Introduction
Langerhans' cell histiocytosis (LCH) is a group of
disorders in various tissues characterized by the proliferation of
Langerhans cells, and its etiopathogenesis is unknown. Although LCH
has been known for approximately a century, its etiology has yet to
be elucidated. It is rarely observed in adults (1). Langerhans cells are dendritic cells that
express cluster of differentiation 1a (CD1a) and S100 protein, and
contain Birbeck granules. One possible etiological cause is a
reactive proliferation of Langerhans cells following chemotherapy
or radiotherapy for Hodgkin's disease (HD). Eosinophilic granuloma
is the benign accumulation of histiocytes located primarily in the
bones, but which also affects other organs, including the skin,
lungs and lymph nodes. A number of cases of LCH associated with
malignant lymphoma have been reported previously: It may follow
after the malignant lymphoma (2–8) or occur
with it (9). However, fewer cases
have been reported in which LCH followed after Hodgkin's lymphoma
(HL). In the present report, we present one case patient who was
diagnosed with HD following chemotherapy for LCH.
Case report
Diagnosis of LCH
In March 2013, a 31-year-old man presented with a
one year history of fever, pruritus and left flank bone pain. The
patient's medical history indicated no abnormalities. He was
referred to another clinic hospital for an assessment of the flank
bone diseases. Computed tomographic (CT) scans of the left flank
bone revealed a bone-destructive lesion. The patient underwent
surgery to obtain a diagnosis and to excise the destructive bone.
