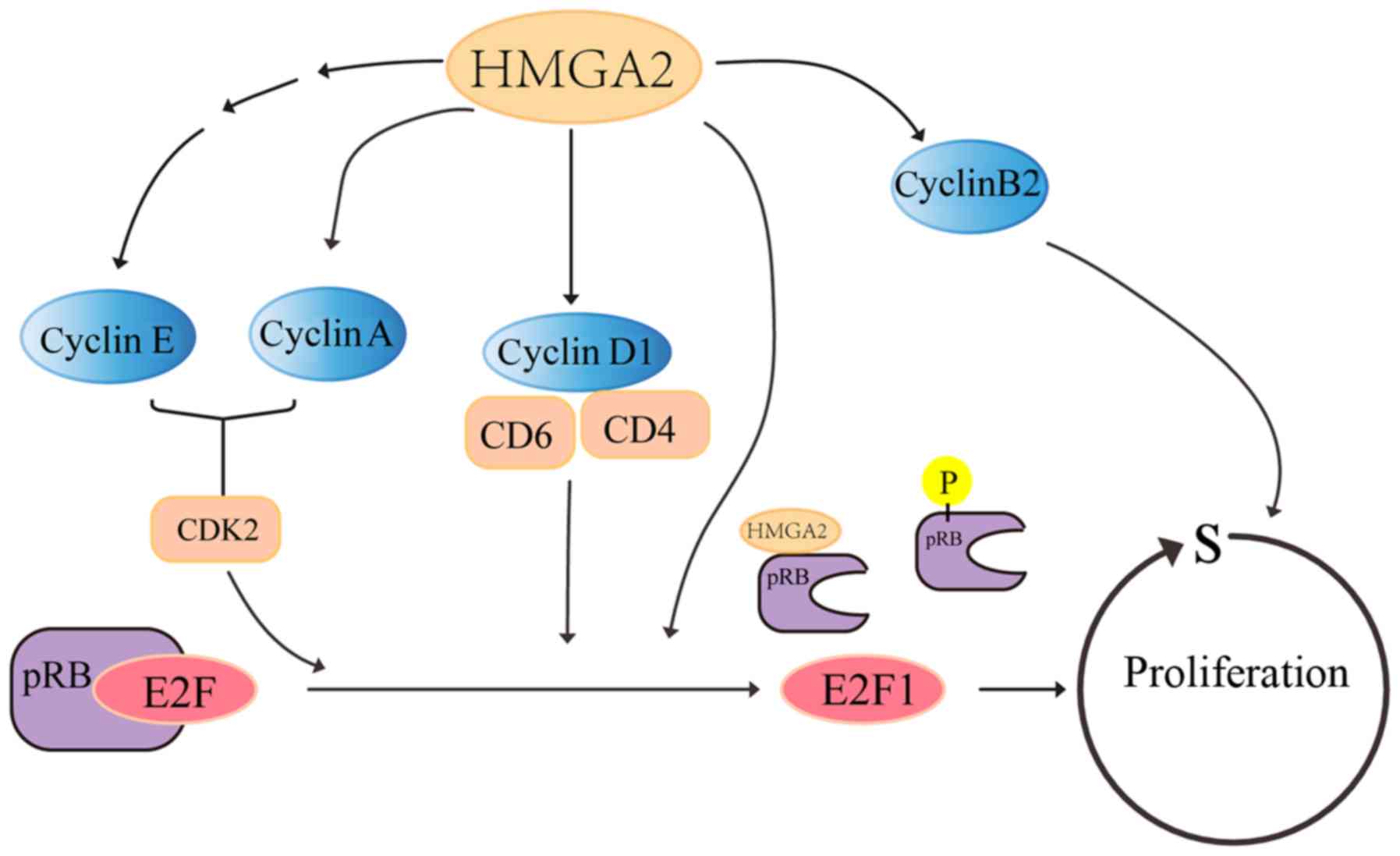
Cell proliferation requires precise progression of
the cell cycle, and an uncontrolled cell cycle gives rise to
malignant behaviour, which is responsible for neoplastic
transformation. As the overexpression of HMGA2 promotes cancer cell
proliferation, HMGA2 is considered to affect cancer cell cycle
progression (Fig. 2). The
knockdown of HMGA2 causes G1 arrest in ovarian cancer cells
(42) and G2/M arrest in leukaemia
cells (43). HMGA2 directly binds
to the cyclic AMP-responsive element of cyclin A2, which displaces
p120E4F-containing complexes from cyclin A2, thus
inducing the expression of cyclin A2 and contributing to cell cycle
progression (44,45). The transcription factor activator
protein-1 (AP1) complexes, composed of members of Jun proteins
(JUN, JUNB and JUND), FOS proteins (FOS, FOSB and FRA1) and FRA2,
are critical in the regulation of cell proliferation (46). In HMGA2-deficient cells, the
expression of JUNB and FRA1 are completely inhibited (47). By contrast, these genes are
upregulated correspondingly when HMGA2 is ectopically overexpressed
(47). HMGA2 also enhances the
expression of cyclin A2 by promoting AP1 transcriptional induction
(47). As a tumour suppressor
protein, retinoblastoma protein (pRB) strictly controls cell cycle
entry into the S phase through its interactions with the E2F1
transcription factors (48).
Before cells enter the S phase, pRB is phosphorylated and
inactivated to release E2F1, resulting in cell cycle progression
(49). It is suggested that HMGA2
displaces histone deacetylase 1 from phosphorylation of the pRB or
to act directly on the E2F-responsive DNA elements, thereby
promoting the activation of E2F1 and resulting in cell cycle
progression (50).
P16INK4A and p21CIP1/WAF1 are two
cyclin-dependent kinase inhibitors that are important in
restricting cell cycle progression by inhibiting the release of
E2F1. The overexpression of HMGA2 directly activates the
phosphatidylinositide 3-kinase (PI3K)/AKT/mTOR/p70S6K signalling
pathway, and subsequently facilitates cyclin E and suppresses the
activity of p16INK4A and p21CIP1/WAF1,
resulting in cell proliferation (51). The activation of cyclin
D1/CDK4/CDK6 is responsible for the phosphorylation of RB. HMGA2
knockout markedly decreases the synthesis of cyclin D1, whereas the
ectopic expression of HMGA2 has the opposite effect, suggesting
that HMGA2 also regulates cell cycle progression by influencing the
cyclin D1/CDK4/CDK6/pRB-E2F1 axis (52,53).
In addition, cyclin B2 protein, coded by the ccnb2 gene, is a
cell-cycle-dependent protein controlling the G2-M transition
(54). The HMGA2 protein is
capable of binding to the ccnb2 promoter and enhancing the
expression of cyclin B2 to increase cell growth (55).
The complete replication of chromosomal DNA is
required for maintaining genome stability. However, DNA damage
frequently occurs by endogenous and exogenous stimuli and induces a
fraction of replication fork arrest in every cell cycle (56). The complex of triple-stranded RecA
then forms on the nascent DNA to stabilise forks until the
replication is resumed (57,58).
HMGA2 can serve as a complement of the RecA complex and create a
protective scaffold with branched DNA at arrest forks to reduce
replication recovery times (59).
In response to the DNA damage response (DDR), an array of DNA
repair pathways, such as non-homologous end-joining (NHEJ), base
excision repair (BER) and nucleotide excision repair (NER), occur
in a multiple-step process HMGA2, as a transcriptional regulation
factor, is responsible for the regulation of several DNA
repair-related proteins and influences the DNA repair process
(Fig. 3). It is reasonable to
hypothesise that, during the early stage of carcinogenesis, HMGA2
inhibits DDR, resulting in increased DNA mutation and promoting
tumour development. In tumour treatment, HMGA2 protects tumour
cells from chemoradiotherapy damage by facilitating the DNA repair
process.
DNA double-strand breaks (DSBs) are among the most
deleterious forms of DNA damage caused by genotoxic agents
(61). There are two main repair
pathways, the homologous recombination (HR) and the NHEJ pathways,
in response to DSBs (62). The
NHEJ pathway is the major DSB repair system in homologue absence
during the S/G2 phase of the cell cycle (63,64).
When DSBs occur in vertebrates, Ku protein binds to the damaged DNA
end to form a Ku:DNA complex, serving as a scaffold that not only
recruits kinases to the sites of DNA damage but also serves a major
role in activating, other PI3K-related kinase family kinases,
including ataxia-telangiectasia-mutated (ATM) and ataxia
telangiectasia and Rad3-related (ATR), respectively (65). Subsequently, the DNA-dependent
protein kinase catalytic subunit (DNA-PKcs) associates with Ku in a
DNA-dependent manner to form an active DNA-PK holoenzyme (66). The phosphorylation of H2AX is a
sensor of DSBs, facilitating the DNA repair process by altering
chromatin structure surrounding the DNA lesion and allowing DNA
repair proteins to access the damaged regions (67). Finally, DNA ligase IV, X-ray repair
cross-complementing 4 (XRCC4) and XRCC4-like factor are recruited
for the final joining of the DNA strands As a tumour promotor,
HMGA2 causes more spontaneous chromosome aberrations in eukaryotes
(69). Li et al observed
that there were considerably more DNA lesions in cells
overexpressing HMGA2 (69). Cells
with tetraploidy (4n DNA), a phenotype of NHEJ-impaired cells, are
frequently observed in HMGA2-overexpressing cells (70), suggesting that HMGA2 is associated
with the NHEJ process. Notably, HMGA2 has been suggested to
interact with NHEJ-related proteins and serve as a negative
regulator of the DNA-PK pathway, thereby impairing the NHEJ process
and rendering cells more susceptible to DNA damage (71). The prolonged presence of DNA-PKcs
phosphorylation at Ser-2056 and Thr-2609, and accumulation of
γ-H2AX before and after laser damage have been observed in
HMGA2-overexpressing cancer cells. The release of DNA-PKcs from DSB
sites and the steady-state of the Ku80 complex in the nuclear and
DNA ends were significantly decreased when cells ectopically
overexpressed HMGA2, indicating that an appropriate NHEJ repair
mechanism did not occur in time in the HMGA2-overexpressing cells
(72). Additionally, ATM is
difficult to activate effectively following doxorubicin treatment
in HMGA2-overexpressing cancer cells, which renders cells more
susceptible to doxorubicin-induced genotoxicity (73). These observations suggest that
HMGA2 has a negative effect in the regulation of NHEJ process,
which may specifically render precancerous cells more susceptible
to harmful stimuli, but may render cancer cells more sensitive to
chemotherapeutics. By contrast, HMGA2 may also promote the NHEJ
pathway. A positive feedback loop of ATM activation during the DNA
repair process is dependent on the presence of HMGA2, and the
phosphorylation of ATM at serine 1981 is reduced in HMGA2-knockout
cells in response to DSB, causing cells to be more sensitive to
infrared exposure (74).
