Introduction
Deletion of the long arm of chromosome 5 is the most
common cytogenetic abnormality in patients with myelodysplastic
syndrome (MDS), occurring in ~15% of cases (1). However, loss of the chromosome 5q31.3 to
5q33.3 region, as a sole chromosomal abnormality, is observed in 5%
of MDS patients and characterizes del(5q) MDS, which is a clonal
stem cell disorder (2). This MDS
subtype has a favorable prognosis with transformation to acute
myeloid leukemia occurring in <10% of the cases and it responds
well to lenalidomide treatment (3).
The JAK2 V617F mutation is present in the
majority of patients with myeloproliferative neoplasms (MPN):
>90% of patients with polycythemia vera, and 50% of patients
with primary myelofibrosis and essential thrombocythemia (ET)
(4). The presence of this mutation
has also been described in other hematological malignancies,
however, to a much lesser extent; ~3–5% of patients with MDS and
<5% of patients with acute myeloid leukemia (AML) (5). The coexistence of del(5q) as sole
cytogenetic abnormality and the JAK2 V617F mutation is even
rarer occurring in a small subset of del(5q) MDS cases, and the
optimal therapeutic approach in these patients remains to be
elucidated (6).
The present study reports the case of a patient with
del(5q) MDS and a concomitant JAK2 V617F mutation, who
achieved complete cytogenetic and molecular remission with
lenalidomide and enjoyed durable hematological response that lasts
for >5 years post treatment discontinuation.
Case report
A 77-year-old female patient was referred to the
Department of Haematology, University Hospital of Ioannina
(Ioannina, Greece) in November 2008 for severe macrocytic anemia
(hemoglobin, 6.5 g/dl), leucocytosis [white blood cell count (WBC),
21.4×109/l] with predominance of the neutrophils and
severe eosinophilia, and thrombocytosis (platelets,
774×109/l). In the peripheral blood smear basophilic
stippling and megaloblastoid changes of erythrocytes were observed.
The biochemical tests were unremarkable, with the exception of
increased levels of lactic dehydrogenase. On clinical examination,
mild hepatosplenomegaly was observed, which was confirmed by
ultrasound scan.
The patient underwent diagnostic assessments to
exclude infections, connective tissue disorders and cancer. These
assessments were negative. Computer tomography of the abdomen
revealed mild hepatosplenomegaly. At this point bone marrow
aspiration and biopsy were performed. Karyotyping and molecular
analysis were also performed for the presence of bcr-abl
chimeric transcript and JAK2 V617F mutation, and also
FIP1L1/PDGFRA and TEL/PDGFRB rearrangements, considering MPN as the
most likely diagnosis.
The bone marrow aspirate revealed increased
cellularity, myeloid lineage hyperplasia with a left shift and
increased numbers of hypolobated megakaryocytes, compatible with a
MPN. Molecular analyses were positive for the presence of the
JAK2 V617F mutation and negative for the presence of
bcr-abl transcript. Pending the results from the trephine
biopsy and karyotyping, and on the basis of MPN being the most
possible diagnosis, either ET or the cellular phase of primary
myelofibrosis, a clinical decision was made to initiate treatment.
The patient was started on interferon-α, low-dose aspirin and
recombinant erythropoietin as a result of a constant requirement
for blood transfusions and her age, which defined her as a
high-risk patient. A month later, WBC count and platelets were
normalized, however, the patient remained transfusion-dependent,
requiring at least two packed red blood cell units per month.
