Introduction
Non-small cell lung cancer (NSCLC) is a type of lung
cancer that severely threatens the health and life of individuals
worldwide (1,2). Although researchers have made
significant progress in early diagnostic and treatment methods in
recent years, the prognosis of patients with NSCLC remains
unsatisfactory (3,4). Chemotherapy is one of the main
approaches used in the treatment of NSCLC, and cisplatin (DDP) is a
common drug used in the treatment of NSCLC (5,6).
However, the acquisition of chemoresistance severely hinders the
effects of chemotherapy (7).
Therefore, it is particularly essential to explore the mechanisms
responsible for drug resistance in NSCLC.
Long non-coding RNAs (lncRNAs) are a set of
non-coding RNAs (ncRNAs) of >200 nucleotides (nts) in length,
which do not have protein-coding potential (8). Recently, an increasing number of
lncRNAs have been elaborated, and studies have demonstrated that
diverse lncRNAs play crucial roles in drug resistance in NSCLC. For
instance, Ge et al indicated that FOXD2 adjacent opposite
strand RNA 1 (FOXD2-AS1) was aberrantly expressed in drug-resistant
NSCLC and that its absence suppressed cisplatin resistance in
cisplatin-resistant NSCLC cells (9). Liu et al demonstrated that HOX
transcript antisense RNA (HOTAIR) was upregulated in
cisplatin-resistant NSCLC patients and the deficiency of HOTAIR
improved cisplatin sensitivity in cisplatin-resistant NSCLC cells
(10). However, Wang et al
found that the maternally expressed 3 (MEG3) level was decreased in
patients with cisplatin-resistant NSCLC and that the elevation of
MEG3 enhanced the sensitivity of NSCLC cells to cisplatin (11). These studies suggest that lncRNAs
play dual roles in regulating drug resistance in NSCLC. The present
study focused on the function of lung cancer-associated transcript
1 (LUCAT1) in cisplatin resistance in NSCLC.
MicroRNAs (miRNAs or miRs), a family of ncRNAs of
approximately 22 nts in length, which modulate gene expression by
recognizing the 3′-untranslated region (3′UTR) of target messenger
RNAs (mRNAs) (12). An increasing
number of miRNAs have been confirmed to function as vital
media-tors of drug resistance in human tumors, including NSCLC. For
example, miR-197 has been shown to be weakly expressed in patients
with platinum-resistant NSCLC and miR-197 inhibition has been shown
to enhance drug resistance and tumor growth (13). It has also been demonstrated that
the upregulation of miR-451 suppresses the resistance of A549 cells
to DDP by inhibiting cell growth and inducing cell apoptosis
(14). The deficiency of
miR-138-5p also contributes to the resistance of NSCLC cells to
gefitinib (15). Nevertheless, to
the best of our knowledge, there are no studies available to date
on the role of miR-514a-3p in DDP resistance in NSCLC.
Uncoordinated-51-like kinase 1 (ULK1) is an
autophagy-related gene which has been revealed to play a role in
the progression of drug resistance in diverse human cancers, such
as hepatocellular carcinoma (HCC) (16), breast cancer (17) and colorectal cancer (18). Moreover, Zhao et al proved
that claudin 1 (CLDN1) enhanced drug resistance via the
phosphorylation of ULK1 in NSCLC (19), indicating that ULK1 plays a vital
role in drug resistance in NSCLC.
In the present study, the expression levels of
LUCAT1, miR-514a-3p and ULK1 in cisplatin-resistant NSCLC cells
were investigated. Furthermore, the functions and underlying
mechanisms of LUCAT1 in the resistance of NSCLC cells to DDP were
explored.
Materials and methods
Tissue collection
After the patients received DDP treatment, a total
of 30 DDP-resistant NSCLC tissues, 30 DDP-sensitive NSCLC tissues
and 30 tumor-adjacent normal tissues were harvested from patients
with NSCLC who were resistant or sensitive to DDP at the First
Hospital of China Medical University between October, 2015 and
June, 2017. All patients with NSCLC received DDP-based treatment
for 6 cycles. The clinicopathological characteristics of the
patients with NSCLC are presented in Table I. The samples were immediately
placed in liquid nitrogen and preserved at -80°C until use. The
sample collection was conducted under the supervision of the Ethics
Committee of the First Hospital of China Medical University.
Written informed consent forms were signed by the patients.
 | Table IClinicopathological characteristics
of the patients with NSCLC. |
Table I
Clinicopathological characteristics
of the patients with NSCLC.
| Parameter | Case | NSCLC patients
| P-value |
|---|
| DDP-resistant
(n=30) | DDP-sensitive
(n=30) |
|---|
| Age (years) | | | | |
| ≤65 | 32 | 17 | 15 | 0.6048 |
| >65 | 28 | 13 | 15 | |
| Sex | | | | |
| Male | 32 | 14 | 18 | 0.3006 |
| Female | 28 | 16 | 12 | |
| Lymph node
metastasis | | | | |
| No | 22 | 7 | 15 | 0.0321a |
| Yes | 38 | 23 | 15 | |
| Stage | | | | |
| I+II | 35 | 13 | 22 | 0.0184a |
| III | 25 | 17 | 8 | |
Cells and cell culture
Normal human lung fibroblasts (IMR90) and NSCLC
cells (A549) were purchased from the American Type Culture
Collection (ATCC). To establish DDP-resistant NSCLC cells
(A549/DDP), A549 cells were exposed to gradually increasing
concentrations of cisplatin (Beijing Solarbio Science &
Technology Co., Ltd.) until the cells were able to proliferate
stably. All the cells were cultured in RPMI-1640 medium (Gibco;
Thermo Fisher Scientific, Inc.) supplemented with 10% fetal bovine
serum (FBS; Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin/streptomycin (Beijing Solarbio Science & Technology
Co., Ltd.) in an incubator containing 5% CO2 at
37°C.
Cell transfection
The overexpression vector of LUCAT1 (LUCAT1), the
overexpression vector of ULK1 (ULK1) and their control (Vector),
short hairpin RNA (shRNA) targeting LUCAT1 (sh-LUCAT1), shRNA
targeting ULK1 (sh-ULK1) and their control (sh-NC), mimics of
miR-514a-3p (miR-514a-3p) and its control (miR-NC), inhibitors of
miR-514a-3p (Anti-miR-514a-3p) and its control (Anti-NC) were
synthesized by Guangzhou RiboBio Co., Ltd. 50 nM synthetic
oligonucleotides or 2 µg vectors were transfected into A549
or A549/DDP cells utilizing Lipofectamine 2000 (Invitrogen; Thermo
Fisher Scientific, Inc.) according to the manufacturers'
instructions. Following 48 h of transfection, the cells were
harvested for further experiments. RT-qPCR or western blot analysis
were conducted to determine the transfection efficiency.