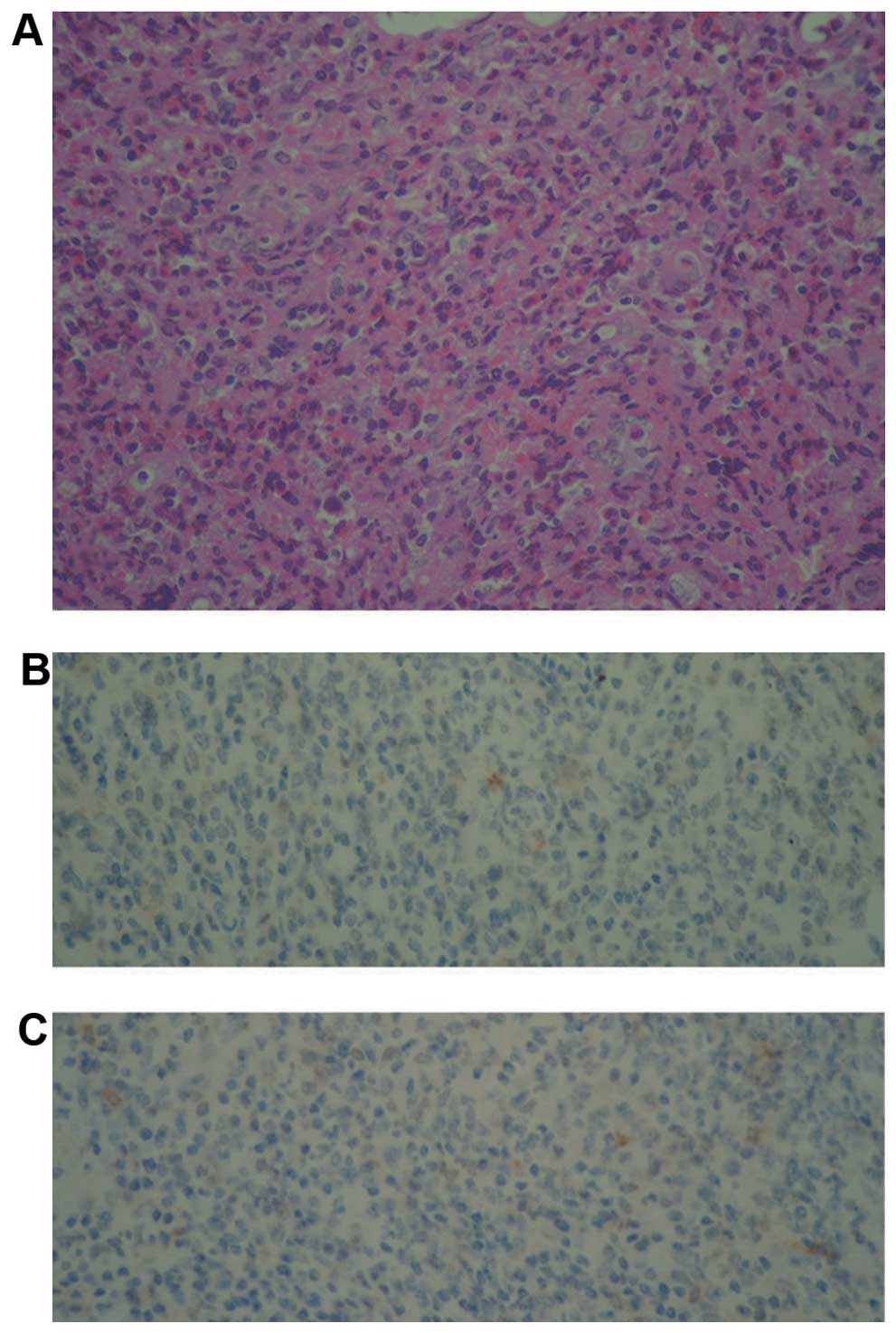
The tissue that was removed during surgery exhibited a histiocytic
cell population, intermixed with eosinophils, neutrophils,
multinuclear giant cells and foamy macrophages, which were observed
in the background. In addition, certain fibrotic bands existed in
this background. The histiocytic cells were positive for CD1a and
CD30 marker proteins, as revealed by immunohistochemistry staining.
However, S100 as a marker was not available for immunohistochemical
analysis (Fig. 1). On the basis of
the morphology and immunohistochemical staining profiles, the
patient was diagnosed with LCH. The patient's treatment commenced
with analgesia, and clinical remission was achieved.
In June 2013, the patient presented to The First
Central Hospital of Tianjin (Tianjin, China) with two sides of
flank bone pain and pruritus, which had been apparent for one week.
Upon admission, a physical examination revealed a rash on his
epipodites. Neither peripheral lymphadenopathy nor
hepatosplenomegaly were detected. The patient had no other B
symptoms. The laboratory tests revealed normal levels of the blood
cell count, C reactive protein, lactate dehydrogenase, hepatic and
renal functions. The results of bone marrow aspirate examination,
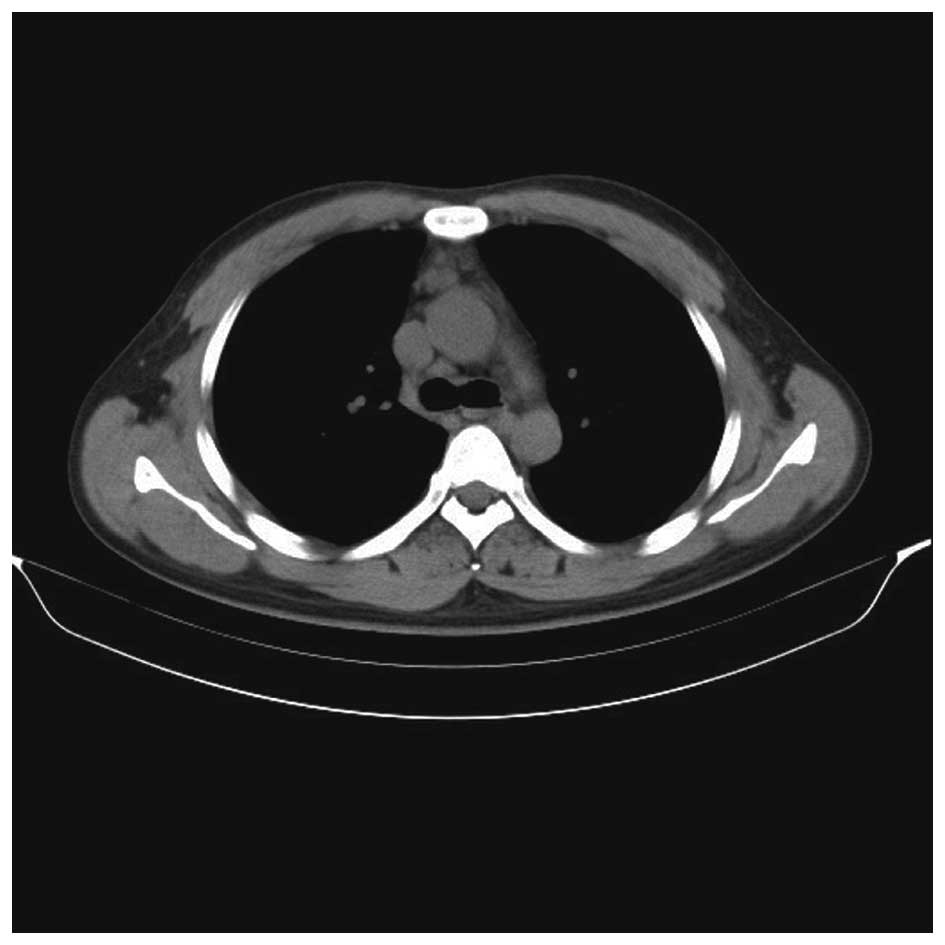
biopsy and flow cytometric analysis were normal. CT scans of the
thorax and abdomen revealed lymph nodes and a large mass in the
anterior and middle mediastinum (Fig.
2). A magnetic resonance imaging scan demonstrated the
