1. Introduction
Abnormal DNA methylation is a hallmark of a
multitude of pathological processes, including neurodevelopmental
imprinting disorders, atherosclerosis, autoimmune diseases, and
cancer. Notably, abnormal DNA hypermethylation of tumor suppressor
genes plays a critical role in the malignant transformation of
myelodysplastic syndrome (MDS) (1)
into acute myeloid leukemia (AML), with the possibility of this
process being slowed or reversed using hypomethylating agents
(HMAs). There is accumulating data that HMAs may have a role to
play as well in the treatment of solid tumors, where they have
primarily been explored in combination with other antineoplastic
drugs, and may act through a variety of possible mechanisms, not
just limited to the re-expression of tumor suppressor genes
(2,3).
Glioblastoma, IDH-wildtype, (GBM) is most frequently
diagnosed in adults in the seventh decade of life, and accounts for
approximately 15% of all intracranial neoplasms and 50% of all
primary malignant brain tumors (4). Despite advances in treatment over the
past twenty years, median overall survival remains well under two
years (5). The current paradigm
for first-line treatment consists of maximal safe surgical
resection when possible, followed by conformal external beam
radiotherapy with concurrent and adjuvant temozolomide (TMZ), an
alkylating chemotherapy agent that crosses the blood-brain barrier.
Approximately 40% of GBMs exhibit elevated methylation levels at
the methylguanine methyltransferase (MGMT) gene promoter, which
generally predicts a favorable response to TMZ, although it is not
the sole determining factor and some MGMT-unmethylated GBMs benefit
from TMZ as well (6). Nonetheless,
the lack of meaningful and lasting responses to TMZ in the majority
of GBM patients emphasizes the critical need to identify and
develop new treatment strategies (7).
This work explores the antineoplastic potential of
current HMAs as well as established data of their preclinical and
clinical effectiveness in GBM and other solid tumors. Although
genome-wide hypermethylation, as seen in IDH-mutant gliomas, is not
characteristic of GBM, a multitude of evidence points to the role
HMAs might have in reversing focal genomic methylation aberrations
that contribute to GBM treatment resistance. Additionally, we
review possible synergies these drugs may have with current and
emerging GBM therapies with a focus on temozolomide and
immunotherapy. Challenges for future clinical trials are also
assessed.
2. Mechanism of HMAs
Epigenetic alterations cause heritable changes in
gene expression without changes in the DNA nucleotide sequence
(8). Thus, an epigenetic mechanism
can be thought of as a system for selectively using genetic
information to turn ‘on’ and ‘off’ various functional genes in
order to carry out key processes during normal embryonal
development (9), including
chromosome X inactivation (10),
the maintenance of genomic stability (11), and transcriptional regulation
(12). Since the early 1980s, DNA
methylation has been recognized as one such epigenetic mechanism
that plays a significant role in controlling cellular
differentiation states (13,14).
DNA methylation is a tightly regulated gene
silencing or activation process mediated through DNA
methyltransferases (DNMTs). The DNMT family consists of 5 members,
including DNMT1, DNMT2, DNMT3A, DNMT3B, and DNMT3L, of which DNMT1
is the best characterized (Table
I). DNMT1, also referred to as maintenance DNMT, binds to newly
synthesized DNA and acts to maintain the methylation pattern of the
template DNA strand after replication. DNMT1 is also recruited to
sites of DNA damage, including base mismatches and double-strand
breaks, to prevent loss of DNA methylation and gene dysregulation
after DNA repair (15). DNMT2 is
primarily a tRNA methyltransferase, which acts to protect tRNA
against fragmentation. DNMT3A, DNMT3B, and DNMT3L, also referred to
as de novo DNMTs, can establish new methylation patterns
during normal development and in response to environmental cues.