Furthermore, HMGA2 promotes the DNA repair pathway by sustaining
the phosphorylation of ATR, also rendering cells more resistant to
the genotoxic agent hydroxyurea (75).
The base excision repair (BER) system serves a
critical role in removing the lesions and mutations of single
bases. During the BER process, damaged DNA bases are recognised and
removed by DNA glycosylases, such that apyrimidinic/apurinic (AP)
sites are formed. These AP sites are then incised by AP
endonuclease 1 (APE1) to create 5'-dRP and 3'-OH strand break
products. Finally, the single nucleotide gaps are filled by Polβ
and the XRCC1/LIG3α complex (76).
Of note, HMGA2 possesses intrinsic dRP site cleavage activity
residing within the AT-hook 3, and the lysine at the N-terminus of
the hook of HMGA2 is responsible for recognizing and cleaving DNA
containing AP sites. In addition, HMGA2 can physically interact
with APE1 and enhance the BER process, thereby reducing the number
of genomic DNA strand breaks and conferring resistance to AP
site-inducing genotoxicants (77).
Poly(ADP-ribose) polymerase 1 (PARP-1) is a eukaryotic nucleus
enzyme that can bind to DNA damage AP sites and DNA strand breaks
by zinc-finger of binding of the PARP-1 N-terminal DNA-binding
domain, and the C-terminal catalytic domain of PARP-1 is involved
in the poly ADP-ribosylation (PARylation) of DNA binding proteins,
thus contributing to the DNA damage repair process. HMGA2 has been
reported to function as an antagonist of PARP1 inhibitors in human
cancer cells (78). Specifically,
HMGA2 colocalises and interacts with PARP1 to increase the activity
of PARP1. The AT-hooks of HMGA2 are required for PARylation upon
DNA damage during BER. As a result, HMGA2 increases cell survival
and reduces sensitivity to PARP inhibitors in cancer cells, and
targeting HMGA2 in combination with a PARP inhibitor may be a
promising therapeutic approach (78).
Nucleotide excision repair (NER) is a main DNA
repair pathway when cells confront broad helix-distorting adducts
as a result of UV-light or chemical mutagens (79). During NER, the complex of
xeroderma-pigmentosum C (XPC)-RAD23B-centrin 2 initially recognise
DNA lesions, following which the TFIIH complex, helicases XPB and
XPD, and endonucleases XPG and excision repair cross complementing
group 1 (ERCC1)-XPF complex are recruited to lesions in an orderly
manner, thus opening the DNA double helix and performing cleavage
to remove the aberrant bases (80). Therefore, ERCC1 serves a critical
role in the NER pathway, and the high expression of ERCC1 is
considered a marker for NER activity (81). A microarray experiment revealed
that the ERCC1 gene was transcriptionally regulated by HMGA2.
Furthermore, the HMGA2 protein has a high affinity to the ERCC1
promoter, which enhances its expression (82). Luciferase promoter assays showed
that the wild-type HMGA2 formed 1:1 stoichiometry binding to the
ERCC1 promoter, while the truncated HMGA2 formed 2:1 complexes with
ERCC1 but without transcriptional activity (82). Together, HMGA2 may promote the NER
pathway by increasing the transcription of ERCC1, resulting in the
resistance of cancer cells to chemotherapeutic treatment.
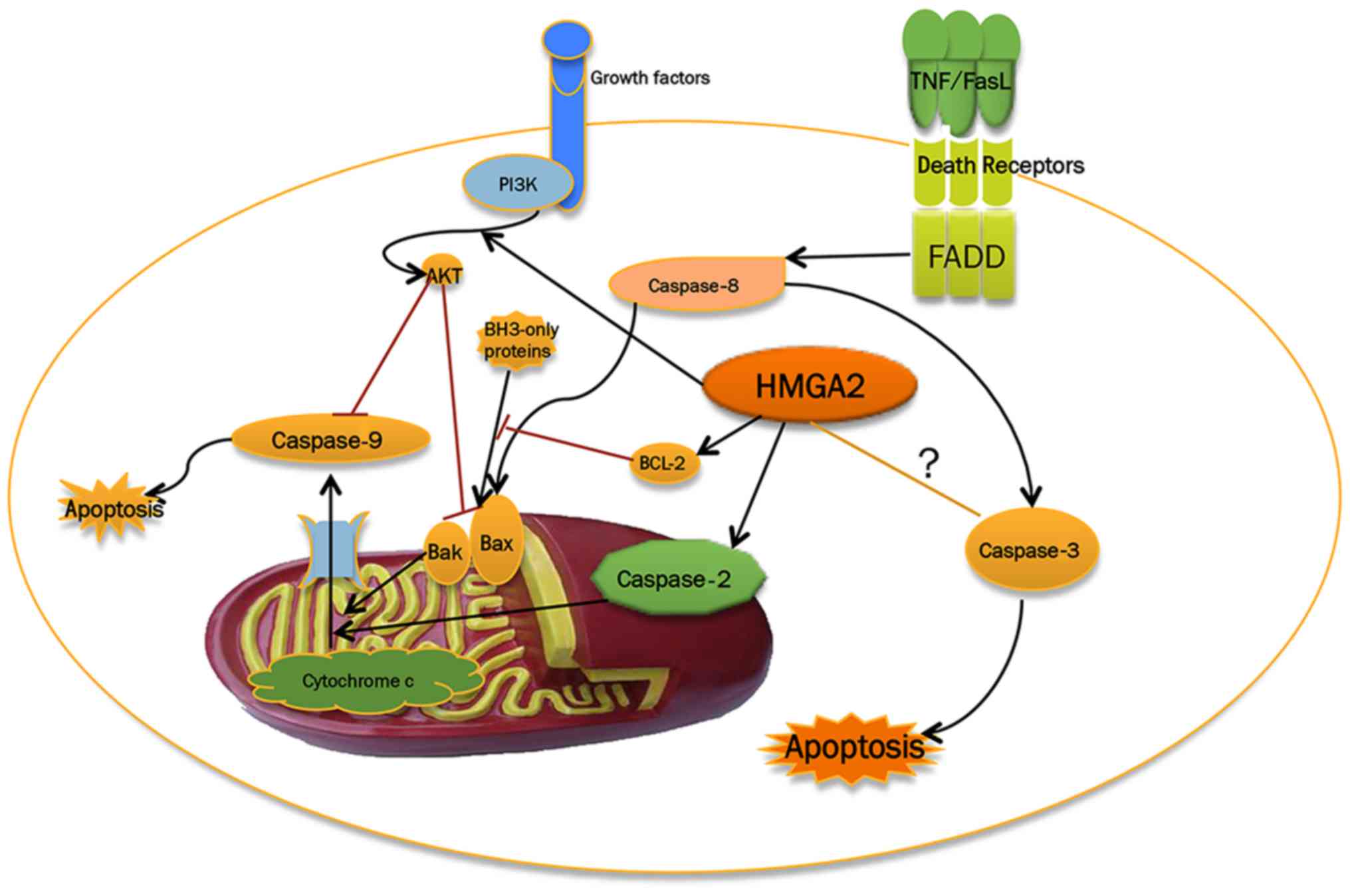
Apoptosis is a crucial process in multicellular
organisms, where it eliminates unnecessary and abnormal cells,
thereby preventing unwanted immune responses, to maintain a healthy
balance between cell survival and cell death (83,84).
Tumours deficient in apoptosis are prone to progression and lead to
a poor prognosis (85). Inducing
the apoptosis of tumour cells is considered an effective way to
prevent tumour progression (86).
As the 'guardian of the genome', p53 protein is essential for the
maintenance of genome stability. This protein induces growth arrest
and promotes DNA repair following the appearance of soft DNA
damage. Extensive DNA damage induces prolonged activated p53,
thereby initiating cellular apoptosis (87). There are two major pathways, the
extrinsic pathway and the intrinsic pathway, contributing to
apoptosis (88). The interaction
between HMGA2 and these two pathways is described in Fig. 4.
Considering the malignant property of HMGA2, it is
suggested that HMGA2 can prevent tumour cells from undergoing
apoptosis and contribute to tumour growth. Accumulating evidence
supports that tumour cells with a low expression of HMGA2 present
with more apoptosis and growth inhibition compared with
HMGA2-overexpressing cells (89-91).