A549 cells were transfected with LUCAT1, Vector,
Anti-NC, Anti-miR-514a-3p, Anti-miR-514a-3p + sh-NC,
Anti-miR-514a-3p + sh-LUCAT1, ULK1, ULK1 + miR-NC, ULK1 +
miR-514a-3p, LUCAT1+miR-NC or LUCAT1 + miR-514a-3p and then treated
with or without cisplatin. A549/DDP cells were transfected with
sh-LUCAT1, sh-NC, miR-NC, miR-514a-3p, miR-514a-3p + Vector,
miR-514a-3p + LUCAT1, sh-ULK1, sh-ULK1 + Anti-NC or sh-ULK1 +
Anti-miR-514a-3p and then treated with or without cisplatin.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from the tissues and cells
using RNAiso Plus (Takara Biotechnology Co., Ltd.). The abundance
of RNA samples was measured using a NanoDrop 2000c
spectrophotometer (Thermo Fisher Scientific, Inc.). Reverse
transcription was then conducted using the PrimeScript™ RT reagent
kit (Takara Biotechnology Co., Ltd.) or the miRNA 1st Strand cDNA
Synthesis kit (Vazyme Biotech Co., Ltd.). Subsequently, qPCR was
performed with AceQ Universal SYBR qPCR Master Mix (Vazyme Biotech
Co., Ltd.) under the following thermocycling conditions: i) 95°C
for 5 min; ii) 40 cycles of 95°C for 10 sec and 60°C for 30 sec;
iii) 95°C for 15 sec, 60°C for 60 sec and 95°C for 15 sec. The
levels of LUCAT1, miR-514a-3p and ULK1 were evaluated using the
2−ΔΔCq method (20).
Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used to
normalize the expression of LUCAT1 and ULK1, while small nuclear
RNA U6 was used to normalize the expression of miR-514a-3p. The
primers used were as follows: LUCAT1 forward, 5′-ACC AGC TGT CCC
TCA GTG TTC T-3′ and reverse, 5′-AGG CCT TTA TCC TCG GGT TGC CT-3′;
miR-514a-3p forward, 5′-ATT GAC ACT TCT GTG AGT AGA-3′ and reverse,
5′-CAG TGC GTG TCG TGG AGT-3′); ULK1 forward, 5′-TGC CCC TGG TTG
AAT GTT CT-3′ and reverse, 5′-ACA CCA GCC CAA CAA TTC CA-3′; GAPDH
forward, 5′-GGT CTC CTC TGA CTT CAA CA-3′ and reverse, 5′-GTG AGG
GTC TCT CTC TTC CT-3′); and U6 forward, 5′-TGC GGG TGC TCG CTT CGG
CAG C-3′ and reverse, 5′-CCA GTG CAG GGT CCG AGG T-3′.
Cell Counting kit-8 (CCK-8) assay
For the analysis of cisplatin resistance, A549 and
A549/DDP cells were seeded into 96-well plates and exposed to
various concentrations of cisplatin (0.5, 1.5 or 2.0 µg/ml;
Beijing Solarbio Science & Technology Co., Ltd.) for 24 h.
Subsequently, 20 µl CCK-8 solution (Beyotime Institute of
Biotechnology, Inc.) was added to each well and cultured for a
further 2 h at 37°C. The absorbance was examined at 450 nm using a
microplate reader (Bio-Rad Laboratories, Inc.). The half maximal
inhibitory concentration (IC50) was the concentration of
cisplatin that induced 50% growth inhibition and determined by the
relative survival curve. For the analysis of cell viability, CCK-8
(Beyotime Institute of Biotechnology, Inc.) was added at 0, 24, 48
and 72 h. The other steps were the same as those described
above.
Flow cytometric analysis
The Annexin V-fluorescein isothiocyanate
(FITC)/propidium iodide (PI) Apoptosis Detection kit (Beyotime
Institute of Biotechnology, Inc.) was utilized for the analysis of
cell apoptosis. In brief, cells were harvested, washed,
resuspended, and then 5 µl AnnexinV-FITC and 10 µl PI
were added and maintained for 15 min in the dark to stain the
cells. The stained cells were analyzed using a flow cytometry (BD
Biosciences).
Western blot analysis
Total protein was isolated from the cells using RIPA
buffer (Beyotime Institute of Biotechnology, Inc.). The proteins
were quantified using a NanoDrop 2000c spectrophotometer (Thermo
Fisher Scientific, Inc.). A total of 20 µg proteins were
separated by 10% sodium dodecyl sulfonate-polyacrylamide gel
(SDS-PAGE; Beijing Solarbio Science & Technology Co., Ltd.) and
then transferred onto polyvinylidene difluoride membranes (PVDF;
Pall Corporation). The membranes were blocked in skim milk for 1 h
at room temperature and incubated with primary antibodies against
microtubule-associated protein light chain 3-I/II (LC3-I/II;
ab192890; 1:2,000; Abcam), p62 (ab56416; 1:2,000; Abcam), ULK1
(ab167139; 1:1,000; Abcam) or GAPDH (ab9485; 1:2,500; Abcam)
overnight at 4°C followed by incubation with corresponding
secondary antibody (ab150117; 1:2,000; Abcam) for 2 h at room
temperature. The protein levels were analyzed using an enhanced
chemiluminescence kit (Vazyme Biotech Co., Ltd.). The results were
analyzed using software ImageJ v1.8.0 (National Institutes of
Health).
Transwell assay
Transwell chambers (Corning, Inc.) coated with (for
cell invasion assay) or without (for cell migration assay) Matrigel
(Beijing Solarbio Science & Technology Co., Ltd.) were employed
to examine cell invasion and migration. Briefly, following relevant
transfection and treatment, the A549 or A549/DDP cells in
serum-free medium were added to the upper chamber and culture
medium supplemented with 10% FBS (Gibco; Thermo Fisher Scientific,
Inc.) was added to the bottom chamber. After 24 h, the migrated or
invaded cells were fixed with methanol and stained with 0.1%
crystal violet (Beijing Solarbio Science & Technology Co.,
Ltd.) for 20 min at room temperature. The stained cells were
observed under an inverted microscope (Olympus Corporation).
Dual-luciferase reporter assay
The potential binding sites between miR-514a-3p and
LUCAT1 or ULK1 were predicated by StarBase 3.0 (http://starbase.sysu.edu.cn/tutorialAPI.php) and then
verified by dual-luciferase reporter assay. In brief, the sequences
of LUCAT1 or 3′UTR of ULK1 containing the potential binding sites
of miR-514a-3p were amplified and introduced into the XhoI
and XbaI sites of the downstream of Firefly luciferase gene
in the pmirGLO vector (Promega Corporation) to generate luciferase
reporter vectors LUCAT1-WT and ULK1-3′UTR-WT, respectively. The
sequences of mutant type LUCAT1 or 3′UTR of ULK1 lacking the
binding sites of miR-514a-3p were also cloned into the pmirGLO
vector to construct the reporter vectors, LUCAT1-MUT and
ULK1-3′UTR-MUT, respectively. The A549 cells and A549/DDP cells
were seeded into 24-well plates and 100 ng of the indicated
luciferase reporter vector were then transfected into the cells in
combination with 50 nM miR-514a-3p or miR-NC using Lipofectamine
2000 (Invitrogen; Thermo Fisher Scientific, Inc.). After 48 h, the
cells were collected and the luciferase activity was determined
using a Dual-Luciferase Reporter Assay kit (Promega Corporation).