These de novo DNMTs also form part of chromatin-remodeling
complexes and help complete the process of establishing and
maintaining cell-specific methylation arrangements (16,17).
 | Table ISummary of the function and role of
DNMTs in human diseases. |
Table I
Summary of the function and role of
DNMTs in human diseases.
| Gene | Function | Role in human
disease |
|---|
| DNMT1 | De novo
methylating activity; maintains DNA methylation patterns during
embryo development. | Missense mutations
linked to hereditary sensory and autonomic neuropathy type 1E;
knockout in mouse models causes embryonic lethality. |
| DNMT2 | Also known as
TRDMT1; mainly a tRNA methyltransferase; protects tRNA against
fragmentation; restricts the activation of cryptic promoters. | Possible links to
aberrant hematopoiesis; knockout mice are viable and fertile. |
| DNMT3A/B | De novo
methylating activity; methylates previously unmethylated regions of
the genome in a non-selective manner; role in transcription
activation at enhancers. | DNMT3A mutations
are common in AML; DNMT3A mutations linked to Tatton-Brown-Rahman
syndrome; DNMT3B mutations linked to immunodeficiency, centromeric
instability and facial anomalies (ICF syndrome). |
| DNMT3L | Stimulates
methyltransferase activity of DNMT3A and DNMT3B; cofactor for
retrotransposon methylation in male individuals; expressed during
brain development and in the thymus in adulthood. | Reduced activity
linked to AML. |
DNMTs covalently transfer a methyl group to the C-5
position of cytosine residues within CpG dinucleotides (18). DNA regions with a high frequency of
CpG sites are called CpG islands, which can range from 200 to 3,000
base pairs and are typically associated with gene promoters
(19). In the normal mammalian
cell, CpG islands are usually hypomethylated and have activating
histone modifications, which allows for unhindered DNA
accessibility and facilitated gene expression (20,21).
DNA methylation can take place in the promoter of a gene, generally
resulting in the repression of gene transcription, or in the gene
body, where the usual result is promotion of gene transcription
(14,22,23).
Gene promoter methylation has several known effects. It may prevent
RNA polymerase and transcription factors from binding to active
regulatory sites. Alternatively, methylation can lure methyl-CpG
binding domain proteins that recruit histone deacetylases, leading
to the removal of gene-activating acetylation marks and chromatin
condensation (24). Gene silencing
from methylation-induced heterochromatization also results from the
recruitment of polycomb repressor complexes (25) and nucleosome complexes (26-28).
On the other hand, methylation of CpG islands within
the gene body may promote normal gene transcription through several
interrelated mechanisms, including slowing the kinetics of RNA
polymerase II for proper splice site recognition and inhibiting
spurious transcription from ectopic promoters. Recent studies have
shown that aberrant gene body methylation may have varying effects
depending on cell type and differentiation state. The effects may
also be gene specific. For example, aberrant gene body
hypermethylation of the stem cell lineage marker brachyury has been
associated with precancerous intestinal metaplasia, while global
hypomethylation occurs when these same cells undergo neoplastic
transformation into gastric adenocarcinoma (29). Contrastingly, hypomethylation can
have antitumor effects when it occurs within the gene body of
oncogenes, where a physiologic level of CpG methylation may act to
promote the expression of oncogenic factors (30). Overall, the normal role of
methylation and the consequences of aberrant methylation in gene
bodies is not fully understood and is an active area of
investigation.
3. Types of HMAs
HMAs are pharmacological agents that can inhibit
methylation by trapping DNMTs, resulting in the expression of a
previously hypermethylated silenced gene (31,32),
and possibly also repression or modification of transcription at
sites within the gene body. Developed beginning in the 1960s
(33,34), HMAs currently in use include
decitabine, azacytidine, guadecitabine and ASTX727(35), all of which have demonstrated
effects on cell cycle control, DNA repair, cell signaling,
apoptosis and metastasis (36). In
general, these agents are cytosine analogs that exert their effects
once they have integrated into newly synthesized DNA or RNA.
Decitabine (5-aza-2-deoxycytidine) acts as a
cytosine analogue, replacing cytosine in the CpG dinucleotide pair,
which is the typical target of DNMTs. Unlike cytosine, decitabine
possesses a nitrogen molecule instead of carbon at the fifth carbon
position, preventing the transfer of a methyl group to this site.