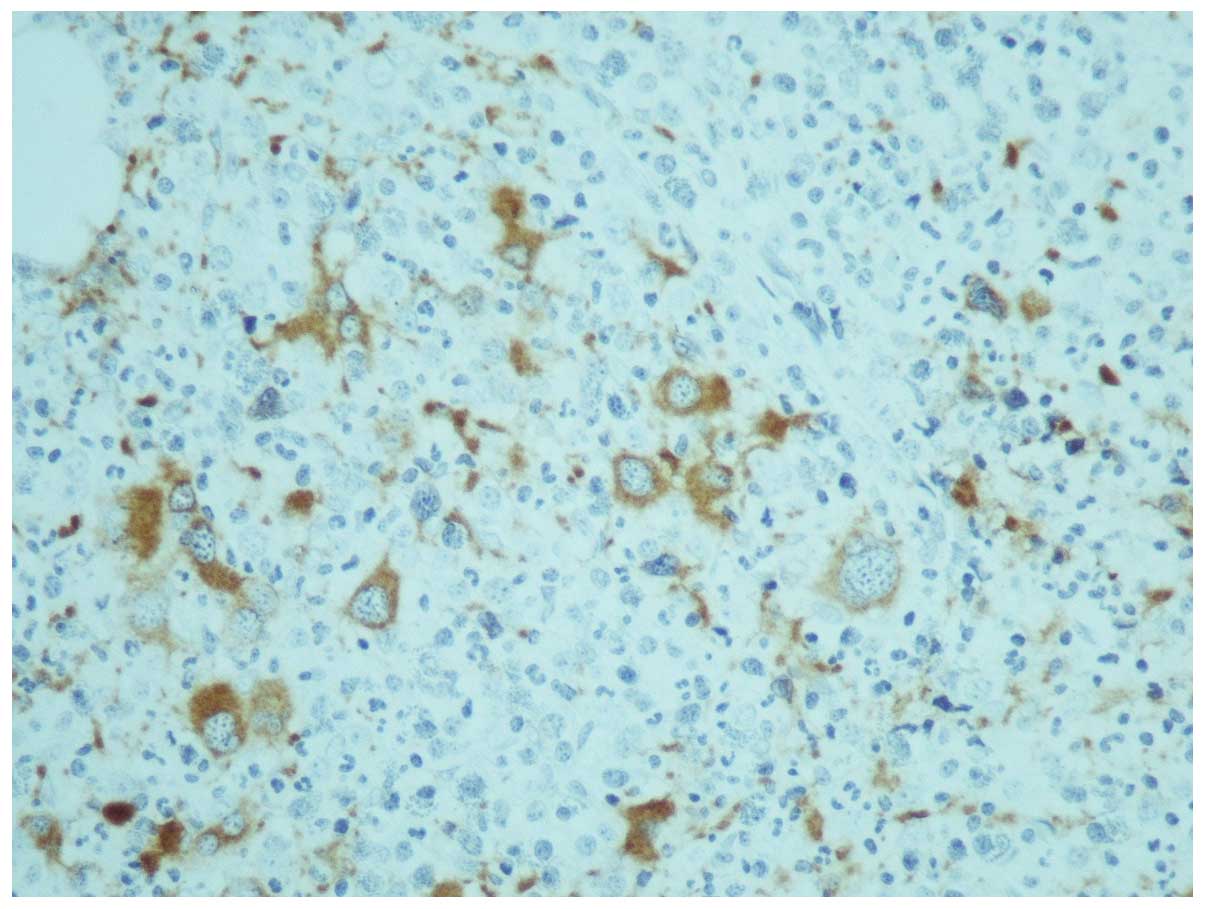
One month later, the results of karyotyping and the
trephine biopsy were acquired. The latter was reported as having
increased cellularity (95%) with concomitant hypoplasia of the
erythroid lineage; the myeloid lineage was hyperplastic with a left
shift and the megakaryocytes were increased in number, decreased in
size and their nuclei were hypolobulated, while micromegakaryocytes
were also observed (Fig. 1). An
abnormal karyotype was observed in all twenty metaphases that
showed a deletion of the long arm of chromosome 5, involving the
chromosome region 5q13-5q33 [46, XX, del (5) (q13 q33)], as the sole cytogenetic
abnormality.
Therefore, the diagnosis, according to the WHO
classification of hematologic neoplasms (7), was MDS with an isolated deletion of 5q
chromosome and a concomitant JAK2 V617F mutation,
categorizing her to the low risk group, according to the
International Prognostic Scoring System (anemia, blasts <5%,
favorable karyotype) (8). On the
basis of definite diagnosis, previous therapy was discontinued and
the patient was initiated on 10 mg lenalidomide daily for 21 days
in a 28-day-cycle since the patient was transfusion-dependent.