Renilla luciferase activity was used to normalize Firefly
luciferase activity.
RNA immunoprecipitation (RIP) assay
RIP assay was conducted using a Magna RIP™ RNA
Binding Protein Immunoprecipitation kit (EMD Millipore). In brief,
the A549 cells or A549/DDP cells were lysed with RIP lysis buffer
and then incubated overnight at 4°C with magnetic beads conjugated
with antibody against Argonaute2 (Anti-Ago2; ab32381; 1:2,000;
Abcam) or immunoglobulin G (Anti-IgG; ab109489; 1:5,000; Abcam).
The cells were incubated with Proteinase K (Beijing Solarbio
Science & Technology Co., Ltd.) for 30 min at 55°C. Finally,
the enrichment of LUCAT1, miR-514a-3p and ULK1 was measured by
RT-qPCR following the purification of the RNA as described
above.
In vivo experiment
A total of 28 male nude mice (age: 5 weeks old;
weight: 16-23 g) were obtained from Shanghai SLAC Laboratory
Animals Co., Ltd. and divided into 4 groups (n=7/group): Vector +
Cisplatin, LUCAT1 + Cisplatin, sh-NC + Cisplatin and sh-LUCAT1 +
Cisplatin. All the mice were housed at 27°C in pathogen-free
conditions with 45% humidity and 12 h light/dark cycle and fed
sterile fodder and drinking water. For LUCAT1 + Cisplatin and
Vector + Cisplatin groups, the A549 cells were transfected with
LUCAT1 or Vector and then injected subcutaneously into the flanks
of the mice. For sh-LUCAT1 + Cisplatin and sh-NC + Cisplatin
groups, the A549/DDP cells were transfected with sh-LUCAT1 or
sh-NC, and then injected subcutaneously into the flanks of the
mice. After 1 week, all mice were administered cisplatin (5 mg/kg;
Beijing Solarbio Science & Technology Co., Ltd.) each week for
5 weeks. Tumor volume was detected each week and calculated using
the formula: (length x width2)/2. After 6 weeks, the
mice were euthanized by cervical dislocation and tumors were
harvested, weighed and preserved at −80°C for analysis by RT-qPCR.
The criteria for judging the death of the mice were continuous
involuntary breathing for 2-3 min and no blinking reflex. The
animal experiments were approved by the Ethics Committee of Animal
Research of the First Hospital of China Medical University. The
humane endpoints established in the present study for the animal
experiments were as follows: A tumor burden >10% of the body
weight and a tumor which did not exceed 20 mm in any one
dimension.
Statistical analysis
All data were obtained from 3 independent
experiments and are presented as the means ± standard deviation
(SD). GraphPad Prism 7 software (GraphPad Inc.) was employed to
analyze the collected data. A paired/unpaired Student's t-test or
one-way analysis of variance (ANOVA) followed by Tukey's test were
utilized to determine significant differences between groups. The
clinicopathological features were analyzed using an χ2
test. A value of <0.05 was considered to indicate a
statistically significant difference.
Results
LUCAT1 is highly expressed in
DDP-resistant NSCLC tissues and cells
In order to investigate the potential role of LUCAT1
in DDP resistance in NSCLC, the expression of LUCAT1 in
DDP-resistant NSCLC tissues and cells was first determined. The
results of RT-qPCR revealed that LUCAT1 was markedly upregulated in
NSCLC tissues (DDP-resistant NSCLC tissues and DDP-sensitive NSCLC
tissues) and cells (A549 cells and A549/DDP cells) compared to that
in normal tissues and cells; furthermore, LUCAT1 expression was
higher in DDP-resistant NSCLC tissues and cells when compared to
DDP-sensitive NSCLC tissues and cells (Fig. 1A and B). In addition, the
IC50 value of cisplatin evidently increased in the
A549/DDP compared to the A549 cells, indicating that cisplatin
resistance was established in the A549/DDP cells (Fig. 1C). Moreover, the cells were treated
with various concentrations of cisplatin for 24 h and CCK-8 assay
was then performed. The results demonstrated that the viability of
the A549/DDP cells was increased compared to that of the A549 cells
(Fig. 1D). Cells treated with 2
µg/ml cisplatin were used in the subsequent experiments. All
these data indicated that LUCAT1 may play a role in the regulation
of DDP resistance in NSCLC.
LUCAT1 enhances cisplatin resistance by
inhibiting the apoptosis, and promoting the viability, autophagy,
migration and invasion in NSCLC cells
In view of the fact that LUCAT1 expression was
higher in the A549/DDP cells compared to the A549 cells, LUCAT1
overexpression vector was transfected into A549 cells and sh-LUCAT1
was transfected into A549/DDP cells to explore the functional role
of LUCAT1 in the regulation of the cisplatin resistance of NSCLC
cells. As shown in Fig. 2A, LUCAT1
overexpression led to a marked increase in LUCAT1 expression in the
A549 cells and sh-LUCAT1 transfection led to a marked decrease in
LUCAT1 expression in the A549/DDP cells. CCK-8 assay revealed that
the viability of the A549 cells was promoted following the
overexpression of LUCAT1 and was suppressed in the A549/DDP cells
following the knockdown of LUCAT1 (Fig. 2B). Cisplatin treatment induced the
apoptosis of the A549 cells and A549/DDP cells; however, the
apoptosis of the A549 cells was suppressed following the
overexpression of LUCAT1 and that of the A549/DDP cells was
promoted following the knockdown of LUCAT1, as indicated by flow
cytometric analysis (Fig. 2C).