Decitabine also forms a covalent bond with the methyltransferase
enzyme leading to its inactivation. Covalently trapped DNMTs are
targeted for degradation by the proteasome, leading to a
genome-wide decrease in CpG methylation levels. This may enable the
re-expression of aberrantly repressed genes by preventing the
re-methylation of CpG islands over the course of multiple cell
cycles. It is critical to note that incorporation of decitabine
into DNA requires the transition to S-phase of the cell cycle in
the target cell (37,38); it has a limited effect on CpG
methylation in non-proliferating cells, thus making it useful as an
antineoplastic agent (39).
DAC reaches a maximum plasma concentration of about
65-77 ng/ml when given at standard intravenous dosing of 15
mg/m2 every 8 h in patients with AML and MDS (40). Cellular uptake of the drug is
dependent on the nucleoside-specific transport mechanism. Rapid
equilibration between the intra- and extracellular compartments
results in a short alpha half-life of 5 min. In plasma, the drug is
quickly inactivated by high levels of cytidine deaminase in the
liver, spleen, intestinal epithelia, and blood, which accounts for
its short plasma beta half-life of 15 to 25 min. Pharmacokinetic
studies in rabbits and dogs show that DAC crosses the blood-brain
barrier (41). Human
pharmacokinetic studies have not been performed, but data from
clinical trials of DAC in MDS indicate rates of neurological and
psychiatric adverse reactions suggestive of CNS activity (42). Upon cellular entry, the prodrug
form (5-AZA-CdR) undergoes phosphorylation by a series of kinases
into its final active triphosphate form (5-AZA-CtR), which acts as
a substrate for DNA polymerase (Fig.
1). 5-AZA-CtR is then incorporated into the cell DNA and
asserts its effects. At lower dosages, the drug induces DNA
hypomethylation and reactivation of genes leading to cell
differentiation. Conversely, higher concentrations of the drug lead
to a cytotoxic effect by blocking DNA synthesis.
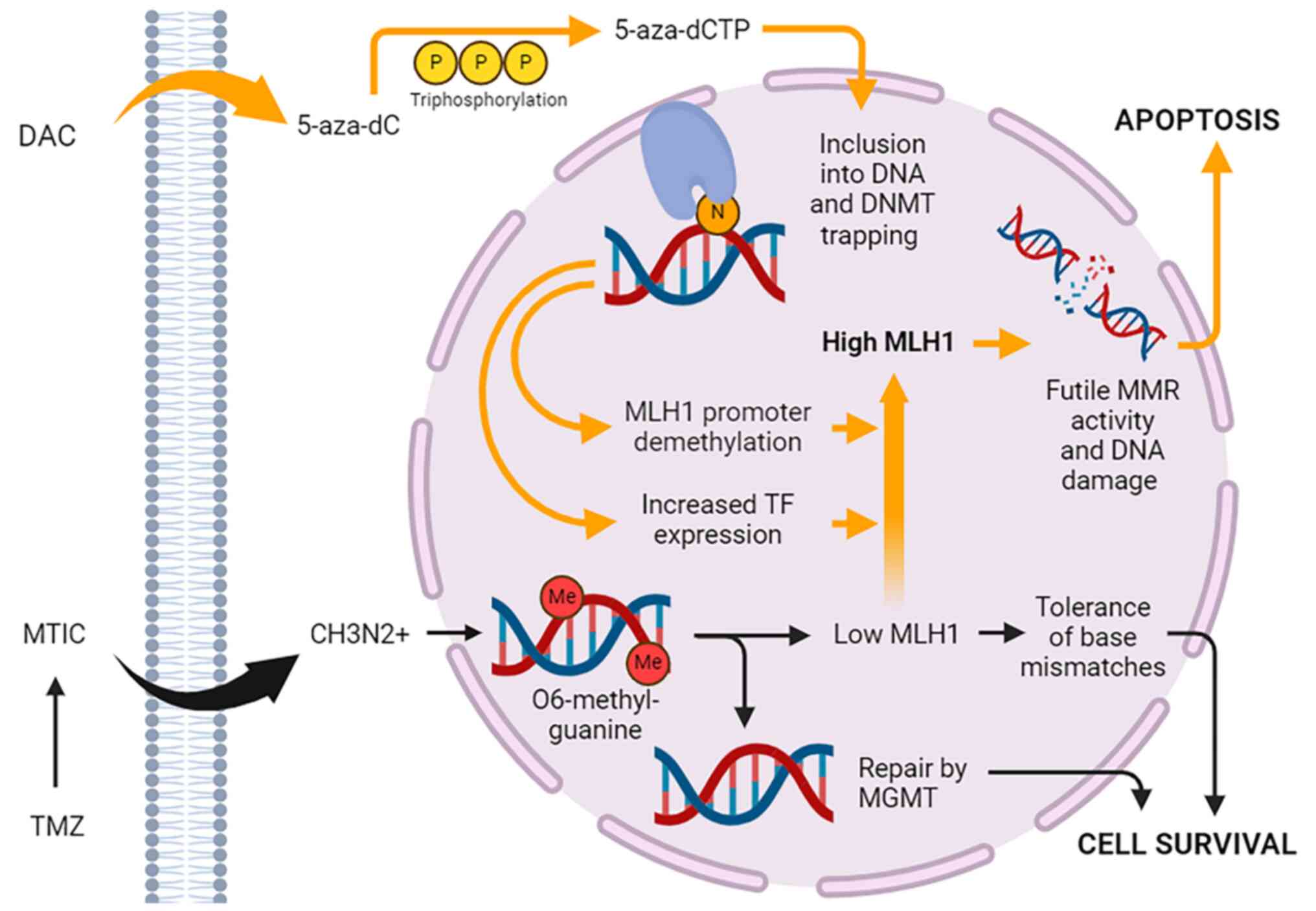 | Figure 1Proposed mechanism of action of DAC