However, the mechanism of how HMGA2 regulates cellular apoptosis
remains to be fully elucidated to date. The Bcl-2 protein serves as
an anti-apoptotic factor that negatively regulates the apoptotic
pathway. The knockdown of HMGA2 in epithelial ovarian carcinoma
cells enhances cellular apoptosis with decreased expression of
Bcl-2 (92). In addition, HMGA2
derepresses the expression of Bcl-2 by inhibiting miR-34a, thereby
promoting an anti-apoptotic pathway (91). In thyroid cells overexpressing
Bcl-2 by infection with a Bcl-2 retroviral vector, the expression
of HMGA2 was correspondingly increased and apoptosis was inversely
decreased (93). The PI3K/Akt
signalling pathway is always hyperactivated in human cancer, which
is a key contributor to resistance to apoptosis (94). Activated Akt is sufficient to
inhibit the activation of caspase-9 and Bad, leading to the
inhibition of cellular apoptosis (95,96).
Notably, HMGA2 can initiate activation of the PI3K/Akt pathway and
impair the activation of caspase-9 and Bad, thus suppressing
apoptosis (97). Taken together,
the above evidence supports that HMGA2 is able to protect cancer
cells from apoptosis. However, variations also indicate that the
overexpression of HMGA2 may lead to cellular apoptosis. Caspase-2
contributes to the leakage of cytochrome c from
mitochondria, which is an essential step in apoptosis (98). The ectopic expression of HMGA2 in
WI38 cells was shown to significantly induce apoptosis, accompanied
by the activation of caspase-2 (99). The HMGA2 protein also promotes
apoptosis triggered by O6-methylguanine-induced DNA damage
(100). In the process of
apoptosis, the phosphorylation of ATR/CHK1 is significantly
reduced, and the activation of caspase-9 is repressed by inhibiting
HMGA2, which results in the inhibition of apoptosis (101). Otherwise, interrupting the
apoptotic pathway by the knockdown of TNF-related
apoptosis-inducing ligand-R2 significantly increased the level of
let-7 and decreased the expression of HMGA2 (102). Taken together, these findings
indicate that HMGA2 serves multifactorial roles in apoptosis, and
the anti-apoptotic effect of HMGA2 exacerbates tumour growth and
enhances resistance to chemotherapy. However, HMGA2 can also
promote apoptosis, and the contradictory results may be associated
with the different expression levels of HMGA2 in cells (99).
Cellular senescence was originally defined as the
state of proliferative arrest accompanied by replicative exhaustion
of cultured human cells due to the shorter telomere (103). Generally, it is well known that
diverse stress-induced senescence is the outcome of DDR (104,105). Developmental senescence is
initiated without DDR during embryonic development (106,107). The senescence response serves a
pivotal role in maintaining genome integrity and stability,
protecting cells with dysfunctional telomeres from malignant
transformation (108). From a
certain point of view, cellular senescence can be equated to
cellular apoptosis, acting as a tumour-suppressive mechanism to
remove cells with a mutation (109,110). There are two mainly primary
pathways that are governed by proteins p53 and pRB, contributing to
the senescence process (111).
p14AFR causes premature senescence by neutralizing the
ability of MDM2, resulting in p53 stabilization and activation
(112,113). The overexpression of
p16INK4a induces an allosteric change of CDK4/6, which
dephosphorylates the Rb protein (114), leading to cellular senescence
(115,116). Notably, HMGA2 is reported to
directly bind to the p14AFR/p16INK4a locus
and negatively regulate the expression of p14AFR and
p16INK4a (117,118),
thus restraining the cellular senescence process. p14AFR
also acts as an upstream repressor of HMGA2; p14AFR
reduces the expression of HMGA2 and results in senescence (119). In addition, miRNA profiling and
microarray analysis have revealed that miR-10A and miR-21 are two
critical miRNAs that can regulate senescence. The inhibition of
miR-10A and miR-21 induced the expression of HMGA2, which
subsequently led to the downregulation of p16INK4a and
senescence-associated β -ga lactosid ase (SA-β -ga l), thus
rejuvenating senescence (120).
In a transgenic mouse model, mouse embryonic fibroblasts (MEFs)
from HMGA1/HMGA2-null mice were more susceptible to senescence than
MEFs from HMGA1-null mice and wild-type mice, on account of
SA-β-gal activity and increased levels of p16INK4a
(121). By contrast, HMGA2 has
been shown to induced the formation of senescence-associated
heterochromatin foci (SAHF) in the nuclei and repress
proliferation-associated genes (122,123). The HMGA2 protein colocalises with
SAHF by binding to the minor groove of AT-rich DNA, serving as an
essential component of SAHF. If the expression of HMGA2 is
depleted, SAHF may be dissolved. However,
p16INK4-knockout did not affect the morphology of
HMGA2-induced SAHF, suggesting that HMGA2 is indispensable for
senescence establishment and maintenance (123). Furthermore, emerging evidence has
indicated that the PI3K/Akt pathway serves a critical role in
endothelial senescence (124).
AKT protects against stressed-induced premature senescence
(125), and suppression of the
PI3K/Akt pathway by an Akt inhibitor triggers cellular senescence
efficiently (126). Further
mechanistic studies have revealed that senescence-induced Akt
inhibition is mediated by HMGA2, the colocalization of which to the
nucleus into SAHF is required for senescence. The knockdown of
HMGA2 significantly decreases the formation of SAHF bodies in
response to an Akt inhibitor (127).
Epithelial-mesenchymal transition (EMT), a process
that comprises the transdifferentiation of epithelial cells into
motile mesenchymal cells, is important in embryonic development,
wound healing, stem cell behaviour and cancer progression (128). The changes in gene expression
contributing to repression of the epithelial phenotype and
activation of the mesenchymal phenotype involve several master
regulators, including Snail1, Snail2, Twist, E47, E2 and
zinc-finger E-box-binding transcription factors. During EMT,
epithelial proteins, including E-cadherin and zonula 1, are
downregulated, whereas mesenchymal proteins, including vitamin and
fibronectin, are upregulated (129). The detailed molecular mechanisms
of EMT have been reviewed previously (130,131). Accumulating studies have shown
that the decrease in epithelial characteristics and enhanced
expression of mesenchymal markers accompanied by the overexpression
of HMGA2 are present in several cancer cells (132-137), which suggests that HMGA2 is
involved in the regulation of EMT. Morishita et al suggested
that HMGA2 was upstream of the TGFβ/Smads pathway, and the
expression of TGFβRII and phosphorylation of Smad3 were
significantly increased by HMGA2, which activated the TGFβ pathway
and subsequently induced EMT (137). TGFβ signalling in the regulation
of EMT also includes non-SMAD pathways, such as the PI3K/Akt
signalling pathway. The depletion of HMGA2 represses activation of
the PI3K/AKT pathway by attenuating high glucose-induced EMT in HK2
cells (138). Endogenous HMGA2 is
essential, but not indispensable, for TGFβ-induced EMT, as the
depletion of HMGA2 by RNA interference is not sufficient to
completely prevent EMT (14).
HMGA2 can form a complex with Smads or directly bind to the
critical element of endogenous Twist1 and Snail promoters to induce
target protein expression (135,136,139), resulting in EMT (140). Aberrant HMGA2 directly binds to
the proximal E-cadherin gene (Cdh1) promoter, together with DNA
methyltransferase 3A, which leads to silencing of the expression of
E-cadherin, contributing to EMT (141). RAS/RAF/MEK/ERK signalling is
another pathway contributing to EMT (142). Treating tumour cells with the
MEK1/2 inhibitor U0126, reverses the HMGA2-induced expression of
Snail and impairs HMGA2-induced EMT (135). The interaction between HMGA2 and
the canonical Wnt signalling is presented at different stages
during lung development (143).
Upon activation of the canonical Wnt pathway, a β-catenin-T-cell
factor transcriptional complex is formed to trigger the EMT,
depending on the Axin2-GSK3β-Snail1 axis (139). The Wnt/β-catenin pathway serves
an epistatic role in the regulation of HMGA2 (15). The upregulation of HMGA2 and
downregulation of WIF1 in HMGA2/WIF1 fusion transcript-expressing
cells activates the Wnt/β-catenin pathway (144), suggesting that HMGA2 contributes
to EMT via interaction with the Wnt/β-catenin pathway (139). It has been demonstrated that
HMGA2 interacts with pRb and enhances E2F1 to bind with the
forkhead box protein L2 (FOXL2) promoter, resulting in enhanced
transcription of FOXL2 and contributing to EMT (145). Taken together, these data support
the function of HMGA2 as a tumour promoter, which can directly or
indirectly enhance the formation of EMT (Fig. 5).
Telomeres are the non-coding DNA sequences located
at the ends of the linear chromosomes. The DNA sequence of
telomeres is similar in all vertebrates, which is usually a repeat
of six bases (TTAGGG) (146). The
loss of the coding sequences observed as a result of DNA
replication occurring in a semiconservative manner can be prevented
from degradation by telomeres. Without telomere restoration, cell
senescence and apoptosis are initiated when the limit of the short
telomeres is reached (147).
Telomerase enzyme, a critical complex for telomere restoration,
contains several components, including a catalytic protein subunit
telomerase reverse transcriptase (hTERT), human telomerase RNA
(hTR), human telomerase-associated protein 1 (hTP1), HSP90, P23 and
dyskerin. Almost 90% of cancer cases exhibit of telomerase
hyperactivation, which is a critical step in carcinogenesis
(148). HMGA2 knockdown in HepG2
cells results in telomere erosion, reducing the tumourigenic
ability (149). Furthermore,
HMGA2 can directly localise at telomeres and maintain telomere
stability (150). The expression
of HMGA2 and hTERT are at lower levels in adipose-derived stem
cells, and at higher levels in lipoma-derived mesenchymal stem
cells (151). However, the
mechanism underlying how HMGA2 regulates the expression of hTERT is
elusive. It is suggested that HMGA2 derepress H3-K9
hyperacetylation in a protein-to-protein manner, which can
subsequently stimulate hTERT activation and promote telomere
restoration. TRF2 is a key regulator in telomere protection, it can
directly bind to the tandem array of duplex TTAGGG repeats of
telomeres, executing its functions in chromosomal end-protection
via a two-step mechanism (152).