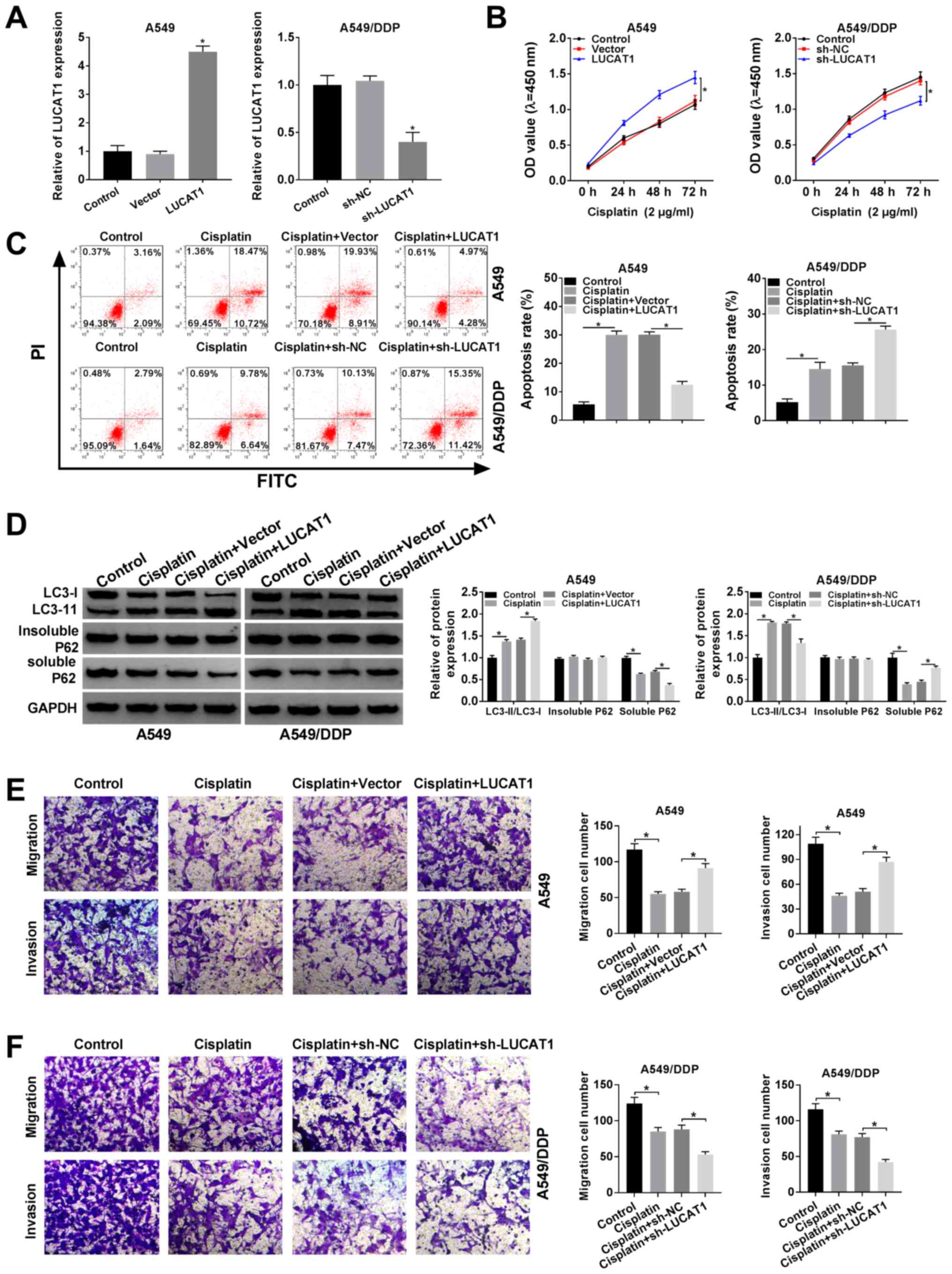 | Figure 2LUCAT1 plays a role in the regulation
of cisplatin resistance by modulating the apoptosis, autophagy,
migration and invasion of NSCLC cells. (A and B) A549 cells were
transfected with Vector or LUCAT1, A549/DDP cells were transfected
with sh-NC or sh-LUCAT1, and untransfected A549 cells and A549/DDP
cells were used as the control groups. (A) The level of LUCAT1 in
A549 cells and A549/DDP cells was measured by RT-qPCR. (B) The
viability of A549 cells and A549/DDP cells was assessed through
CCK-8 assay. (C-F) A549 cells were treated with cisplatin,
cisplatin + Vector or cisplatin + LUCAT1, A549/DDP cells were
treated with cisplatin, cisplatin + sh-NC or cisplatin + sh-LUCAT1,
and untreated A549 cells and A549/DDP cells were used as the
control group. (C) The apoptosis of A549 cells and A549/DDP cells
was evaluated by flow cytometric analysis. (D) The ratio of
LC3-II/LC3-I and the levels of insoluble p62 and soluble p62 in
A549 cells and A549/DDP cells were determined by western blot
analysis. (E and F) The migration and invasion of A549 and A549/DDP
cells were examined by Transwell assay. *P<0.05 vs.
respective control. LUCAT1, lung cancer-associated transcript 1;
DDP, cisplatin; NSCLC, non-small cell lung cancer. |
If soluble p62 is decreased, insoluble p62 is
stable, and the ratio of LC3-II/LC3-I is increased, indicating the
activation of autophagy. Thus, the levels of autophagy-related
proteins in the A549 cells and A549/DDP cells were determined by
western blot analysis. It was observed that cisplatin treatment
increased the ratio of LC3-II/LC3-I and decreased the level of
soluble p62 in the A549 cells and A549/DDP cells; more-over, the
elevated expression of LUCAT1 further enhanced the LC3-II/LC3-I
ratio and decreased the soluble p62 level in the A549 cells;
however, the silencing of LUCAT1 decreased the LC3-II/LC3-I ratio
and increased the soluble p62 level in the A549/DDP cells (Fig. 2D). However, the level of insoluble
p62 was not altered when the A549 and A549/DDP cells were subjected
to the above-mentioned treatments (Fig. 2D). When autophagy was enhanced, the
ratio of LC3-II/LC3-I was increased and the level of p62 was
decreased.
The results of Transwell assay indicated that
cisplatin treatment markedly suppressed the migration and invasion
of the A549 and A549/DDP cells, whereas these effects were
abrogated in the A549 cells following LUCAT1 overexpression. In
addition, the migration and invasion of the A549/DDP cells were
further suppressed following transfection with sh-LUCAT1,
indicating that LUCAT1 knockdown counter-acted the resistance of
NSCLC cells to cisplatin, at least to a certain extent (Fig. 2E and F). Thus, these results
demonstrate that LUCAT1 can promote cisplatin resistance by
suppressing the apoptosis, and promoting the viability, autophagy
and metastasis of NSCLC cells.
LUCAT1 negatively regulates miR-514a-3p
expression by directly targeting miR-514a-3p
To determine the underlying mechanisms of LUCAT1,
the online software StarBase 3.0 was searched and it was found that
miR-514a-3p was a target of LUCAT1 (Fig. 3A). To verify this, a
dual-luciferase reporter assay and RIP assay were carried out. As
shown in Fig. 3B, co-transfection
with LUCAT1-WT and miR-514a-3p resulted in an evident inhibition of
the luciferase activity in the A549 cells and A549/DDP cells
compared with that in the cells co-transfected with LUCAT1-WT and
miR-NC; however, the luciferase activity was not affected in the
LUCAT1-MUT group.
To act as miRNA sponges, lncRNAs need to be
predominantly enriched in the cytoplasm and effectively accessible
to RNA-induced silencing complex (RISC) (21,22).
In the present study, RIP assay revealed that LUCAT1 and
miR-514a-3p were enriched in Anti-Ago2 immunoprecipitation
complexes compared with Anti-lgG immunoprecipitates in the A549
cells and A549/DDP cells, indicating that LUCAT1 and miR-514a-3p
existed in the RISC, further confirming the association between
LUCAT1 and miR-514a-3p (Fig. 3C).