and TMZ. Once DAC enters the cell, it undergoes triphosphorylation,
converting it to 5-aza-dCTP, which is incorporated into DNA during
S-phase in place of cytosine. 5-aza-dCTP traps and inactivates
DNMTs, causing exome-wide changes in gene expression mediated by
promoter demethylation, gene body demethylation and changes in TF
expression. MLH1 is upregulated, increasing levels of
O6-methylguanine produced by TMZ and futile MMR activity. This
ultimately results in enhanced cytotoxicity due to DNA
double-strand break formation, cell cycle arrest and apoptosis. The
effects of TMZ are exerted through its spontaneous decarboxylation
to MTIC, which is unstable and degrades into the reactive CH3N2+.
5-aza-dC, 5-aza-2'-deoxycytidine; 5-aza-dCTP,
5-aza-2'-deoxycytidine-5'-triphosphate; CH3N2+, methyldiazonium
ion; DAC, decitabine; DNMT, DNA methyltransferase; Me, methyl
group; MGMT, methylguanine methyltransferase; MLH1, mutL homolog 1;
MMR, mismatch repair; MTIC, 5-(3-methyltriazen-1-yl)
imidazole-4-carboxamide; TF, transcription factor; TMZ,
temozolomide. |
The antileukemic effect of DAC was first
demonstrated in 1968 in mouse models, which prompted further
investigation into the drug's clinical potential in decades to
follow. Early phase 1 clinical trials in the early 1990s determined
a maximally tolerated dose of 2,250 mg/m2 with
myelosuppression as the primary adverse effect. This was followed
by several single-arm phase 2 trials that demonstrated good
responses in AML, MDS, and chronic myelomonocytic leukemia (CMML)
even at low dosing schedules of 20 mg/m2/day for 5 days
(43). Finally, a North American
phase 3 trial comparing DAC to supportive care in patients with
intermediate- and high-risk MDS demonstrated a significant survival
benefit at a dose of 45 mg/m2/day, leading to approval
in the US of DAC for the treatment MDS in 2006(44). An oral form of DAC in combination
with cedazuridine, a novel cytidine deaminase inhibitor that
prevents drug inactivation in the digestive tract, was approved in
the US in 2020 (ASTX727) (42).