Natarajan et al revealed that HMGA2 directly binds to the
TRF2 promoter via its AT-hooks and protects telomeres from damage
(151).
Chemotherapeutic resistance is one of the main
causes of treatment failure in human cancer. In response to
chemotherapeutic stimuli, cancer cells initiate a series of
mechanisms to protect themselves from death and cause
chemoresistance. Tumours with DSB-repair-deficiency exhibit more
sensitive to chemotherapies, whereas cancer cells with enhanced DNA
repair potential show resistance to chemotherapy agents (153). To the best of our knowledge,
HMGA2 can promote DNA repair processes, thereby contributing to
cancer chemoresistance (74,75,82).
By contrast, HMGA2 can impair the DNA repair system and also cause
tumour cells to be more sensitive to chemotherapy (73). Cellular senescence serves a
critical role in regulating antitumour effects (110), and tumour defects in cellular
senescence result in drug resistance (154). Therefore, HMGA2 causes
chemoresistance by inducing cancer cell senescence. The process of
EMT can be hijacked by cancer cells, thus contributing to
therapeutic resistance. Sunitinib, a tyrosine kinase inhibitor, is
widely used in the treatment of renal cell carcinoma,
gastrointestinal stromal tumour and lung cancer. It has been
reported that cancer cells with a high expression of HMGA2 exhibit
increased resistance to sunitinib, partly due to the HMGA2-induced
EMT (155). In addition, the
collagen-rich microenvironment in human pancreatic ductal
adenocarcinoma increases the expression of HMGA2 through the MT1
–MMP pathway (156), and HMGA2
upregulates the expression of HATs to limit the effectiveness of
gemcitabine (157). Retinoic acid
(RA) is a potent inducer of neuroblastoma (NB) cell differentiation
and inhibits NB cell growth, however, the exogenous expression of
HMGA2 is associated with a phenotype that is resistant to RA
(158). With increasing
understanding of the association between the expression of HMGA2
and the efficacy of chemotherapy, detecting the expression of HMGA2
in cancer patients may help to guide rational clinical therapy.
HMGA2 serves a key role in the process of
embryogenesis, however, it becomes an oncoprotein when expressed in
adult cells. HMGA2 is highly expressed in a various types of human
cancer and serves as a prognostic marker (15,40,41,133,135,145,159-177) (Table
I). Almost a decade ago, Fusco and Fedele reviewed the
functions of HMGA proteins, including HMGA1 and HMGA2, in human
neoplastic diseases, and suggested that the detection of HMGA be
introduced as a routine procedure in clinical tumour analysis
(7). Increasingly, evidence has
suggested that HMGA2 is an independent prognostic factor of several
malignant tumours, and that the expression level of HMGA2 is
associated with the therapeutic efficacy of certain
chemotherapeutic agents. Therefore, the detection of HMGA2 in
cancer may provide important prognostic data or other information
for clinicians. However, questions remain that warrant further
investigation, including what standard experimental method to use
to detect the expression of HMGA2, how to make the positive
standard, and how the expression of HMGA2 affects the prognosis of
cancer patients. Although HMGA2-targeting drugs have not been
developed, preclinical experimental studies have demonstrated that
inhibiting HMGA2 protein synthesis via an antisense methodology can
inhibit cancer cell growth and prevent neoplastic transformation
(89,178). Therefore, targeting of the
expression of HMGA2 may be a promising approach for cancer
treatment in the future.
Not applicable.
This study was supported by project grants from the
Zhejiang Provincial Natural Science Foundation of China (grant no.
Y19H160283).
Not applicable.
SZ, QM and XW were involved in the conception of the
study. SZ and QM were involved in writing the article. XW
critically revised the manuscript. All authors read and approved
the final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Bustin M and Reeves R: High-mobility-group
chromosomal proteins: Architectural components that facilitate
chromatin function. Prog Nucleic Acid Res Mol Biol. 54:35–100.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Goodwin GH, Sanders C and Johns EW: A new
group of chromatin-associated proteins with a high content of
acidic and basic amino acids. Eur J Biochem. 38:14–19. 1973.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bustin M: Revised nomenclature for high
mobility group (HMG) chromosomal proteins. Trends Biochem Sci.
26:152–153. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wood LJ, Maher JF, Bunton TE and Resar LM:
The oncogenic properties of the HMG-I gene family. Cancer Res.
60:4256–4261. 2000.PubMed/NCBI
|
|
5
|
De Martino I, Visone R, Fedele M, Petrocca
F, Palmieri D, Martinez Hoyos J, Forzati F, Croce CM and Fusco A:
Regulation of microRNA expression by HMGA1 proteins. Oncogene.
28:1432–1442. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Martinez Hoyos J, Fedele M, Battista S,
Pentimalli F, Kruhoffer M, Arra C, Orntoft TF, Croce CM and Fusco
A: Identification of the genes up- and down-regulated by the high
mobility group A1 (HMGA1) proteins: Tissue specificity of the
HMGA1-dependent gene regulation. Cancer Res. 64:5728–5735. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fusco A and Fedele M: Roles of HMGA
proteins in cancer. Nat Rev Cancer. 7:899–910. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fedele M, Battista S, Kenyon L,
Baldassarre G, Fidanza V, Klein-Szanto AJ, Parlow AF, Visone R,
Pierantoni GM, Outwater E, et al: Overexpression of the HMGA2 gene
in transgenic mice leads to the onset of pituitary adenomas.
Oncogene. 21:3190–3198. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Huth JR, Bewley CA, Nissen MS, Evans JN,
Reeves R, Gronenborn AM and Clore GM: The solution structure of an
HMG-I(Y)-DNA complex defines a new architectural minor groove
binding motif. Nat Struct Biol. 4:657–665. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Thanos D and Maniatis T: The high mobility
group protein HMG I(Y) is required for NF-kappa B-dependent virus
induction of the human IFN-beta gene. Cell. 71:777–789. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tallini G and Dal Cin P: HMGI(Y) and
HMGI-C dysregulation: A common occurrence in human tumors. Adv Anat
Pathol. 6:237–246. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rustighi A, Mantovani F, Fusco A,
Giancotti V and Manfioletti G: Sp1 and CTF/NF-1 transcription
factors are involved in the basal expression of the Hmgi-c proximal
promoter. Biochem Biophys Res Commun. 265:439–447. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ayoubi TA, Jansen E, Meulemans SM and Van
de Ven WJ: Regulation of HMGIC expression: An architectural
transcription factor involved in growth control and development.
Oncogene. 18:5076–5087. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Thuault S, Valcourt U, Petersen M,
Manfioletti G, Heldin CH and Moustakas A: Transforming growth
factor-beta employs HMGA2 to elicit epithelial-mesenchymal
transition. J Cell Biol. 174:175–183. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wend P, Runke S, Wend K, Anchondo B,
Yesayan M, Jardon M, Hardie N, Loddenkemper C, Ulasov I, Lesniak
MS, et al: WNT10B/β-catenin signalling induces HMGA2 and
proliferation in metastatic triple-negative breast cancer. EMBO Mol
Med. 5:264–279. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lam K, Muselman A, Du R, Harada Y, Scholl
AG, Yan M, Matsuura S, Weng S, Harada H and Zhang DE: Hmga2 is a
direct target gene of RUNX1 and regulates expansion of myeloid
progenitors in mice. Blood. 124:2203–2212. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lu J, Getz G, Miska EA, Alvarez-Saavedra
E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA,
et al: MicroRNA expression profiles classify human cancers. Nature.
435:834–838. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shell S, Park SM, Radjabi AR, Schickel R,
Kistner EO, Jewell DA, Feig C, Lengyel E and Peter ME: Let-7
expression defines two differentiation stages of cancer. Proc Natl
Acad Sci USA. 104:11400–11405. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lee WY, Tzeng CC and Chou CY: Uterine
leiomyosarcomas coexistent with cellular and atypical leiomyomata
in a young woman during the treatment with luteinizing
hormone-releasing hormone agonist. Gynecol Oncol. 52:74–79. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Guo L, Chen C, Shi M, Wang F, Chen X, Diao
D, Hu M, Yu M, Qian L and Guo N: Stat3-coordinated
Lin-28-let-7-HMGA2 and miR-200-ZEB1 circuits initiate and maintain
oncostatin M-driven epithelial-mesenchymal transition. Oncogene.
32:5272–5282. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Newman MA, Thomson JM and Hammond SM:
Lin-28 interaction with the Let-7 precursor loop mediates regulated
microRNA processing. RNA. 14:1539–1549. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dröge P and Davey CA: Do cells let-7
determine stemness? Cell Stem Cell. 2:8–9. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Copley MR, Babovic S, Benz C, Knapp DJ,
Beer PA, Kent DG, Wohrer S, Treloar DQ, Day C, Rowe K, et al: The
Lin28b-let-7-Hmga2 axis determines the higher self-renewal
potential of fetal haematopoietic stem cells. Nat Cell Biol.
15:916–925. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dangi-Garimella S, Yun J, Eves EM, Newman
M, Erkeland SJ, Hammond SM, Minn AJ and Rosner MR: Raf kinase
inhibitory protein suppresses a metastasis signalling cascade
involving LIN28 and let-7. EMBO J. 28:347–358. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sun M, Gomes S, Chen P, Frankenberger CA,
Sankarasharma D, Chung CH, Chada KK and Rosner MR: RKIP and HMGA2
regulate breast tumor survival and metastasis through lysyl oxidase
and syndecan-2. Oncogene. 33:3528–3537. 2014. View Article : Google Scholar :
|
|
26
|
Lin Y, Liu AY, Fan C, Zheng H, Li Y, Zhang
C, Wu S, Yu D, Huang Z, Liu F, et al: MicroRNA-33b inhibits breast
cancer metastasis by targeting HMGA2, SALL4 and Twist1. Sci Rep.