In addition, as was expected, the expression of miR-514a-3p was
decreased in the A549 cells compared to the IMR90 cells; moreover,
miR-514a-3p expression was lower in the A549/DDP cells than in the
A549 cells (Fig. 3D).
Furthermore, the level of miR-514a-3p in the A549
cells transfected with the LUCAT1 overexpression vector and in the
sh-LUCAT1-transfected A549/DDP cells was examined by RT-qPCR. The
results revealed that the miR-514a-3p level was markedly decreased
in the A549 cells transfected with the LUCAT1 overexpression
vector, and was increased in the A549/DDP cells transfected with
sh-LUCAT1 (Fig. 3E). Taken
together, these results demonstrate that LUCAT1 functions as an
miR-514a-3p sponge to alter miR-514a-3p expression in A549 and
A549/DDP cells.
The inhibitory effect of miR-514a-3p on
cisplatin resistance of NSCLC cells was reversed by LUCAT1
Since LUCAT1 could directly interact with
miR-514a-3p and negatively regulate miR-514a-3p expression, we
hypothesized that LUCAT1 could improve cisplatin resistance via
targeting miR-514a-3p in NSCLC. As shown in Fig. 4A, transfection with
anti-miR-514a-3p led to a marked decrease in miR-514a-3p expression
in the A549 cells, and transfection with miR-514a-3p mimics led to
an evident increase in miR-514a-3p expression in the A549/DDP
cells. Subsequently, the A549 cells were treated with cisplatin +
Anti-NC, cisplatin + Anti-miR-514a-3p, cisplatin + Anti-miR-514a-3p
+ sh-NC or cisplatin + Anti-miR-514a-3p + sh-LUCAT1, and the
A549/DDP cells were treated with cisplatin + miR-NC, cisplatin +
miR-514a-3p, cisplatin + miR-514a-3p + Vector or cisplatin +
miR-514a-3p + LUCAT1. CCK-8 assay revealed that cell viability was
markedly promoted by miR-514a-3p inhibition and LUCAT1 knockdown
reversed this effect in the A549 cells. Moreover, cell viability
was evidently suppressed following the upregulation of miR-514a-3p
and LUCAT1 overexpression reversed this effect in the A549/DDP
cells (Fig. 4B).
 | Figure 4LUCAT1 overexpression reverses the
inhibitory effects of miR-514a-3p overexpression on cisplatin
resistance in NSCLC cells. (A) A549 cells were transfected with
Anti-NC or Anti-miR-514a-3p, A549/DDP cells were transfected with
miR-NC or miR-514a-3p, and untransfected A549 cells and A549/DDP
cells were used as the control groups. The the level of miR-514a-3p
was then detected by RT-qPCR. (B-F) The A549 cells were assigned to
the control (1), cisplatin +
Anti-NC (2), cisplatin +
Anti-miR-514a-3p (3), cisplatin +
Anti-miR-514a-3p + sh-NC (4) and
cisplatin + Anti-miR-514a-3p + sh-LUCAT1 (5) groups, and the A549/DDP cells were
assigned to the control (I), cisplatin + miR-NC (II), cisplatin +
miR-514a-3p (III), cisplatin + miR-514a-3p + Vector (IV) and
cisplatin + miR-514a-3p + LUCAT1 (V) groups. (B) The viability of
the A549 and A549/DDP cells was analyzed by CCK-8 assay. (C) The
apoptosis of the A549 and A549/DDP cells was evaluated by flow
cytometric analysis. (D) The ratio of LC3-II/LC3-I and the levels
of insoluble p62 and soluble p62 were examined by western blot
analysis. (E and F) The migration and invasion of A549 and A549/DDP
cells were analyzed by Transwell assay. *P<0.05 vs.
respective control. LUCAT1, lung cancer-associated transcript 1;
DDP, cisplatin; NSCLC, non-small cell lung cancer. |
The results of flow cytometric analysis demonstrated
that Anti-miR-514a-3p suppressed the apoptosis of the A549 cells
and sh-LUCAT1 abolished this suppression. Moreover, miR-514a-3p
evidently accelerated A549/DDP cell apoptosis and LUCAT1
overexpression effectively attenuated this effect (Fig. 4C). As demonstrated by western blot
analysis, the LC3-II/LC3-I ratio was increased and the soluble p62
level was decreased in the A549 cells transfected with
Anti-miR-514a-3p; these effects were partly reversed after the
cells were transfected with sh-LUCAT1; however, no change was
observed in the insoluble p62 expression level (Fig. 4D, upper panels). In the A549/DDP
cells, miR-514a-3p overexpression markedly inhibited the
LC3-II/LC3-I ratio and promoted the soluble p62 level, and LUCAT1
overexpression abolished these effects; however, the insoluble p62
level was not altered after these treatments (Fig. 4D, lower panels).
Transwell assay also revealed that A549 cell
migration and invasion were promoted by miR-514a-3p inhibition,
while these effects were abolished by LUCAT1 knockdown (Fig. 4E). Transwell assay also revealed
that A549/DDP cell migration and invasion were suppressed by
miR-514a-3p overexpression, whereas the overexpression of LUCAT
attenuated these effects (Fig.
4F). These, these findings demonstrate that LUCAT1 promotes
cisplatin resistance by targeting miR-514a-3p in NSCLC cells.
miR-514a-3p negatively modulates ULK1
expression by direct interaction
Based on the above-mentioned data, StarBase 3.0 was
further searched and it was found that ULK1 was a target gene of
miR-514a-3p; their binding sites are shown in Fig. 5A. As suggested by dual-luciferase
reporter assay, the luciferase activity was markedly suppressed in
the A549 cells and A549/DDP cells co-transfected with miR-514a-3p
and ULK1-WT compared with that in the cells co-transfected with
miR-NC and ULK1-WT, whereas the luciferase activity was not altered
in the ULK1-MUT group (Fig.
5B).
RIP assay revealed that the enrichment of ULK1 and
miR-514a-3p in the A549 cells and A549/DDP cells was markedly
increased in the Anti-Ago2 RIP group compared with the Anti-IgG RIP
group, suggesting that ULK1 could bind to the RISC consisting
miR-514a-3p (Fig. 5C).
Subsequently, Anti-NC or Anti-miR-514a-3p were transfected into the
A549 cells, and miR-NC or miR-514a-3p were transfected into the
A549/DDP cells to further investigate the association between ULK1
and miR-514a-3p. As shown in Fig.
5D, Anti-miR-514a-3p transfection resulted in a marked increase
in the ULK1 protein level in the A549 cells, and transfection with
miR-514a-3p mimic led to an effective decrease in ULK1 protein
expression in the A549/DDP cells. In addition, the protein level of
ULK1 in THE IMR90, A549 and A549/DDP cells was determined. As was
expected, the protein level of ULK1 was increased in the A549 cells
compared with the IMR90 cells, and ULK1 expression in the A549/DDP
cells was higher than that in the A549 cells (Fig. 5E). All these data indicated that
ULK1 was negatively regulated by miR-514a-3p in NSCLC cells.