destroyed sclerotin in the left flank bone and two sides of the
acetabular bones, and soft tissue in the left articulatio
sacroiliaca. The anterior mediastinum biopsy by CT-guided needle
puncture revealed the presence of lymphocytes, epithelial cell
nests (cytokeratin-positive), fibrous tissue and adipose tissue,
indicating thoracic gland tissue.
As previously, the patient was diagnosed with LCH
and treated with a combination of chemotherapeutic agents in two
cycles of cyclophosphamide, vincristine and prednisolone (COP).
However, the bone pain and fever continued. A CT scan of the thorax
revealed that the tumor mass in the mediastinum was unchanged.
Subsequently, the patient was treated with a combination of various
chemotherapeutic agents in two cycles of cyclophosphamide,
doxorubicin, vincristine, and prednisolone (CHOP). The patient went
into remission as far as the bone pain and fever were concerned,
and the CT scan of the thorax revealed a marked reduction in the
size of the tumor mass in the mediastinum. Following six cycles of
treatment with CHOP as a maintenance therapy, the patient achieved
a successful clinical remission.
Diagnosis of HD
One year after the patient entered clinical
remission, the patient once again developed bone pain and fever. A
physical examination revealed peripheral lymph node enlargement,
although no hepatosplenomegaly. The laboratory tests were normal,
as identified previously. CT scans of the thorax and abdomen
revealed augmentation of the tumor mass in the mediastinum. An
excisional biopsy of the right inguinal lymph node was performed.