5:99952015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kim TH, Song JY, Park H, Jeong JY, Kwon
AY, Heo JH, Kang H, Kim G and An HJ: miR-145, targeting
high-mobility group A2, is a powerful predictor of patient outcome
in ovarian carcinoma. Cancer Lett. 356B:937–945. 2015. View Article : Google Scholar
|
|
28
|
Emmrich S, Katsman-Kuipers JE, Henke K,
Khatib ME, Jammal R, Engeland F, Dasci F, Zwaan CM, den Boer ML,
Verboon L, et al: miR-9 is a tumor suppressor in pediatric AML with
t(8;21). Leukemia. 28:1022–1032. 2014. View Article : Google Scholar
|
|
29
|
Liu S, Patel SH, Ginestier C, Ibarra I,
Martin-Trevino R, Bai S, McDermott SP, Shang L, Ke J, Ou SJ, et al:
MicroRNA93 regulates proliferation and differentiation of normal
and malignant breast stem cells. PLoS Genet. 8:e10027512012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ye ZH and Gui DW: miR-539 suppresses
proliferation and induces apoptosis in renal cell carcinoma by
targeting high mobility group A2. Mol Med Rep. 17:5611–5618.
2018.PubMed/NCBI
|
|
31
|
Li T, Yang XD, Ye CX, Shen ZL, Yang Y,
Wang B, Guo P, Gao ZD, Ye YJ, Jiang KW, et al: Long noncoding RNA
HIT000218960 promotes papillary thyroid cancer oncogenesis and
tumor progression by upregulating the expression of high mobility
group AT-hook 2 (HMGA2) gene. Cell Cycle. 16:224–231. 2017.
View Article : Google Scholar :
|
|
32
|
Boque-Sastre R, Soler M, Oliveira-Mateos
C, Portela A, Moutinho C, Sayols S, Villanueva A, Esteller M and
Guil S: Head-to-head antisense transcription and R-loop formation
promotes transcriptional activation. Proc Natl Acad Sci USA.
112:5785–5790. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhou X, Benson KF, Ashar HR and Chada K:
Mutation responsible for the mouse pygmy phenotype in the
developmentally regulated factor HMGI-C. Nature. 376:771–774. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Weedon MN, Lettre G, Freathy RM, Lindgren
CM, Voight BF, Perry JR, Elliott KS, Hackett R, Guiducci C, Shields
B, et al Diabetes Genetics Initiative; Wellcome Trust Case Control
Consortium: A common variant of HMGA2 is associated with adult and
childhood height in the general population. Nat Genet.
39:1245–1250. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Abi Habib W, Brioude F, Edouard T, Bennett
JT, Lienhardt-Roussie A, Tixier F, Salem J, Yuen T, Azzi S, Le Bouc
Y, et al: Genetic disruption of the oncogenic HMGA2-PLAG1-IGF2
pathway causes fetal growth restriction. Genet Med. 20:250–258.
2018. View Article : Google Scholar
|
|
36
|
Zaidi MR, Okada Y and Chada KK:
Misexpression of full-length HMGA2 induces benign mesenchymal
tumors in mice. Cancer Res. 66:7453–7459. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Efanov A, Zanesi N, Coppola V, Nuovo G,
Bolon B, Wernicle-Jameson D, Lagana A, Hansjuerg A, Pichiorri F and
Croce CM: Human HMGA2 protein overexpressed in mice induces
precursor T-cell lymphoblastic leukemia. Blood Cancer J.
4:e2272014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Schoenmakers EF, Wanschura S, Mols R,
Bullerdiek J, Van den Berghe H and Van de Ven WJ: Recurrent
rearrangements in the high mobility group protein gene, HMGI-C, in
benign mesenchymal tumours. Nat Genet. 10:436–444. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Dreux N, Marty M, Chibon F, Vélasco V,
Hostein I, Ranchère-Vince D, Terrier P and Coindre JM: Value and
limitation of immunohistochemical expression of HMGA2 in
mesenchymal tumors: about a series of 1052 cases. Mod Pathol.
23:1657–1666. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Marquis M, Beaubois C, Lavallée VP,
Abrahamowicz M, Danieli C, Lemieux S, Ahmad I, Wei A, Ting SB,
Fleming S, et al: High expression of HMGA2 independently predicts
poor clinical outcomes in acute myeloid leukemia. Blood Cancer J.
8:682018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang X, Liu X, Li AY, Chen L, Lai L, Lin
HH, Hu S, Yao L, Peng J, Loera S, et al: Overexpression of HMGA2
promotes metastasis and impacts survival of colorectal cancers.
Clin Cancer Res. 17:2570–2580. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Malek A, Bakhidze E, Noske A, Sers C,
Aigner A, Schäfer R and Tchernitsa O: HMGA2 gene is a promising
target for ovarian cancer silencing therapy. Int J Cancer.
123:348–356. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Tan L, Wei X, Zheng L, Zeng J, Liu H, Yang
S and Tan H: Amplified HMGA2 promotes cell growth by regulating Akt
pathway in AML. J Cancer Res Clin Oncol. 142:389–399. 2016.
View Article : Google Scholar
|
|
44
|
Tessari MA, Gostissa M, Altamura S, Sgarra
R, Rustighi A, Salvagno C, Caretti G, Imbriano C, Mantovani R, Del
Sal G, et al: Transcriptional activation of the cyclin A gene by
the architectural transcription factor HMGA2. Mol Cell Biol.
23:9104–9116. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Li Y, Peng L and Seto E: Histone
deacetylase 10 regulates the cell cycle G2/M phase transition via a
novel Let-7-HMGA2-cyclin A2 pathway. Mol Cell Biol. 35:3547–3565.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Shaulian E and Karin M: AP-1 as a
regulator of cell life and death. Nat Cell Biol. 4:E131–E136. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Vallone D, Battista S, Pierantoni GM,
Fedele M, Casalino L, Santoro M, Viglietto G, Fusco A and Verde P:
Neoplastic transformation of rat thyroid cells requires the junB
and fra-1 gene induction which is dependent on the HMGI-C gene
product. EMBO J. 16:5310–5321. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Evan GI, Brown L, Whyte M and Harrington
E: Apoptosis and the cell cycle. Curr Opin Cell Biol. 7:825–834.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Seville LL, Shah N, Westwell AD and Chan
WC: Modulation of pRB/E2F functions in the regulation of cell cycle
and in cancer. Curr Cancer Drug Targets. 5:159–170. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Fedele M, Visone R, De Martino I, Troncone
G, Palmieri D, Battista S, Ciarmiello A, Pallante P, Arra C,
Melillo RM, et al: HMGA2 induces pituitary tumorigenesis by
enhancing E2F1 activity. Cancer Cell. 9:459–471. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yu KR, Park SB, Jung JW, Seo MS, Hong IS,
Kim HS, Seo Y, Kang TW, Lee JY, Kurtz A, et al: HMGA2 regulates the
in vitro aging and proliferation of human umbilical cord
blood-derived stromal cells through the mTOR/p70S6K signaling
pathway. Stem Cell Res (Amst). 10:156–165. 2013. View Article : Google Scholar
|
|
52
|
Zhang H, Tang Z, Deng C, He Y, Wu F, Liu O
and Hu C: HMGA2 is associated with the aggressiveness of tongue
squamous cell carcinoma. Oral Dis. 23:255–264. 2017. View Article : Google Scholar
|
|
53
|
Xie H, Wang J, Jiang L, Geng C, Li Q, Mei
D, Zhao L and Cao J: ROS-dependent HMGA2 upregulation mediates
Cd-induced proliferation in MRC-5 cells. Toxicol In Vitro.
34:146–152. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Minshull J, Blow JJ and Hunt T:
Translation of cyclin mRNA is necessary for extracts of activated
Xenopus eggs to enter mitosis. Cell. 56:947–956. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Liu WD, Tan L, Xiong XF, Liang YP and Tan
H: The effects of lentivirus-mediated RNA interference silencing
HMGA2 on proliferation and expressions of cyclin B2 and cyclin A2
in HL-60 cells. Zhonghua Xue Ye Xue Za Zhi. 33:448–452. 2012.in
Chinese. PubMed/NCBI
|
|
56
|
Branzei D and Foiani M: Maintaining genome
stability at the replication fork. Nat Rev Mol Cell Biol.
11:208–219. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Masai H, Tanaka T and Kohda D: Stalled
replication forks: Making ends meet for recognition and
stabilization. BioEssays. 32:687–697. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Courcelle J, Donaldson JR, Chow KH and
Courcelle CT: DNA damage-induced replication fork regression and
processing in Escherichia coli. Science. 299:1064–1067. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Yu H, Lim HH, Tjokro NO, Sathiyanathan P,
Natarajan S, Chew TW, Klonisch T, Goodman SD, Surana U and Dröge P:
Chaperoning HMGA2 protein protects stalled replication forks in
stem and cancer cells. Cell Rep. 6:684–697. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Iyama T and Wilson DM III: DNA repair
mechanisms in dividing and non-dividing cells. DNA Repair (Amst).
12:620–636. 2013. View Article : Google Scholar
|
|
61
|
Bartkova J, Rajpert-De Meyts E, Skakkebaek
NE, Lukas J and Bartek J: DNA damage response in human testes and
testicular germ cell tumours: Biology and implications for therapy.