Silencing of miR-514a-3p attenuates the
inhibitory effect of ULK1 knockdown on the resistance of NSCLC
cells to cisplatin
To reveal the function of miR-514a-3p and ULK1 in
regulating the resistance of NSCLC cells to cisplatin, ULK1 or
Vector were transfected into the A549 cells, and sh-NC or sh-ULK1
were transfected into the A549/DDP cells. The results of western
blot analysis revealed that the protein expression of ULK1 was
markedly increased in the A549 cells following transfection with
ULK1 overexpression vector, and was notably decreased in the
A549/DDP cells following transfection with sh-ULK1 (Fig. 6A). It was then demonstrated that
ULK1 overexpression markedly enhanced the viability of the A549
cells and this enhancement was suppressed by miR-514a-3p
overexpression; moreover, ULK1 knockdown evidently suppressed the
viability of the A549/DDP cells and this suppression was attenuated
by transfection with Anti-miR-514a-3p, as illustrated by CCK-8
assay (Fig. 6B).
 | Figure 6miR-514a-3p inhibition abolishes the
effects of ULK1 knockdown on cisplatin resistance in NSCLC cells.
(A) A549 cells were transfected with Vector or ULK1, A549/DDP cells
were transfected with sh-NC or sh-ULK1, and untransfected A549
cells and A549/DDP cells were used as the control groups; the
protein level of ULK1 was measured by western blot analysis. (C-F)
A549 cells were assigned to the control (1), cisplatin + Vector (2), cisplatin + ULK1 (3), cisplatin + ULK1 + miR-NC (4) and cisplatin + ULK1 + miR-514a-3p
(5) groups; the A549/DDP cells
were assigned to the control (I), cisplatin + sh-NC (II), cisplatin
+ sh-ULK1 (III), cisplatin + sh-ULK1 + Anti-NC (IV) and cisplatin +
sh-ULK1 + Anti-miR-514a-3p (V) groups. (B) The viability of the
A549 and A549/DDP cells was examined by CCK-8 assay. (C) The
apoptosis of the A549 and A549/DDP cells was assessed via flow
cytometric analysis. (D) The ratio of LC3-II/LC3-I and the levels
of insoluble p62 and soluble p62 were measured using western blot
analysis. (E and F) The migration and invasion of A549 and A549/DDP
cells were determined by Transwell assay. *P<0.05 vs.
the respective control. ULK1, uncoordinated-51-like kinase 1; DDP,
cisplatin; NSCLC, non-small cell lung cancer. |
In addition, the results of flow cytometric analysis
indicated that the decreased cell apoptosis induced by ULK1
overexpression was evidently increased in the A549 cells following
the overexpression of miR-514a-3p, and the increased cell apoptosis
mediated by sh-ULK1 was significantly inhibited in the A549/DDP
cells following the downregulation of miR-514a-3p (Fig. 6C). It was also demonstrated that
the ratio of LC3-II/LC3-I was elevated and the protein level of
soluble p62 was decreased following the overexpression of ULK1 in
the A549 cells, and these effects were restored following
miR-514a-3p upregulation; however, the insoluble p62 level was not
altered following these treatments, indicating that the enhancement
of autophagy caused by ULK1 was reversed by miR-514a-3p
overexpression (Fig. 6D, upper
panels). In the A549/DDP cells, ULK1 downregulation markedly
decreased the ratio of LC3-II/LC3-I and induced the level of
soluble p62, whereas the inhibition of miR-514a-3p partly restored
these effects; however, the level of insoluble p62 was stable
following these treatments (Fig.
6D, lower panels).
Furthermore, Transwell assay indicated that ULK1
over-expression promoted the migration and invasion of the A549
cells, while the overexpression of miR-514a-3p reversed these
effects (Fig. 6E). The silencing
of ULK1 markedly impeded the migration and invasion of the A549/DDP
cells, while miR-514a-3p inhibition reversed these effects
(Fig. 6F). Collectively, these
data indicated tht miR-514a-3p bound to ULK1 to regulate cisplatin
resistance in NSCLC cells.
LUCAT1 positively regulates ULK1
expression by sponging miR-514a-3p in NSCLC cells
In order to determine the association between
LUCAT1, miR-514a-3p and ULK1, the A549 cells were assigned to the
control, Vector, LUCAT1, LUCAT1 + miR-NC and LUCAT1 + miR-514a-3p
groups, and the A549/DDP cells were assigned to the control, sh-NC,
sh-LUCAT1, sh-LUCAT1 + Anti-NC and sh-LUCAT1 + Anti-miR-514a-3p
groups. It was found that the mRNA and protein levels of ULK1 were
increased in the A549 cells following the overexpression of LUCAT1,
while miR-514a-3p upregulation partially suppressed this increase
(Fig. 7A and B). The knockdown of
LUCAT1 decreased the mRNA and protein levels of ULK1 in the
A549/DDP cells, whereas miR-514a-3p inhibition abolished these
effects (Fig. 7C and D). Thus, it
was demonstrated that LUCAT1 upregulated the expression of ULK1 by
targeting miR-514a-3p in NSCLC cells.
 | Figure 7LUCAT1 alters ULK1 expression via
acting as a sponge of miR-514a-3p in NSCLC cells. (A and B) A549
cells were transfected with Vector, LUCAT1, LUCAT1 + miR-NC or
LUCAT1 + miR-514a-3p and then the mRNA and protein levels of ULK1
were examined by RT-qPCR and western blot analysis, respectively.
(C and D) A549/DDP cells were transfected with sh-NC, sh-LUCAT1,
sh-LUCAT1 + Anti-NC or sh-LUCAT1 + Anti-miR-514a-3p and the mRNA
and protein levels of ULK1 were then examined by RT-qPCR and
western blot analysis, respectively. *P<0.05 vs. the
respective control. LUCAT1, lung cancer-associated transcript 1;
ULK1, uncoordinated-51-like kinase 1; DDP, cisplatin; NSCLC,
non-small cell lung cancer. |
LUCAT1 knockdown suppresses tumorigenesis
and cisplatin resistance in vivo
To investigate the role of LUCAT1 in vivo,
A549 cells transfected with Vector or LUCAT1 and A549/DDP cells
transfected with sh-NC or sh-LUCAT1 were injected into mice, and
the tumor volume was then examined weekly, and tumor weight was
examined after 6 weeks. The data indicated that LUCAT1 upregulation
evidently promoted tumor growth and tumor weight compared to the
Vector group (Fig. 8A). The
downregulation of LUCAT1 notably suppressed tumor growth and tumor
weight compared with the sh-NC group (Fig. 8B). Furthermore, the levels of
LUCAT1, miR-514a-3p and ULK1 in the collected tumor tissues were
measured. The results revealed that the expression levels of
LUCAT1, ULK1 mRNA and ULK1 protein were markedly upregulated, and
the expression level of miR-514a-3p was notably downregulated in
the tumor tissues from the LUCAT1 group; however, the opposite
results were observed in the tumor tissues from the sh-LUCAT1 group
(Fig. 8C and D). These data
suggested that the knockdown of LUCAT1 inhibited tumor growth and
cisplatin resistance in vivo.