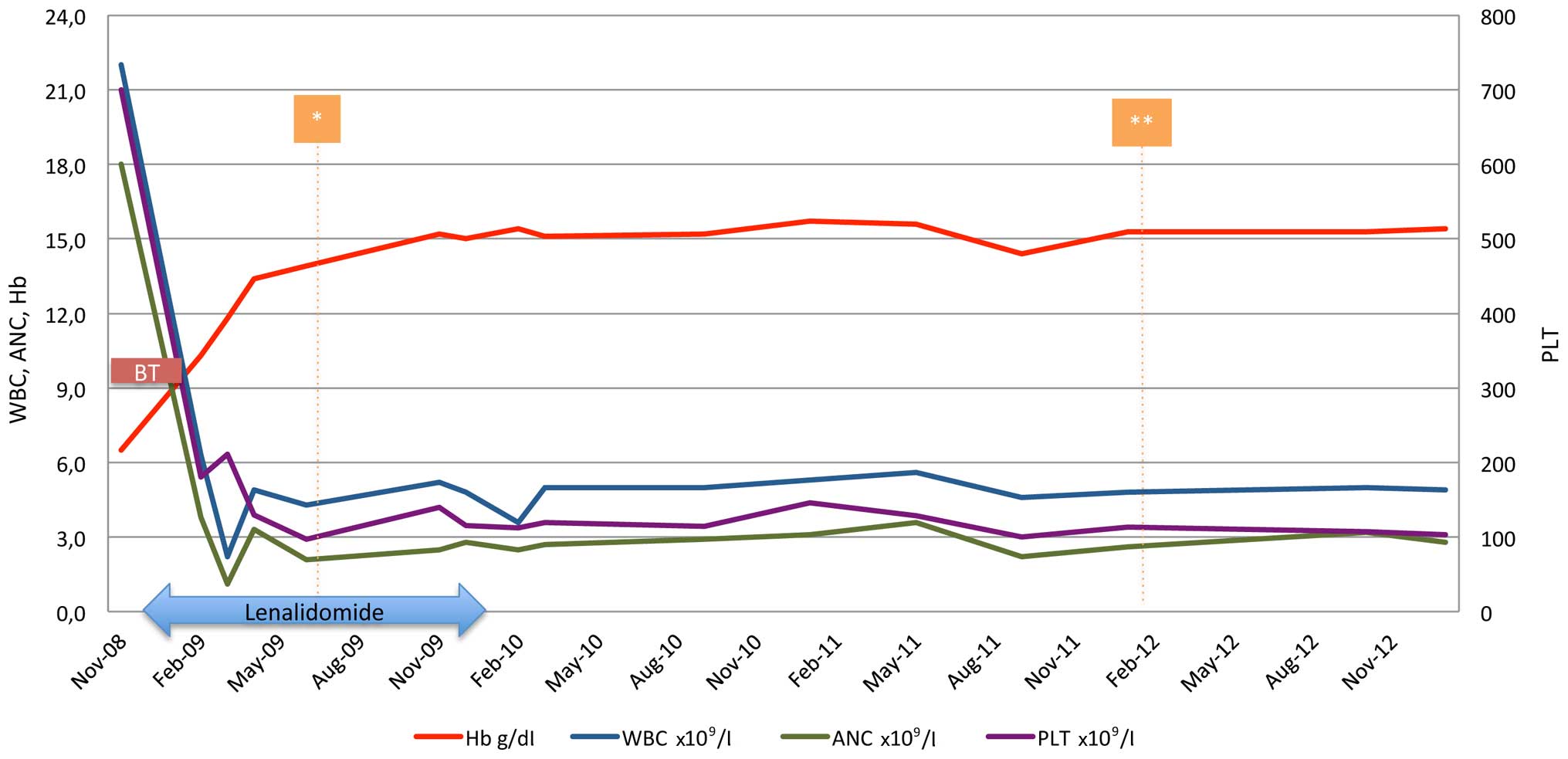
Within the first month of treatment the patient
became transfusion-independent. The only toxicity observed was
grade 3 neutropenia, and was managed by treatment withhold until
resolution and one dose level reduction on restart (5 mg). On
lenalidomide (5 mg) in cycles of 21/28 days, the patient achieved
both cytogenetic and molecular complete response (normal karyotype
and undetected JAK2 V617F mutation) within 6 months of
treatment (Fig. 2) and remained
transfusion-independent, maintaining hemoglobin levels of ~15 g/dl
for the next 6 months. However, 12 months post-treatment initiation
and while on hematological, cytogenetic and molecular response, the
patient was unwilling to continue with treatment and lenalidomide
was discontinued. The patient remained in complete response for a
further 23 months when a new bone marrow aspirate and cytogenetic
analysis was performed and the karyotype of the diagnosis [46, XX,
del (5) (q13 q33)] re-occurred, but
in one of the twenty-four metaphases examined. However, the patient
preserved the hematological and molecular response, and the
transfusion-independency, therefore, treatment was not
re-initiated. Currently, 70 months following diagnosis and 55
months without treatment, the patient is in excellent condition and
transfusion-independent, maintaining a hematocrit level of 42%.