Azacytidine (5-azacitidine) (AZA), in contrast to
DAC, is a cytidine analogue that integrates preferentially into RNA
after entering the cell via nucleoside transporters, although
10-20% is reduced by ribonucleotide reductase into DAC and
incorporated into DNA (45,46).
Once incorporated into RNA by RNA polymerase, AZA interferes with
gene expression and protein synthesis by hampering RNA stability
and correct folding (47),
ultimately promoting apoptosis in tumor cells.
AZA absorption into tissues is fast and complete
after IV or subcutaneous administration, with a peak concentration
in 30 min and about 90% bioavailability (48). Unlike DAC, it does not cross the
blood-brain barrier, limiting its potential for use in CNS cancers.
It is metabolized in the liver and excretion is mostly through the
kidneys with a half-life of 4 h (49).
In a 2004 randomized open-label, phase 3 multicenter
trial conducted by the Cancer and Leukemia Group B, which included
patients with all five MDS subtypes, treatment with AZA resulted in
an overall response rate of 15.7%, compared to 0% response in the
observation arm. Responses included partial or complete
normalization of blood cells and bone marrow structure. On the
basis of this study and two other smaller single-arm trials, AZA
received approval in the US for all MDS subtypes (48).
Guadecitabine (SGI-110) is a second-generation HMA
currently being investigated as an alternative to DAC and AZA in
MDS and acute myeloid leukemia (50). Because the incorporation of DAC
into DNA is S-phase dependent, its relatively short half-life of 20
min due to degradation by cytidine deaminase limits its ability to
enter a large proportion of tumor cells after a single IV dose,
necessitating three doses every 8 h. As a dinucleotide of DAC and
deoxyguanosine linked by a 3'-5' phosphodiester bond, guadecitabine
is resistant to cytidine deaminase (51). After subcutaneous administration,
guadecitabine is cleaved into DAC in a slow, sustained fashion,
resulting in a prolonged exposure period and better tolerated
toxicity profile (52).
4. HMAs and tumor suppressors
Aberrant DNA methylation can provide survival
benefits to cancer cells by silencing essential genes for
anti-tumor activity, known as tumor suppressors. DNA demethylating
agents therefore provide a possible means of reactivating silenced
tumor suppressor genes and epigenetically reprogramming neoplastic
cells to therapeutic advantage (53). In MDS, aberrant methylation
patterns due to loss-of-function mutations in a number of
epigenetic regulators, including DNMT3A and TET2, result in
ineffective hematopoiesis and peripheral blood cytopenias by
disrupting hematopoietic stem cell differentiation homeostasis
(54,55). The sequestration of
methyltransferases using HMAs may counteract the development of
these aberrant patterns, as well as induce the re-expression of
various tumor suppressor genes often silenced in MDS, including
p15INK4B and p16INK4A (56),
TP53(57) and DAPK1(58). Overall, this yields
anti-proliferative and pro-apoptotic effects. Genes without
CpG-island-containing promoters have also been shown to be
upregulated by DAC in MDS and AML cells, emphasizing the role
methylation-independent effects may have as well (59).
In a number of solid cancers, investigators have
similarly demonstrated the ability of HMAs to re-express silenced
tumor suppressor genes with favorable effects on tumor cell growth
and gene expression profile. These include the Ras association
domain family 1A gene (RASSF1A) in lung cancer (60), the DNA double-strand break repair
gene BRCA1 in breast cancer (61),
and the homeobox transcription factor HOXA10 in ovarian cancer
(62). In GBM, several tumor
suppressor genes have been found to be hypermethylated at their
promoter regions and downregulated, including the microtubule
associated tumor suppressor gene (MTUS1) (63), esophageal-cancer related gene
(ECRG4) (64), epithelial membrane
protein 3 (EMP3) (65), and
SOCS1/3(66). In various GBM cell
lines, the dampened expression of these genes was associated with
increased cellular survival, invasion, proliferation and reduced
apoptosis. DAC treatment was able to reverse hypermethylation and
re-express these genes both at the mRNA and protein level.