Int J Androl. 30:282–291; discussion 291. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Shrivastav M, De Haro LP and Nickoloff JA:
Regulation of DNA double-strand break repair pathway choice. Cell
Res. 18:134–147. 2008. View Article : Google Scholar
|
|
63
|
Lieber MR: The mechanism of double-strand
DNA break repair by the nonhomologous DNA end-joining pathway. Annu
Rev Biochem. 79:181–211. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Arnoult N, Correia A, Ma J, Merlo A,
Garcia-Gomez S, Maric M, Tognetti M, Benner CW, Boulton SJ,
Saghatelian A, et al: Regulation of DNA repair pathway choice in S
and G2 phases by the NHEJ inhibitor CYREN. Nature. 549:548–552.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Meek K, Dang V and Lees-Miller SP: DNA-PK:
The means to justify the ends? Adv Immunol. 99:33–58. 2008.
View Article : Google Scholar
|
|
66
|
Uematsu N, Weterings E, Yano K,
Morotomi-Yano K, Jakob B, Taucher-Scholz G, Mari PO, van Gent DC,
Chen BP and Chen DJ: Autophosphorylation of DNA-PKCS regulates its
dynamics at DNA double-strand breaks. J Cell Biol. 177:219–229.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Downs JA, Lowndes NF and Jackson SP: A
role for Saccharomyces cerevisiae histone H2A in DNA repair.
Nature. 408:1001–1004. 2000. View Article : Google Scholar
|
|
68
|
Nick McElhinny SA, Snowden CM, McCarville
J and Ramsden DA: Ku recruits the XRCC4-ligase IV complex to DNA
ends. Mol Cell Biol. 20:2996–3003. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Li AY, Boo LM, Wang SY, Lin HH, Wang CC,
Yen Y, Chen BP, Chen DJ and Ann DK: Suppression of nonhomologous
end joining repair by overexpression of HMGA2. Cancer Res.
69:5699–5706. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Kühne C, Tjörnhammar ML, Pongor S, Banks L
and Simoncsits A: Repair of a minimal DNA double-strand break by
NHEJ requires DNA-PKcs and is controlled by the ATM/ATR checkpoint.
Nucleic Acids Res. 31:7227–7237. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Bullerdiek J and Rommel B: Comment re:
HMGA2 is a negative regulator of DNA-PK pathway. Cancer Res.
70:1742author reply 1742. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Cleynen I and Van de Ven WJ: The HMGA
proteins: A myriad of functions (Review). Int J Oncol. 32:289–305.
2008.PubMed/NCBI
|
|
73
|
Boo LM, Lin HH, Chung V, Zhou B, Louie SG,
O'Reilly MA, Yen Y and Ann DK: High mobility group A2 potentiates
genotoxic stress in part through the modulation of basal and DNA
damage-dependent phosphatidylinositol 3-kinase-related protein
kinase activation. Cancer Res. 65:6622–6630. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Palmieri D, Valentino T, D'Angelo D, De
Martino I, Postiglione I, Pacelli R, Croce CM, Fedele M and Fusco
A: HMGA proteins promote ATM expression and enhance cancer cell
resistance to genotoxic agents. Oncogene. 30:3024–3035. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Natarajan S, Hombach-Klonisch S, Dröge P
and Klonisch T: HMGA2 inhibits apoptosis through interaction with
ATR-CHK1 signaling complex in human cancer cells. Neoplasia.
15:263–280. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Jackson SP and Bartek J: The DNA-damage
response in human biology and disease. Nature. 461:1071–1078. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Summer H, Li O, Bao Q, Zhan L, Peter S,
Sathiyanathan P, Henderson D, Klonisch T, Goodman SD and Dröge P:
HMGA2 exhibits dRP/AP site cleavage activity and protects cancer
cells from DNA-damage-induced cytotoxicity during chemotherapy.
Nucleic Acids Res. 37:4371–4384. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Hombach-Klonisch S, Kalantari F, Medapati
MR, Natarajan S, Krishnan SN, Kumar-Kanojia A, Thanasupawat T,
Begum F, Xu FY, Hatch GM, et al: HMGA2 as a functional antagonist
of PARP1 inhibitors in tumor cells. Mol Oncol. 13:153–170. 2019.
View Article : Google Scholar :
|
|
79
|
Alekseev S and Coin F: Orchestral
maneuvers at the damaged sites in nucleotide excision repair. Cell
Mol Life Sci. 72:2177–2186. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
de Laat WL, Jaspers NG and Hoeijmakers JH:
Molecular mechanism of nucleotide excision repair. Genes Dev.
13:768–785. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Westerveld A, Hoeijmakers JH, van Duin M,
de Wit J, Odijk H, Pastink A, Wood RD and Bootsma D: Molecular
cloning of a human DNA repair gene. Nature. 310:425–429. 1984.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Borrmann L, Schwanbeck R, Heyduk T,
Seebeck B, Rogalla P, Bullerdiek J and Wisniewski JR: High mobility
group A2 protein and its derivatives bind a specific region of the
promoter of DNA repair gene ERCC1 and modulate its activity.
Nucleic Acids Res. 31:6841–6851. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Cotter TG: Apoptosis and cancer: The
genesis of a research field. Nat Rev Cancer. 9:501–507. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Taylor RC, Cullen SP and Martin SJ:
Apoptosis: Controlled demolition at the cellular level. Nat Rev Mol
Cell Biol. 9:231–241. 2008. View Article : Google Scholar
|
|
85
|
Ma C, Nong K, Zhu H, Wang W, Huang X, Yuan
Z and Ai K: H19 promotes pancreatic cancer metastasis by
derepressing let-7's suppression on its target HMGA2-mediated EMT.
Tumour Biol. 35:9163–9169. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Jia J, Yang M, Chen Y, Yuan H, Li J, Cui X
and Liu Z: Inducing apoptosis effect of caffeic acid
3,4-dihydroxy-phenethyl ester on the breast cancer cells. Tumour
Biol. 35:11781–11789. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Sionov RV and Haupt Y: The cellular
response to p53: The decision between life and death. Oncogene.
18:6145–6157. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Meier P and Vousden KH: Lucifer's
labyrinth - ten years of path finding in cell death. Mol Cell.
28:746–754. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Pentimalli F, Dentice M, Fedele M,
Pierantoni GM, Cito L, Pallante P, Santoro M, Viglietto G, Dal Cin
P and Fusco A: Suppression of HMGA2 protein synthesis could be a
tool for the therapy of well differentiated liposarcomas
overexpressing HMGA2. Cancer Res. 63:7423–7427. 2003.PubMed/NCBI
|
|
90
|
Kaur H, Hütt-Cabezas M, Weingart MF, Xu J,
Kuwahara Y, Erdreich-Epstein A, Weissman BE, Eberhart CG and Raabe
EH: The chromatin-modifying protein HMGA2 promotes atypical
teratoid/rhabdoid cell tumorigenicity. J Neuropathol Exp Neurol.
74:177–185. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Mansoori B, Mohammadi A, Shirjang S and
Baradaran B: HMGI-C suppressing induces P53/caspase-9 axis to
regulate apoptosis in breast adenocarcinoma cells. Cell Cycle.
15:2585–2592. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Gao X, Dai M, Li Q, Wang Z, Lu Y and Song
Z: HMGA2 regulates lung cancer proliferation and metastasis. Thorac
Cancer. 8:Jul 28–2017.Epub ahead of print. View Article : Google Scholar
|
|
93
|
Basolo F, Fiore L, Fusco A, Giannini R,
Albini A, Merlo GR, Fontanini G, Conaldi PG and Toniolo A:
Potentiation of the malignant phenotype of the undifferentiated ARO
thyroid cell line by insertion of the bcl-2 gene. Int J Cancer.
81:956–962. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Sos ML, Fischer S, Ullrich R, Peifer M,
Heuckmann JM, Koker M, Heynck S, Stückrath I, Weiss J, Fischer F,
et al: Identifying genotype-dependent efficacy of single and
combined PI3K- and MAPK-pathway inhibition in cancer. Proc Natl
Acad Sci USA. 106:18351–18356. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Cardone MH, Roy N, Stennicke HR, Salvesen
GS, Franke TF, Stanbridge E, Frisch S and Reed JC: Regulation of
cell death protease caspase-9 by phosphorylation. Science.
282:1318–1321. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Wang XT, Pei DS, Xu J, Guan QH, Sun YF,
Liu XM and Zhang GY: Opposing effects of Bad phosphorylation at two
distinct sites by Akt1 and JNK1/2 on ischemic brain injury. Cell
Signal. 19:1844–1856. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Wei CH, Wei LX, Lai MY, Chen JZ and Mo XJ:
Effect of silencing of high mobility group A2 gene on gastric
cancer MKN-45 cells. World J Gastroenterol. 19:1239–1246. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Danial NN and Korsmeyer SJ: Cell death:
Critical control points. Cell. 116:205–219. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Shi X, Tian B, Ma W, Zhang N, Qiao Y, Li
X, Zhang Y, Huang B and Lu J: A novel anti-proliferative role of
HMGA2 in induction of apoptosis through caspase 2 in primary human
fibroblast cells. Biosci Rep. 35:e001692015. View Article : Google Scholar
|
|
100
|
Fujikane R, Komori K, Sekiguchi M and
Hidaka M: Function of high-mobility group A proteins in the DNA
damage signaling for the induction of apoptosis. Sci Rep.
6:317142016. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Wang WY, Cao YX, Zhou X, Wei B, Zhan L and
Fu LT: HMGA2 gene silencing reduces epithelial-mesenchymal
transition and lymph node metastasis in cervical cancer through
inhibiting the ATR/Chk1 signaling pathway. Am J Transl Res.