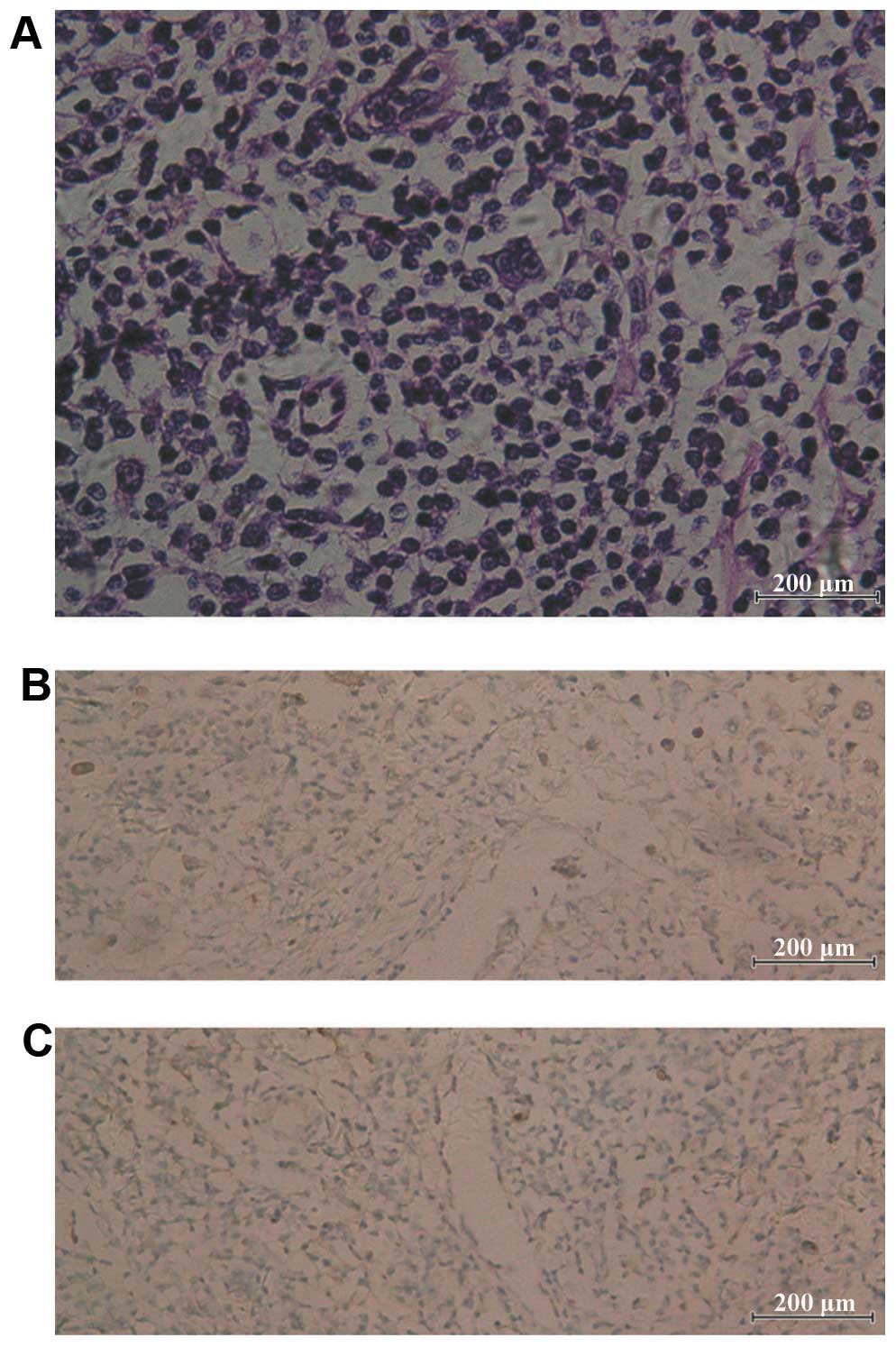
Microscopic examination revealed that the atypical cells were
either mononucleated or binucleated, composed of lymphocytes,
plasma cells and a few eosinophils. Immunohistochemical staining
revealed CD30- and CD15-positive cells. Immunostaining for CD1a
protein was positive, as determined previously (Fig. 3). The patient was diagnosed with
nodular sclerosing HD (stage IIIA, with spleen involvement),
secondary to LCH. Subsequently, the patient was treated with a
combination of various chemotherapeutic agents of adriamycin,
bleomycin, vinblastine and dacarbazine (ABVD). The patient received
four cycles of ABVD chemotherapy. Following the chemotherapy, the
patient obtained a remission of the bone pain, fever and pruritus,
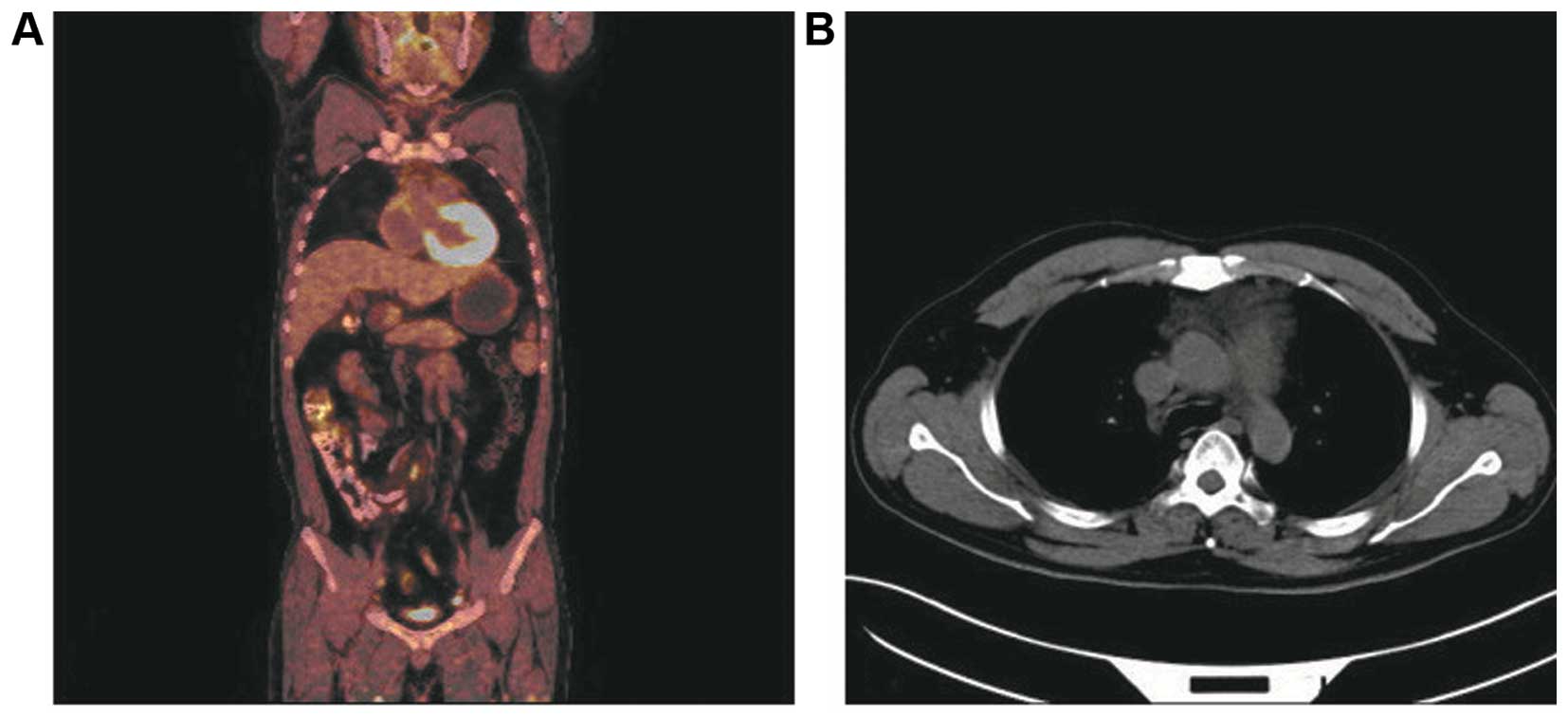
and a whole-body positron emission tomographic (PET) CT scan
revealed a decrease in the tumor mass in the mediastinum (Fig. 4). To date, the patient remains in
treatment, and the prognosis has yet to be fully determined.
Discussion
LCH is a group of disorders, with the proliferation
of Langerhans cells accumulating in various tissues. LCH is a rare
disease that may occur at any age (10). Due to having identical
clinicopathological features, it is a group of disorders
characterized by a clonal neoplastic proliferation of
Langerhans-type cells that express CD1a, langerin and S100 protein,
and show evidence of Birbeck granules on ultrastructural
examination. LCH is also known as a histiocytosis X, a term
proposed by Lichtenstein in 1953 (11). It has been classified into three
clinical variants: Eosinophilic granuloma, Hand-Schüller-Christian
disease and Letterer-Siwe disease (12). Most organs or systems of the body may
be affected, however, the skeleton, skin and pituitary are involved
more frequently. Other organs involved are the liver, spleen, lymph
nodes, lungs, the hematopoietic system and mucocutaneous tissues,
and the central nervous system.