HMAs can also silence pro-oncogenic pathways through
demethylation of gene body CpGs. In colorectal carcinoma, DAC
downregulated genes involved in the regulation of c-MYC signaling
pathways, leading to a suppression of tumor growth; this effect
reversed after withdrawal of treatment (67). In GBM, Sanaei and Kavoosi (66) demonstrated that DAC treatment
significantly downregulated expression of the anti-apoptotic Bcl-2
protein, mirroring a key mechanism of TMZ-induced cytotoxicity
(68) and suggesting DAC could
have a cytotoxicity potentiating effect. DAC also inhibited
JAK/STAT signaling, leading to reductions in cell proliferation and
growth.
The expression of Promonin-1 or CD133, a known
marker for GBM-initiating cancer stem cells (GSCs), correlates with
increased WHO grade in gliomas and exhibits abnormal, but often
variable, promoter methylation patterns even among different cell
subpopulations within the same GBM (69). Through promoter demethylation, DAC
treatment has been shown to upregulate CD133 promoter expression in
multiple GBM cell lines (70),
which could suggest a possible undesired pro-tumorigenic
effect.
Although there is accumulating preclinical evidence
that HMAs have the ability to alter the expression of tumor
suppressors and oncogenes in a way that, on balance, could yield
overall antitumor effects, further investigation will be required
to translate these findings into the clinic. AZA has been tested
clinically in recurrent IDH-mutant gliomas, but did not produce
measurable clinical responses as a single agent (71), possibly due to lack of CNS
penetration; to our knowledge, monotherapy with DAC or another HMA
has not yet been tested clinically in GBM. There are several other
challenges that could curtail therapeutic responses to HMAs,
including relatively lower penetrance into solid tumors compared to
hematologic malignancies (72),
unpredictable off-target effects on other gene networks, and the
intrinsic heterogeneity of GBM as opposed to the clonal nature of
hematologic neoplasms. Demethylation alone may also be insufficient
to reliably re-express a tumor suppressor gene if, for example, the
required activating transcription factor is not expressed.
5. HMA-mediated chemosensitization
For the past 20 years, the alkylating agent
temozolomide has remained the mainstay systemic agent used for GBM
and other diffuse gliomas. In light of this, a large number of
patients would potentially benefit from the identification of a
subset of GBM patients where the unique gene expression-modifying
properties of HMAs could act to potentiate the cytotoxic effects of
this chemotherapy. The strategy of chemosensitization using
epigenetic agents is an active line of investigation in a number of
solid cancers, most notably ovarian cancer, where low-dose DAC was
used successfully in a phase 2 clinical trial to overcome
resistance to platinum-based chemotherapy. Methylation array
profiling of patients in this study with progression-free survival
(PFS) greater than 6 months compared to those less than 6 months
suggested that demethylation of MLH1, RASSF1A, HOXA10, and HOXA11
were associated with longer PFS (73). Hypothesized mechanisms of this
effect include the reactivation of genes involved in mitochondrial
apoptosis, MAPK signaling, and membrane transporter pumps (74). Although not yet clinically tested
in bladder cancer, experiments using urothelial carcinoma cells
have also implicated HMA-induced reactivation of the tumor
suppressor RASSF1A and consequent downstream activation of the
Hippo pathway, which acts to slow cell proliferation, inhibit
cancer stem cell maintenance, and augment sensitivity to cisplatin
and doxorubicin (75).
Unfortunately, many systemically active
chemotherapeutic agents, including cisplatin, are unable to cross
the blood-brain barrier efficiently enough to penetrate into
gliomas without dosing at levels that would be toxic to other end
organs. Thus, the orally bioavailable and well-tolerated TMZ is
strongly favored. The cytotoxic effects of TMZ are primarily
mediated by the formation of O6-methylguanine adducts in
DNA. The DNA repair enzyme MGMT acts to reverse these adducts, but
if expressed at low levels or silenced, O6-methylguanine
will mispair with thymine during DNA replication, triggering the
cell's DNA mismatch repair (MMR) machinery, of which MLH1 is an
essential player. MMR complexes excise the mispaired base, but
because O6-methylguanine will continue to mispair with
thymine, a futile loop of attempted mismatch repair is initiated,
ultimately triggering DNA double-strand break formation, DNA damage
checkpoints, and apoptosis (Fig.