Discussion
Currently, an increasing number of researchers are
focusing on the effects of lncRNAs on tumor development and drug
resistance in human cancers (23,24).
In the present study, the effect of LUCAT1 on cisplatin resistance
in NSCLC was investigated. It was found that the LUCAT1 level was
elevated in the A549/DDP cells, and LUCAT1 knockdown enhanced the
sensitivity of A549/DDP cells to cisplatin by regulating the
miR-514a-3p/ULK1 axis.
Zheng et al demonstrated that the LUCAT1
level was elevated in cisplatin-resistant ovarian cancer cells
(25). Han and Shi suggested that
LUCAT1 was highly expressed in methotrexate-resistant osteosarcoma
(OS) cells, and the inhibition of LUCAT1 markedly hampered
methotrexate resistance, cell growth and metastasis in OS (26). Furthermore, Wang et al
indicated that LUCAT1 expression was higher in A549/DDP cells than
in A549 cells; moreover, LUCAT1 elevation suppressed cell apoptosis
and cisplatin sensitivity in NSCLC cells (27). Consistently, in the present study,
it was observed that the LUCAT1 level was markedly increased in
DDP-resistant NSCLC tissues and cells. The knockdown of LUCAT1
evidently suppressed the viability and motility, and induced the
apoptosis of DDP-resistant NSCLC cells. Autophagy is a process of
cell self-degradation used to remove damaged or redundant proteins
and organelles, and can be observed in a number of physiological
and pathological processes (28).
It has been reported that autophagy is associated with drug
resistance in malignant tumors. For example, Yan et al
demonstrated that HOTAIR silencing relieved drug resistance by
suppressing the activation of autophagy in NSCLC (29). Xiong et al elucidated that
HULC improved the chemoresistance of HCC cells by activating
autophagy (30). To the best of
our knowledge, the present study was the first to examine the
effects of LUCAT1 on the protein levels of autophagy regulators and
it was found that LUCAT1 knock-down decreased the LC3-II/LC3-I
ratio and increased the p62 level in A549/DDP cells, suggesting
that autophagy was suppressed. These data suggest that LUCAT1 plays
a positive role in cisplatin resistance in NSCLC.
Subsequently, the present study explored the
underlying mechanisms of LUCAT1 in the regulation of the drug
sensitivity of NSCLC cells. LUCAT1 was identified to function as a
sponge of miR-514a-3p and the miR-514a-3p level was decreased in
A549/DDP cells. miR-514a-3p has been confirmed to be weakly
expressed and to play a tumor-suppressive role in renal cell
carcinoma by suppressing cell growth and inducing cell apoptosis
(31). Here, we determined the
role of miR-514a-3p in drug resistance for the first time. The
present study found that the upregulation of miR-514a-3p suppressed
cell viability and metastasis, facilitated cell apoptosis and
inactivated autophagy in cisplatin-resistant NSCLC cells, whereas
these effects were partly abolished following LUCAT1
overexpression.
Moreover, ULK1 was confirmed to be a target of
miR-514a-3p. ULK1 is a regulator of autophagy and plays a role part
in the regulation of drug resistance in human tumors (32). For example, Tang et al
proved that ULK1 knockdown suppressed cell growth and cisplatin
resistance by modulating the apoptosis and autophagy of NSCLC cells
(33). Yang et al suggested
that HOTAIR downregulation improved crizotinib sensitivity by
hindering cell viability and autophagy and accelerating cell
apoptosis in NSCLC through decreasing ULK1 expression (29). Consistently, the present study
observed that the ULK1 level was decreased in A549/DDP cells, and
ULK1 deficiency inhibited cisplatin resistance by inhibiting cell
viability and metastasis, promoting cell apoptosis and blocking
autophagy in cisplatin-resistant NSCLC cells, whereas inhibitors of
miR-514a-3p abolished this inhibition.
However, the present study also contains certain
shortcomings. For example, TUNEL staining assay was not performed
to examine the cell apoptotic ability. Moreover,
immunocy-tochemistry and immunofluorescence microscopy were not
conducted to examine the intracellular localization of LC3-II and
the localization of different forms of p62 in cells. The authors
thus aim to conduct these experiments in the future.
In conclusion, the present study demonstrated that
the LUCAT1 level was evidently increased in DDP-resistant NSCLC
cells. LUCAT1 silencing enhanced cisplatin sensitivity by inducing
cell apoptosis, and suppressing autophagy and cell metastasis in
NSCLC. Moreover, LUCAT1 regulated the sensitivity of NSCLC cells to
cisplatin by upregulating ULK1 via sponging miR-514a-3p. These
findings provide a novel regulatory network of the
LUCAT1/miR-514a-3p/ULK1 axis in regulating the chemoresistance of
NSCLC. These findings suggest that LUCAT1 may be a potential target
which may be used to counteract the resistance of NSCLC to DDP.
Acknowledgments
Not applicable.
Funding
No funding was received.
Availability of data and materials
The analyzed data sets generated during the study
are available from the corresponding author on reasonable
request.
Authors' contributions
QS conceived the study, and ZX and SX designed the
study. QMS was involved in data collection. ZX performed the
statistical analysis and prepared the figures. QS drafted the
manuscript. SX contributed substantially to the revision of the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The study protocol was approved by the Ethics
Committee of the First Hospital of China Medical University.
Written informed consent forms were signed by the patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fitzmaurice C, Akinyemiju TF, Al Lami FH,
Alam T, Alizadeh-Navaei R, Allen C, Alsharif U, Alvis-Guzman N,
Amini E and Anderson BO: Global, regional, and national cancer
incidence, mortality, years of life lost, years lived with
disability, and disability-adjusted life-years for 29 cancer
groups, 1990 to 2016: A systematic analysis for the global burden
of disease study. JAMA Oncol. 4:1553–1568. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Heist RS and Engelman JA: SnapShot:
Non-small cell lung cancer. Cancer Cell. 21:448.e22012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hirsch FR, Suda K, Wiens J and Bunn PA Jr:
New and emerging targeted treatments in advanced non-small-cell
lung cancer. Lancet. 388:1012–1024. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fennell D, Summers Y, Cadranel J, Benepal
T, Christoph D, Lal R, Das M, Maxwell F, Visseren-Grul C and Ferry
D: Cisplatin in the modern era: The backbone of first-line
chemotherapy for non-small cell lung cancer. Cancer Treat Rev.
44:42–50. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Arriagada R, Bergman B, Dunant A, Le
Chevalier T, Pignon JP and Vansteenkiste J; International Adjuvant
Lung Cancer Trial Collaborative Group: Cisplatin-based adjuvant
chemotherapy in patients with completely resected non-small-cell
lung cancer. N Engl J Med. 350:351–360. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Herbst RS, Morgensztern D and Boshoff C:
The biology and management of non-small cell lung cancer. Nature.