10:3036–3052. 2018.PubMed/NCBI
|
|
102
|
Haselmann V, Kurz A, Bertsch U, Hübner S,
Olempska-Müller M, Fritsch J, Häsler R, Pickl A, Fritsche H,
Annewanter F, et al: Nuclear death receptor TRAIL-R2 inhibits
maturation of let-7 and promotes proliferation of pancreatic and
other tumor cells. Gastroenterology. 146:278–290. 2014. View Article : Google Scholar
|
|
103
|
Hayflick L: The limited in vitro lifetime
of human diploid cell strains. Exp Cell Res. 37:614–636. 1965.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
d'Adda di Fagagna F: Living on a break:
Cellular senescence as a DNA-damage response. Nat Rev Cancer.
8:512–522. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Matsumura T, Zerrudo Z and Hayflick L:
Senescent human diploid cells in culture: Survival, DNA synthesis
and morphology. J Gerontol. 34:328–334. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Muñoz-Espín D, Cañamero M, Maraver A,
Gómez-López G, Contreras J, Murillo-Cuesta S, Rodríguez-Baeza A,
Varela-Nieto I, Ruberte J, Collado M, et al: Programmed cell
senescence during mammalian embryonic development. Cell.
155:1104–1118. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Storer M, Mas A, Robert-Moreno A, Pecoraro
M, Ortells MC, Di Giacomo V, Yosef R, Pilpel N, Krizhanovsky V,
Sharpe J, et al: Senescence is a developmental mechanism that
contributes to embryonic growth and patterning. Cell.
155:1119–1130. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Artandi SE and DePinho RA: A critical role
for telomeres in suppressing and facilitating carcinogenesis. Curr
Opin Genet Dev. 10:39–46. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Michaloglou C, Vredeveld LC, Soengas MS,
Denoyelle C, Kuilman T, van der Horst CM, Majoor DM, Shay JW, Mooi
WJ and Peeper DS: BRAFE600-associated senescence-like cell cycle
arrest of human naevi. Nature. 436:720–724. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Chen Z, Trotman LC, Shaffer D, Lin HK,
Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, et al:
Crucial role of p53-dependent cellular senescence in suppression of
Pten-deficient tumorigenesis. Nature. 436:725–730. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Bringold F and Serrano M: Tumor
suppressors and oncogenes in cellular senescence. Exp Gerontol.
35:317–329. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Dimri GP, Itahana K, Acosta M and Campisi
J: Regulation of a senescence checkpoint response by the E2F1
transcription factor and p14(ARF) tumor suppressor. Mol Cell Biol.
20:273–285. 2000. View Article : Google Scholar
|
|
113
|
Sharpless NE: INK4a/ARF: A multifunctional
tumor suppressor locus. Mutat Res. 576:22–38. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Kim WY and Sharpless NE: The regulation of
INK4/ARF in cancer and aging. Cell. 127:265–275. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Krishnamurthy J, Torrice C, Ramsey MR,
Kovalev GI, Al-Regaiey K, Su L and Sharpless NE: Ink4a/Arf
expression is a biomarker of aging. J Clin Invest. 114:1299–1307.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Collado M and Serrano M: The power and the
promise of oncogene-induced senescence markers. Nat Rev Cancer.
6:472–476. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Markowski DN, Bartnitzke S, Belge G,
Drieschner N, Helmke BM and Bullerdiek J: Cell culture and
senescence in uterine fibroids. Cancer Genet Cytogenet. 202:53–57.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Nishino J, Kim I, Chada K and Morrison SJ:
Hmga2 promotes neural stem cell self-renewal in young but not old
mice by reducing p16Ink4a and p19Arf
expression. Cell. 135:227–239. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Markowski DN, Winter N, Meyer F, von Ahsen
I, Wenk H, Nolte I and Bullerdiek J: p14Arf acts as an
antagonist of HMGA2 in senescence of mesenchymal stem
cells-implications for benign tumorigenesis. Genes Chromosomes
Cancer. 50:489–498. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Zhu S, Deng S, Ma Q, Zhang T, Jia C, Zhuo
D, Yang F, Wei J, Wang L, Dykxhoorn DM, et al:
MicroRNA-10A* and microRNA-21 modulate endothelial
progenitor cell senescence via suppressing high-mobility group A2.
Circ Res. 112:152–164. 2013. View Article : Google Scholar
|
|
121
|
Federico A, Forzati F, Esposito F, Arra C,
Palma G, Barbieri A, Palmieri D, Fedele M, Pierantoni GM, De
Martino I, et al: Hmga1/Hmga2 double knock-out mice display a
'superpygmy' phenotype. Biol Open. 3:372–378. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Shi X, Tian B, Liu L, Gao Y, Ma C, Mwichie
N, Ma W, Han L, Huang B, Lu J, et al: Rb protein is essential to
the senescence-associated heterochromatic foci formation induced by
HMGA2 in primary WI38 cells. J Genet Genomics. 40:391–398. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Narita M, Narita M, Krizhanovsky V, Nuñez
S, Chicas A, Hearn SA, Myers MP and Lowe SW: A novel role for
high-mobility group a proteins in cellular senescence and
heterochromatin formation. Cell. 126:503–514. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Yentrapalli R, Azimzadeh O, Sriharshan A,
Malinowsky K, Merl J, Wojcik A, Harms-Ringdahl M, Atkinson MJ,
Becker KF, Haghdoost S, et al: The PI3K/Akt/mTOR pathway is
implicated in the premature senescence of primary human endothelial
cells exposed to chronic radiation. PLoS One. 8:e700242013.
View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Kennedy AL, Morton JP, Manoharan I, Nelson
DM, Jamieson NB, Pawlikowski JS, McBryan T, Doyle B, McKay C, Oien
KA, et al: Activation of the PIK3CA/AKT pathway suppresses
senescence induced by an activated RAS oncogene to promote
tumorigenesis. Mol Cell. 42:36–49. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Courtois-Cox S, Genther Williams SM,
Reczek EE, Johnson BW, McGillicuddy LT, Johannessen CM, Hollstein
PE, MacCollin M and Cichowski K: A negative feedback signaling
network underlies oncogene-induced senescence. Cancer Cell.
10:459–472. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Xu X, Lu Z, Qiang W, Vidimar V, Kong B,
Kim JJ and Wei JJ: Inactivation of AKT induces cellular senescence
in uterine leiomyoma. Endocrinology. 155:1510–1519. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesen-chymal transition. J Clin Invest. 119:1420–1428.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Barrallo-Gimeno A and Nieto MA: The Snail
genes as inducers of cell movement and survival: Implications in
development and cancer. Development. 132:3151–3161. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial- mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Kirschmann DA, Seftor EA, Nieva DR,
Mariano EA and Hendrix MJ: Differentially expressed genes
associated with the metastatic phenotype in breast cancer. Breast
Cancer Res Treat. 55:127–136. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Wu J, Zhang S, Shan J, Hu Z, Liu X, Chen
L, Ren X, Yao L, Sheng H, Li L, et al: Elevated HMGA2 expression is
associated with cancer aggressiveness and predicts poor outcome in
breast cancer. Cancer Lett. 376:284–292. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Liu Q, Liu T, Zheng S, Gao X, Lu M,
Sheyhidin I and Lu X: HMGA2 is down-regulated by microRNA let-7 and
associated with epithelial-mesenchymal transition in oesophageal
squamous cell carcinomas of Kazakhs. Histopathology. 65:408–417.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Watanabe S, Ueda Y, Akaboshi S, Hino Y,
Sekita Y and Nakao M: HMGA2 maintains oncogenic RAS-induced
epithelial-mesenchymal transition in human pancreatic cancer cells.
Am J Pathol. 174:854–868. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Thuault S, Tan EJ, Peinado H, Cano A,
Heldin CH and Moustakas A: HMGA2 and Smads co-regulate SNAIL1
expression during induction of epithelial-to-mesenchymal
transition. J Biol Chem. 283:33437–33446. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Morishita A, Zaidi MR, Mitoro A,
Sankarasharma D, Szabolcs M, Okada Y, D'Armiento J and Chada K:
HMGA2 is a driver of tumor metastasis. Cancer Res. 73:4289–4299.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Liu H, Wang X, Liu S and Li H, Yuan X,
Feng B, Bai H, Zhao B, Chu Y and Li H: Effects and mechanism of
miR-23b on glucose-mediated epithelial-to-mesenchymal transition in
diabetic nephropathy. Int J Biochem Cell Biol. 70:149–160. 2016.
View Article : Google Scholar
|
|
139
|
Zha L, Zhang J, Tang W, Zhang N, He M, Guo
Y and Wang Z: HMGA2 elicits EMT by activating the Wnt/β-catenin
pathway in gastric cancer. Dig Dis Sci. 58:724–733. 2013.
View Article : Google Scholar
|
|
140
|
Sakai D, Suzuki T, Osumi N and Wakamatsu
Y: Cooperative action of Sox9, Snail2 and PKA signaling in early
neural crest development. Development. 133:1323–1333. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Tan EJ, Kahata K, Idås O, Thuault S,
Heldin CH and Moustakas A: The high mobility group A2 protein
epigenetically silences the Cdh1 gene during
epithelial-to-mesenchymal transition. Nucleic Acids Res.