1). MMR deficiency from epigenetic silencing or somatic
mutation of component genes is closely linked with TMZ resistance,
as is MGMT overexpression via hypomethylation of its promoter
and/or methylation of its gene body (76). Thus, use of HMAs to reverse TMZ
resistance in GBM has spurred significant interest.
The use of DAC to potentiate the effects of
temozolomide via re-expression of MLH1 was first demonstrated by
Plumb et al (77) in
ovarian and colon cancer xenografts with MMR deficiency due to MLH1
promoter methylation. Subsequently, a phase 1/2 trial in
non-resectable stage IIIB/C or IV melanoma was the first clinical
demonstration that a 14-day regimen of low-dose DAC (0.15 mg/kg/day
or 6 mg/m2/day daily for 5 days) could upregulate MMR
genes and increase TMZ sensitivity (78). Complete responses were seen in 2 of
33 patients, with an overall clinical benefit rate of 61% and a
median overall survival of 12.4 months, suggesting the safety and
potential efficacy of the combination compared to historical
controls. Pre- and post-treatment tumor tissue and peripheral blood
mononuclear cells from 6 participating patients were analyzed for
HMA-induced changes in MGMT and MMR genes, but none were clearly
identified. However, overall gene expression changes were similar
to those seen in MDS, AML, and sickle cell disease patients after
treatment with DAC.
In GBM, using older methylation-specific PCR
techniques, MLH1 promoter methylation has been detected in up to
15% of tumor tissue samples (79-81).
With the advent of highly sensitive next-generation long-read
methylation sequencing techniques, the actual rate is likely
higher, presenting an opportunity to make meaningful improvements
in TMZ response rates in the vast majority of GBM patients who will
ultimately develop TMZ resistance. Work in our own laboratory using
GBM stem cell cultures derived from fresh surgical specimens
provides evidence that DAC increases MLH1 expression in a subset of
GBMs regardless of MGMT methylation status, and that this effect
mediates significant reductions in TMZ IC50.
Interestingly, full DNA methylation sequencing of the MLH1 promoter
region revealed an association between elevated baseline
methylation and a lack of TMZ sensitization, while the absence of
any baseline methylation at the promoter appeared necessary for
sensitization. This suggests that DAC may increase MLH1 levels not
through direct demethylation of the promoter but, rather,
indirectly via induction of an upstream transcription factor
(82) (Fig. 1). MLH1 promoter methylation levels
may therefore serve as a clinically useful predictive biomarker for
GBM patients who might respond well to DAC-based
chemosensitization. Moving forward, biomarker-informed patient
selection will be critical to the success of clinical trials
testing this approach, since the high molecular heterogeneity of
GBM tends to produce negative studies due to underpowering (i.e.,
beta error).
6. HMAs and immunotherapy
To date, cancer immunotherapy has seen the most
success in the treatment of melanoma, lung adenocarcinoma,
colorectal adenocarcinoma, and other malignancies with high
mutational burden (i.e., ‘hot’ tumors). Such malignancies exhibit
relatively high levels of tumor-specific neoantigen expression,
which are potentially detectable by the immune system (83-85).
GBM, however, is notoriously an immunologically ‘cold’ cancer
because of its relatively low mutational burden and low level of
neoantigen expression. Moreover, the immunosuppressive tumor
microenvironment of GBM provides multiple pathways for tumor immune
evasion, leading to low cytotoxic T-cell infiltration and generally
poor responses to immunotherapy (86,87).
Hence, much recent work in the field of GBM immunotherapy focuses
on devising strategies to convert GBMs from ‘cold’ to ‘hot’ in
order to enhance either adaptive or innate immune responses.