553:4462018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schmitz SU, Grote P and Herrmann BG:
Mechanisms of long noncoding RNA function in development and
disease. Cell Mol Life Sci. 73:2491–2509. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ge P, Cao L, Yao YJ, Jing RJ, Wang W and
Li HJ: lncRNA FOXD2-AS1 confers cisplatin resistance of
non-small-cell lung cancer via regulation of miR185-5p-SIX1 axis.
Onco Targets Ther. 12:6105–6117. 2019. View Article : Google Scholar :
|
|
10
|
Liu MY, Li XQ, Gao TH, Cui Y, Ma N, Zhou Y
and Zhang GJ: Elevated HOTAIR expression associated with cisplatin
resistance in non-small cell lung cancer patients. J Thorac Dis.
8:3314–3322. 2016. View Article : Google Scholar
|
|
11
|
Wang P, Chen D, Ma H and Li Y: LncRNA MEG3
enhances cisplatin sensitivity in non-small cell lung cancer by
regulating miR-21-5p/SOX7 axis. Onco Targets Ther. 10:5137–5149.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dragomir MP, Knutsen E and Calin GA:
SnapShot: Unconventional miRNA functions. Cell. 174:1038–1038.e1.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fujita Y, Yagishita S, Hagiwara K,
Yoshioka Y, Kosaka N, Takeshita F, Fujiwara T, Tsuta K, Nokihara H,
Tamura T, et al: The clinical relevance of the
miR-197/CKS1B/STAT3-mediated PD-L1 network in chemoresistant
non-small-cell lung cancer. Mol The. 23:717–727. 2015.
|
|
14
|
Bian B, Pan X, Yang JS, Wang ZX and De W:
Upregulation of microRNA-451 increases cisplatin sensitivity of
non-small cell lung cancer cell line (A549). J Exp Clin Cancer Res.
30:202011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gao Y, Fan X, Li W, Ping W, Deng Y and Fu
X: miR-138-5p reverses gefitinib resistance in non-small cell lung
cancer cells via negatively regulating G protein-coupled receptor
124. Biochem Biophys Res Commun. 446:179–186. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jin F, Wang Y, Li M, Zhu Y, Liang H, Wang
C, Wang F, Zhang CY, Zen K and Li L: MiR-26 enhances
chemosensitivity and promotes apoptosis of hepatocellular carcinoma
cells through inhibiting autophagy. Cell Death Dis. 8:e25402017.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu Z, Shi A, Song D, Han B, Zhang Z, Ma
L, Liu D and Fan Z: Resistin confers resistance to
doxorubicin-induced apoptosis in human breast cancer cells through
autophagy induction. Am J Cancer Res. 7:574–583. 2017.PubMed/NCBI
|
|
18
|
Wang X, Lan Z, He J, Lai Q, Yao X, Li Q,
Liu Y, Lai H, Gu C and Yan Q: LncRNA SNHG6 promotes chemoresistance
through ULK1-induced autophagy by sponging miR-26a-5p in colorectal
cancer cells. Cancer Cell Int. 19:2342019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhao Z, Li J, Jiang Y, Xu W, Li X and Jing
W: CLDN1 increases drug resistance of non-small cell lung cancer by
activating autophagy via up-regulation of ULK1 phosphorylation. Med
Sci Monit. 23:2906–2916. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
21
|
Du Z, Sun T, Hacisuleyman E, Fei T, Wang
X, Brown M, Rinn JL, Lee MG, Chen Y, Kantoff PW and Liu XS:
Integrative analyses reveal a long noncoding RNA-mediated sponge
regulatory network in prostate cancer. Nat Commun. 7:109822016.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kartha RV and Subramanian S: Competing
endogenous RNAs (ceRNAs): New entrants to the intricacies of gene
regulation. Front Genet. 5:82014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Deng H, Zhang J, Shi J, Guo Z, He C, Ding
L, Tang JH and Hou Y: Role of long non-coding RNA in tumor drug
resistance. Tumor Biol. 37:11623–11631. 2016. View Article : Google Scholar
|
|
24
|
Majidinia M and Yousefi B: Long non-coding
RNAs in cancer drug resistance development. DNA Repair (Amst).
45:25–33. 2016. View Article : Google Scholar
|
|
25
|
Zheng ZG, Xu H, Suo SS, Xu XL, Ni MW, Gu
LH, Chen W, Wang LY, Zhao Y and Tian B: The essential role of H19
contributing to cisplatin resistance by regulating glutathione
metabolism in high-grade serous ovarian cancer. Sci Rep.
6:260932016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Han Z and Shi L: Long non-coding RNA
LUCAT1 modulates methotrexate resistance in osteosarcoma via
miR-200c/ABCB1 axis. Biochem Biophys Res Commun. 495:947–953. 2018.
View Article : Google Scholar
|
|
27
|
Wang W, Dong M, Zhang W and Liu T: Long
noncoding LUCAT1 promotes cisplatin resistance of non-small cell
lung cancer by promoting IGF-2. Eur Rev Med Pharmacol Sci.
23:5229–5234. 2019.PubMed/NCBI
|
|
28
|
Galluzzi L, Pietrocola F, Bravo-San Pedro
JM, Amaravadi RK, Baehrecke EH, Cecconi F, Codogno P, Debnath J,
Gewirtz DA, Karantza V, et al: Autophagy in malignant
transformation and cancer progression. EMBO J. 34:856–880. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yang Y, Jiang C, Yang Y, Guo L, Huang J,
Liu X, Wu C and Zou J: Silencing of LncRNA-HOTAIR decreases drug
resistance of Non-small cell lung cancer cells by inactivating
autophagy via suppressing the phosphorylation of ULK1. Biochem
Biophys Res Commun. 497:1003–1010. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xiong H, Ni Z, He J, Jiang S, Li X, Gong
W, Zheng L, Chen S, Li B and Zhang N: LncRNA HULC triggers
autophagy via stabilizing Sirt1 and attenuates the chemosensitivity
of HCC cells. Oncogene. 36:35282017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jin L, Li Y, Zhang Z, He T, Hu J, Liu J,
Chen M, Gui Y, Yang S and Mao X: miR-514a-3p functions as a tumor
suppressor in renal cell carcinoma. Oncol Lett. 14:5624–5630.
2017.PubMed/NCBI
|
|
32
|
Kim J, Kundu M, Viollet B and Guan KL:
AMPK and mTOR regulate autophagy through direct phosphorylation of
Ulk1. Nat Cell Biol. 13:1322011. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tang F, Hu P, Yang Z, Xue C, Gong J, Sun
S, Shi L, Zhang S, Li Z and Yang C: SBI0206965, a novel inhibitor
of Ulk1, suppresses non-small cell lung cancer cell growth by
modulating both autophagy and apoptosis pathways. Oncol Rep.
37:3449–3458. 2017. View Article : Google Scholar : PubMed/NCBI
|


