43:162–178. 2015. View Article : Google Scholar :
|
|
142
|
Huber MA, Kraut N and Beug H: Molecular
requirements for epithelial-mesenchymal transition during tumor
progression. Curr Opin Cell Biol. 17:548–558. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Singh I, Mehta A, Contreras A, Boettger T,
Carraro G, Wheeler M, Cabrera-Fuentes HA, Bellusci S, Seeger W,
Braun T, et al: Hmga2 is required for canonical WNT signaling
during lung development. BMC Biol. 12:212014. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Queimado L, Lopes CS and Reis AM: WIF1, an
inhibitor of the Wnt pathway, is rearranged in salivary gland
tumors. Genes Chromosomes Cancer. 46:215–225. 2007. View Article : Google Scholar
|
|
145
|
Dong J, Wang R, Ren G, Li X, Wang J, Sun
Y, Liang J, Nie Y, Wu K, Feng B, et al: HMGA2-FOXL2 Axis regulates
metastases and epithelial-to-mesenchymal transition of
chemoresistant gastric cancer. Clin Cancer Res. 23:3461–3473. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Bodnar AG: Marine invertebrates as models
for aging research. Exp Gerontol. 44:477–484. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
d'Adda di Fagagna F, Reaper PM,
Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G,
Carter NP and Jackson SP: A DNA damage checkpoint response in
telomere-initiated senescence. Nature. 426:194–198. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Harley CB: Telomerase and cancer
therapeutics. Nat Rev Cancer. 8:167–179. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Li AY, Lin HH, Kuo CY, Shih HM, Wang CC,
Yen Y and Ann DK: High-mobility group A2 protein modulates hTERT
transcription to promote tumorigenesis. Mol Cell Biol.
31:2605–2617. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Natarajan S, Begum F, Gim J, Wark L,
Henderson D, Davie JR, Hombach-Klonisch S and Klonisch T: High
mobility group A2 protects cancer cells against telomere
dysfunction. Oncotarget. 7:12761–12782. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Qian YW, Gao JH, Lu F and Zheng XD: The
differences between adipose tissue derived stem cells and lipoma
mesen-chymal stem cells in characteristics. Zhonghua Zheng Xing Wai
Ke Za Zhi. 26:125–132. 2010.In Chinese. PubMed/NCBI
|
|
152
|
Okamoto K, Bartocci C, Ouzounov I,
Diedrich JK, Yates JR III and Denchi EL: A two-step mechanism for
TRF2-mediated chromosome-end protection. Nature. 494:502–505. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Fojo T: Cancer, DNA repair mechanisms, and
resistance to chemotherapy. J Natl Cancer Inst. 93:1434–1436. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Schmitt CA, Fridman JS, Yang M, Lee S,
Baranov E, Hoffman RM and Lowe SW: A senescence program controlled
by p53 and p16INK4a contributes to the outcome of cancer
therapy. Cell. 109:335–346. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Marijon H, Dokmak S, Paradis V, Zappa M,
Bieche I, Bouattour M, Raymond E and Faivre S:
Epithelial-to-mesenchymal transition and acquired resistance to
sunitinib in a patient with hepato-cellular carcinoma. J Hepatol.
54:1073–1078. 2011. View Article : Google Scholar
|
|
156
|
Dangi-Garimella S, Krantz SB, Barron MR,
Shields MA, Heiferman MJ, Grippo PJ, Bentrem DJ and Munshi HG:
Three-dimensional collagen I promotes gemcitabine resistance in
pancreatic cancer through MT1-MMP-mediated expression of HMGA2.
Cancer Res. 71:1019–1028. 2011. View Article : Google Scholar
|
|
157
|
Dangi-Garimella S, Sahai V, Ebine K, Kumar
K and Munshi HG: Three-dimensional collagen I promotes gemcitabine
resistance in vitro in pancreatic cancer cells through
HMGA2-dependent histone acetyltransferase expression. PLoS One.
8:e645662013. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Giannini G, Di Marcotullio L, Ristori E,
Zani M, Crescenzi M, Scarpa S, Piaggio G, Vacca A, Peverali FA,
Diana F, et al: HMGI(Y) and HMGI-C genes are expressed in
neuroblastoma cell lines and tumors and affect retinoic acid
responsiveness. Cancer Res. 59:2484–2492. 1999.PubMed/NCBI
|
|
159
|
Xia YY, Yin L, Tian H, Guo WJ, Jiang N,
Jiang XS, Wu J, Chen M, Wu JZ and He X: HMGA2 is associated with
epithelial-mesenchymal transition and can predict poor prognosis in
nasopharyngeal carcinoma. OncoTargets Ther. 8:169–176. 2015.
View Article : Google Scholar
|
|
160
|
Davidson B, Holth A, Hellesylt E, Tan TZ,
Huang RY, Tropé C, Nesland JM and Thiery JP: The clinical role of
epithelial-mesen-chymal transition and stem cell markers in
advanced-stage ovarian serous carcinoma effusions. Hum Pathol.
46:1–8. 2015. View Article : Google Scholar
|
|
161
|
Rogalla P, Drechsler K, Kazmierczak B,
Rippe V, Bonk U and Bullerdiek J: Expression of HMGI-C, a member of
the high mobility group protein family, in a subset of breast
cancers: Relationship to histologic grade. Mol Carcinog.
19:153–156. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Lee CT, Wu TT, Lohse CM and Zhang L:
High-mobility group AT-hook 2: An independent marker of poor
prognosis in intrahepatic cholangiocarcinoma. Hum Pathol.
45:2334–2340. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Hristov AC, Cope L, Reyes MD, Singh M,
Iacobuzio-Donahue C, Maitra A and Resar LM: HMGA2 protein
expression correlates with lymph node metastasis and increased
tumor grade in pancreatic ductal adenocarcinoma. Mod Pathol.
22:43–49. 2009. View Article : Google Scholar :
|
|
164
|
Yang GL, Zhang LH, Bo JJ, Hou KL, Cai X,
Chen YY, Li H, Liu DM and Huang YR: Overexpression of HMGA2 in
bladder cancer and its association with clinicopathologic features
and prognosis HMGA2 as a prognostic marker of bladder cancer. Eur J
Surg Oncol. 37:265–271. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Zou Q, Xiong L, Yang Z, Lv F, Yang L and
Miao X: Expression levels of HMGA2 and CD9 and its
clinicopathological signifi-cances in the benign and malignant
lesions of the gallbladder. World J Surg Oncol. 10:922012.
View Article : Google Scholar
|
|
166
|
Lee CT, Zhang L, Mounajjed T and Wu TT:
High mobility group AT-hook 2 is overexpressed in hepatoblastoma.
Hum Pathol. 44:802–810. 2013. View Article : Google Scholar
|
|
167
|
Raskin L, Fullen DR, Giordano TJ, Thomas
DG, Frohm ML, Cha KB, Ahn J, Mukherjee B, Johnson TM and Gruber SB:
Transcriptome profiling identifies HMGA2 as a biomarker of melanoma
progression and prognosis. J Invest Dermatol. 133:2585–2592. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Qian ZR, Asa SL, Siomi H, Siomi MC,
Yoshimoto K, Yamada S, Wang EL, Rahman MM, Inoue H, Itakura M, et
al: Overexpression of HMGA2 relates to reduction of the let-7 and
its relationship to clinicopathological features in pituitary
adenomas. Mod Pathol. 22:431–441. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Belge G, Meyer A, Klemke M, Burchardt K,
Stern C, Wosniok W, Loeschke S and Bullerdiek J: Upregulation of
HMGA2 in thyroid carcinomas: A novel molecular marker to
distinguish between benign and malignant follicular neoplasias.
Genes Chromosomes Cancer. 47:56–63. 2008. View Article : Google Scholar
|
|
170
|
Zhang S, Zhang H and Yu L: HMGA2 promotes
glioma invasion and poor prognosis via a long-range chromatin
interaction. Cancer Med. 7:3226–3239. 2018. View Article : Google Scholar :
|
|
171
|
Na N, Si T, Huang Z, Miao B, Hong L, Li H
and Qiu J and Qiu J: High expression of HMGA2 predicts poor
survival in patients with clear cell renal cell carcinoma.
OncoTargets Ther. 9:7199–7205. 2016. View Article : Google Scholar
|
|
172
|
Günther K, Foraita R, Friemel J, Günther
F, Bullerdiek J, Nimzyk R, Markowski DN, Behrens T and Ahrens W:
The stem cell factor HMGA2 is expressed in non-HPV-associated head
and neck squamous cell carcinoma and predicts patient survival of
distinct subsites. Cancer Epidemiol Biomarkers Prev. 26:197–205.
2017. View Article : Google Scholar
|
|
173
|
Mito JK, Agoston AT, Dal Cin P and
Srivastava A: Prevalence and significance of HMGA2 expression in
oesophageal adeno-carcinoma. Histopathology. 71:909–917. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Sarhadi VK, Wikman H, Salmenkivi K, Kuosma
E, Sioris T, Salo J, Karjalainen A, Knuutila S and Anttila S:
Increased expression of high mobility group A proteins in lung
cancer. J Pathol. 209:206–212. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Di Cello F, Hillion J, Hristov A, Wood LJ,
Mukherjee M, Schuldenfrei A, Kowalski J, Bhattacharya R, Ashfaq R
and Resar LM: HMGA2 participates in transformation in human lung
cancer. Mol Cancer Res. 6:743–750. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Strell C, Norberg KJ, Mezheyeuski A,
Schnittert J, Kuninty PR, Moro CF, Paulsson J, Schultz NA,
Calatayud D, Löhr JM, et al: Stroma-regulated HMGA2 is an
independent prognostic marker in PDAC and AAC. Br J Cancer.
117:65–77. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Motoyama K, Inoue H, Nakamura Y, Uetake H,
Sugihara K and Mori M: Clinical significance of high mobility group
A2 in human gastric cancer and its relationship to let-7 microRNA
family. Clin Cancer Res. 14:2334–2340. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Berlingieri MT, Manfioletti G, Santoro M,
Bandiera A, Visconti R, Giancotti V and Fusco A: Inhibition of
HMGI-C protein synthesis suppresses retrovirally induced neoplastic
transformation of rat thyroid cells. Mol Cell Biol. 15:1545–1553.
1995. View Article : Google Scholar : PubMed/NCBI
|