The etiology of LCH remains unknown, although viral
causes, reactive immunological responses and genetic factors have
been suggested (4). However, the
progressive characteristics of the disease and its response to
cancer therapies depict its neoplastic nature (13). An association between LCH and a
variety of other tumor types has been recognized, and LCH has been
described in association with a variety of other tumor types. The
malignancies may precede, occur concurrently with, or follow the
diagnosis of LCH. The most common associations are with malignant
lymphoma and acute lymphoblastic leukemia (ALL) (14). Lymphoma and ALL more often occur prior
to the diagnosis of LCH, although they may be diagnosed within 5
years following LCH (15). Solid
tumors may occur concurrently, or follow the diagnosis of LCH. Most
of those that followed LCH developed in a previous radiation field.
The association between tumor formation and LCH has yet to be fully
elucidated. Shin et al (16)
discussed the association between the conditions of LCH and HD, and
proposed various possibilities: i) HD induces LCH; ii) radiotherapy
and/or chemotherapy for HD leads to the development of LCH; iii)
LCH may represent a specific cell-mediated immune response to HD;
and iv) a common etiological agent induces HD and LCH.
Egeler et al (15) reported their experience of 39 patients
with LCH and malignant neoplasia. That study revealed that there
was an association between the two conditions. A total of 25
patients were diagnosed with LCH and HL. The conclusion was that
LCH is associated with lymphoma. In certain cases, histological
examination revealed lymphoid cells and Langerhans cells in the
same lymph node. Furthermore, the year-long interval between the
development of LCH and HL suggested an association between the two
diseases that is independent of chemotherapy or radiation
treatments. Such an association may involve a malignant change, or
modulation of the monocyte-histiocyte system. For example,
Langerhans cells may themselves be premalignant, or may provide an
immunological environment conducive to the development, growth and
promotion of malignant cells.
In the present case study, the patient was diagnosed
with LCH on the first occasion, although it was regrettable that
multiple lymph nodes were observed in the mediastinum at that time,
located in the anterior mediastinum and the middle mediastinum.
Effective pathological tissues could not be obtained by fine needle
aspiration, and the damage was too extensive for chest surgery, so
no pathology was performed on the lymph node. The patient was
treated with COP and CHOP chemotherapy. The B symptoms were
controlled, and the diameter of the mediastina lymph nodes was
reduced. A retrospective analysis determined that the COP and CHOP
chemotherapies were not an effective treatment for HL; however, the
B symptoms of the patient were controlled and the diameter of the
lymph nodes was reduced. Perhaps at that time, HD may not have
appeared. One year later, the right inguinal lymph node was
observed to be enlarged in the patient. The diagnosis of HD was
confirmed by a pathological biopsy of the lymph node. Following six
cycles of ABVD, a PET CT scan revealed a decrease in tumor mass in
the mediastinum. However, it was difficult to identify LCH and HD
from a PET CT scan (17,18), therefore it is possible that HD may
appear following LCH.
Acknowledgments
The present study was supported by grants from the
Tianjin Municipal Health Bureau of Science and Technology Fund
(2014KZ021).
References
|
1
|
Pan Z, Sharma S and Sharma P: Primary
langerhans cell histiocytosis of the vulva: Report of a case and
brief review of the literature. Indian J Pathol Microbiol.