Discussion
Approximately 5% of MDS patients present with
isolated del(5q), characterized by severe anemia, preserved or even
elevated platelet count and high response rates to lenalidomide
(3). The concomitant presence of the
5q deletion and a JAK2 V617F mutation in MDS was first shown
in 6/97 patients with MDS with the 5q deletion (6). Patients harboring the JAK2 V617F
mutation compared to those without the mutation, presented with
hypercellular bone marrow and exhibited significantly higher levels
of WBC count and a trend towards higher levels of platelet count
(6). Since the clinical outcome of
this subset of patients remains unclear, the 2008 WHO
classification classifies them as MDS with isolated del(5q), rather
than in the MDS/MPN category. Therefore, more data collection from
patients with such characteristics are required in order to draw
clear conclusions.
The present patient was diagnosed with isolated
del(5q) MDS and a concurrent JAK2 V617F mutation.
Lenalidomide was administered and the patient achieved
transfusion-independence within 4 weeks of treatment initiation,
and complete cytogenetic and molecular response within 6 months.
The patient remains transfusion-independent for the last 70 months,
despite lenalidomide discontinuation after 12 months of therapy.
The del(5q) clone reappeared 23 months later in 1/24 metaphases,
however, without clinical effects.
Beneficial treatment with lenalidomide in isolated
del(5q) and a concomitant JAK2 V617F mutation has been
previously reported in three MDS patients (9–11) and one
patient with AML arising from JAK2 V617F and del(5q) MDS. It
remains unclear whether the lenalidomide effect has a suppressive
role against del(5q) cells and another independent activity against
the JAK2 V617F mutated clone (10), since it has not been delineated
whether the two genetic abnormalities involve the same or two
distinct hematopoietic clones. Sokol et al (12) investigated the clonal origin of the
JAK2 V617F mutation in a patient with del(5q) MDS presenting
with thrombocytosis and normal hemoglobin, and suggested that the
latter is the case.
Currently, the recommendation regarding lenalidomide
treatment in MDS del(5q) patients is to treat until disease
progression (13). Although it was
recently shown that lenalidomide treatment in MDS del(5q) patients
does not appear to increase the risk to AML transformation
(14), other long-term effects of
immunomodulation have not been elucidated and one could argue that
intermittent use of lenalidomide may be prudent is certain cases.
The present case indicated that durable hematological response may
be sustained despite discontinuation of lenalidomide, whereas
others have shown that re-challenge may also be safe and feasible,
albeit with no profound cytogenetic response (15). However, in order to draw safe
conclusions regarding the treatment duration and time of
discontinuation, a randomized clinical trial is required to compare
continuous treatment until progression, vs. treatment
discontinuation after having achieved sustained complete
cytogenetic response.
In conclusion, the present case highlights the
coexistence of a JAK2 V617F mutation in del(5q) MDS and
suggests that lenalidomide treatment is beneficial and effective
for this subset of patients, leading to complete cytogenetic and
molecular response, while durable hematological response may be
sustained even following the discontinuation of the treatment.
References
|
1
|
Bejar R, Levine R and Ebert BL: Unraveling
the molecular pathophysiology of myelodysplastic syndromes. J Clin
Oncol. 29:504–515. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cazzola M: Myelodysplastic syndrome with
isolated 5q deletion (5q-syndrome). A clonal stem cell disorder
characterized by defective ribosome biogenesis. Haematologica.
93:967–972. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
List A, Dewald G, Bennett J, Giagounidis
A, Raza A, Feldman E, Powell B, Greenberg P, Thomas D, Stone R, et
al: Lenalidomide in the myelodysplastic syndrome with chromosome 5q
deletion. N Engl J Med. 355:1456–1465. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Baxter EJ, Scott LM, Campbell PJ, East C,
Fourouclas N, Swanton S, Vassiliou GS, Bench AJ, Boyd EM, Curtin N,
et al: Acquired mutation of the tyrosine kinase JAK2 in human
myeloproliferative disorders. Lancet. 365:1054–1061. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Scott LM, Campbell PJ, Baxter EJ, Todd T,
Stephens P, Edkins S, Wooster R, Stratton MR, Futreal PA and Green
AR: The V617F JAK2 mutation is uncommon in cancers and in myeloid
malignancies other than the classic myeloproliferative disorders.
Blood. 106:2920–2921. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ingram W, Lea NC, Cervera J, Germing U,
Fenaux P, Cassinat B, Kiladjian JJ, Varkonyi J, Antunovic P,
Westwood NB, et al: The JAK2 V617F mutation identifies a subgroup
of MDS patients with isolated deletion 5q and a proliferative bone
marrow. Leukemia. 20:1319–1321. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Swerdlow SH, Campo E, Harris NL, Jaffe ES,
Pileri SA, Stein H, Thiele J and Vardiman JW: WHO Classification of
Tumours of Haematopoietic and Lymphoid Tissues. IARC Press. Lyon,
France: 2008.
|
|
8
|
Greenberg P, Cox C, LeBeau MM, Fenaux P,
Morel P, Sanz G, Sanz M, Vallespi T, Hamblin T, Oscier D, et al:
International scoring system for evaluating prognosis in
myelodysplastic syndromes. Blood. 89:2079–2088. 1997.PubMed/NCBI
|
|
9
|
Azaceta G, Calasanz MJ, Dourdil V,
Bonafonte E, Izquierdo I and Palomera L: Response to lenalidomide
in a patient with myelodysplastic syndrome with isolated del(5q)
and JAK2 V617F mutation. Leuk Lymphoma. 51:1941–1943. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nomdedeu M, Maffioli M, Calvo X,
Martínez-Trillos A, Baumann T, Díaz-Beyá M, Aguilar JL, Rozman M,
Costa D, Esteve J, et al: Efficacy of lenalidomide in a patient
with myelodysplastic syndrome with isolated del(5q) and JAK2V617F
mutation. Leuk Res. 35:1276–1278. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Musto P, Simeon V, Guariglia R, Bianchino
G, Grieco V, Nozza F, La Rocca F, Marziano G, Lalinga AV, Fabiani
E, et al: Myelodysplastic disorders carrying both isolated del(5q)
and JAK2 (V617F) mutation: Concise review, with focus on
lenalidomide therapy. Onco Targets Ther. 7:1043–1050.
2014.PubMed/NCBI
|
|
12
|
Sokol L, Caceres G, Rocha K, Stockero KJ,
Dewald DW and List AF: JAK2 (V617F) mutation in myelodysplastic
syndrome (MDS) with del(5q) arises in genetically discordant
clones. Leuk Res. 34:821–823. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Giagounidis A, Fenaux P, Mufti GJ, Muus P,
Platzbecker U, Sanz G, Cripe L, Von Lilienfeld-Toal M and Wells RA:
Practical recommendations on the use of lenalidomide in the
management of myelodysplastic syndromes. Ann Hematol. 87:345–352.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Adès L, Le Bras F, Sebert M, Kelaidi C,
Lamy T, Dreyfus F, Eclache V, Delaunay J, Bouscary D, Visanica S,
et al: Treatment with lenalidomide does not appear to increase the
risk of progression in lower risk myelodysplastic syndromes with 5q
deletion. A comparative analysis by the groupe francophone des
myelodysplasies. Haematologica. 97:213–218. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pisani F, Orlandi G and Merola R:
Long-term response in a patient with del(5q) myelodysplastic
syndrome who discontinued lenalidomide and obtained a good response
and tolerance to rechallenge. Case Rep Oncol. 7:277–284. 2014.
View Article : Google Scholar : PubMed/NCBI
|