52:65–68. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shanley DJ, Lerud KS and Luetkehans TJ:
Development of pulmonary histiocytosis X after chemotherapy for
Hodgkin disease. AJR Am J Roentgenol. 155:741–742. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
L'Hoste RJ Jr, Arrowsmith WR, Leonard GL
and McGaw H: Eosinophilic granuloma occurring in a patient with
Hodgkin disease. Hum Pathol. 13:592–595. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Adu-Poku K, Thomas DW, Khan MK, Holgate CS
and Smith ME: Langerhans cell histiocytosis in sequential
discordant lymphoma. J Clin Pathol. 58:104–106. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dehkordi NR, Rajabi P, Naimi A and
Heidarpour M: Langerhans cell histiocytosis following Hodgkin
lymphoma: A case report from Iran. J Res Med Sci. 15:58–61.
2010.PubMed/NCBI
|
|
6
|
Lee JS, Ko GH, Kim HC, Jang IS, Jeon KN
and Lee JH: Langerhans cell sarcoma arising from Langerhans cell
histiocytosis: A case report. J Korean Med Sci. 21:577–580. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Egeler RM, Neglia JP, Puccetti DM, Brennan
CA and Nesbit ME: Association of Langerhans cell histiocytosis with
malignant neoplasms. Cancer. 71:865–873. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
de Ibarrola Andrés C, Toscano R, Lahuerta
JJ and Martínez-González MA: Simultaneous occurrence of Hodgkin's
disease, nodal Langerhans' cell histiocytosis and multiple myeloma
IgA(kappa). Virchows Arch. 434:259–262. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Neumann MP and Frizzera G: The coexistence
of Langerhans' cell granulomatosis and malignant lymphoma may take
different forms: Report of seven cases with a review of the
literature. Hum Pathol. 17:1060–1065. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ardekian L, Peled M, Rosen D, Rachmiel A,
Abu el-Naaj I and Laufer D: Clinical and radiographic features of
eosinophilic granuloma in the jaws: Review of 41 lesions treated by
surgery and low-dose radiotherapy. Oral Surg Oral Med Oral Pathol
Oral Radiol Endod. 87:238–242. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lichtenstein L: Histiocytosis X;
integration of eosinophilic granuloma of bone, Letterer-Siwe
disease, and Schüller-Christian disease as related manifestations
of a single nosologic entity. AMA Arch Pathol. 56:84–102.
1953.PubMed/NCBI
|
|
12
|
Piattelli A and Paolantonio M:
Eosinophilic granuloma of the mandible involving the periodontal
tissues. A case report. J Periodontol. 66:731–736. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Haupt R, Minkov M, Astigarraga I, Schäfer
E, Nanduri V, Jubran R, Egeler RM, Janka G, Micic D,
Rodriguez-Galindo C, et al: Langerhans cell histiocytosis (LCH):
Guidelines for diagnosis, clinical work-up, and treatment for
patients till the age of 18 years. Pediatr Blood Cancer.
60:175–184. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Willman CL: Detection of clonal
histiocytes in Langerhans cell histiocytosis: Biology and clinical
significance. Br J Cancer Suppl. 23:S29–S33. 1994.PubMed/NCBI
|
|
15
|
Egeler RM, Neglia JP, Aricò M, Favara BE,
Heitger A, Nesbit ME and Nicholson HS: The relation of Langerhans
cell histiocytosis to acute leukemia, lymphomas, and other solid
tumors. The LCH-malignancy study group of the histiocyte society.
Hematol Oncol Clin North Am. 12:369–378. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shin MS, Buchalter SE and Ho KJ:
Langerhans' cell histiocytosis associated with Hodgkin's disease: A
case report. J Natl Med Assoc. 86:65–69. 1994.PubMed/NCBI
|
|
17
|
Naumann R, Beuthien-Baumann B, Fischer R,
Kittner T, Bredow J, Kropp J, Ockert D and Ehninger G: Simultaneous
occurrence of Hodgkin's lymphoma and eosinophilic granuloma: A
potential pitfall in positron emission tomography imaging. Clin
Lymphoma. 3:121–124. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fotos JS, Flemming DJ and Tulchinsky M:
False-positive PET/CT for lymphoma recurrence secondary to
Langerhans cell. Clin Nucl Med. 36:717–719. 2011. View Article : Google Scholar : PubMed/NCBI
|