Epigenetic alterations and global hypermethylation
contribute to an immunosuppressive landscape in GBM through a
number of mechanisms. Downregulation of MHC class I in GBM cancer
stem cells secondary to promoter methylation by EZH2, a
methyltransferase, results in resistance to NK cell killing and
subsequent innate immune escape (88). EZH2 has also been implicated in
downregulating AP-2a, a transcription factor that blocks PD-L1
expression when bound to its promoter (89). HMAs, by suppressing AP-2a
methylation, may have the ability to reduce levels of PD-L1 in
glioma cells, thereby enhancing immune checkpoint blockade.
Progressive methylation of genes can also impair inflammatory
pathways in GBM, increasingly inhibiting the adaptive immune
response with time. For example, methylation at promoter regions of
the IL-7 gene and its receptor has been shown to be significantly
more elevated in recurrent compared to newly diagnosed GBM
(90).
These observations indicate that HMAs may have the
potential to synergize with tumor immunotherapy via multiple,
parallel mechanisms. Currently, clinical trials testing HMAs in
combination with a variety of immunotherapies are ongoing in
malignances such as AML and MDS. In the following sections, we
discuss their applications in the enhancement of neoantigen
expression, immune checkpoint blockade, cancer vaccines, and innate
immune responses (91).
Neoantigen expression
Similar to their effects on tumor suppressor and
oncogene expression, HMAs may directly modify neoantigen expression
through their inhibition of CpG methylation at the promoters and
bodies of neoantigen genes. Cancer-specific neoantigens, including
those derived from TP53, KRAS, IDH1/2 and MLH1, may be clonal or
highly subclonal throughout a tumor and therefore ideal targets for
cancer immunotherapy. At the same time, undesired effects of the
known oncogenic functions of these gene products must be weighed in
any approach that attempts to increase their expression for
heightened immune detection.
In one recent study, autologous neoepitopes
generated from the mutational hotspot region of TP53, the most
commonly mutated gene across all cancers, were found to be
immunogenic in 39% of patients (92). The development of adoptive cell
therapy using ex vivo expanded tumor-infiltrating T-cells
targeted against TP53 mutations and other public neoepitopes is an
active area of investigation (93), and HMAs may be a promising means of
increasing the efficacy of this approach. In U87 and GBM
patient-derived cell lines, Ma et al (94) demonstrated that DAC induces
upregulation of genes encoding for both HLA-A2-restricted
neoantigens and tumor-associated cancer testis antigens, leading to
an enhanced CD8+ T-cell mediated toxicity response by healthy donor
cells in a TCR:MHC class I-dependent manner. The authors also found
that DAC generally increased the activation of preexisting
cytotoxic T lymphocytes in GBM patients, improving the endogenous
recognition of cancer cells.
Another novel approach that has been explored in GBM
hijacks the ability of HMAs to upregulate certain oncogenes to
instead enhance oncolytic virotherapy. Okemoto et al
(95) used an engineered version
of the neurotropic HSV1 virus that had been placed under the
replicative control of a nestin promoter-enhancer sequence. Nestin
is a glioma-specific intermediate filament known to be
overexpressed in glioma cells and is used as a marker of the cancer
stem cell compartment. The investigators identified several CpG
islands within the nestin promoter that became hypermethylated
after the virus entered glioma cells. AZA was able to reverse this
hypermethylation, significantly improving viral replication both
in vitro and in vivo in an orthotopic mouse xenograft
model. These studies illustrate the innovative ways in which the
wide-ranging effects of HMAs on tumor-associated gene expression
can be channeled into anticancer treatment strategies.
Immune checkpoint inhibition
Immune checkpoint blockade (ICB) is an
immunotherapeutic approach widely recognized for its potential to
produce long-term and deep responses in a subset of cancer
patients. The most potent example of checkpoint inhibition is the
targeting of the programmed cell death protein 1 (PD-1)/programed
cell death ligand 1 (PD-L1) axis and CTLA-4 to unleash a powerful
T-cell response and eliminate cancer cells (96). Checkpoint inhibitors have shown
promising results in melanoma, where PD-1 blockade with the IgG
monoclonal antibody pembrolizumab has improved survival by
inhibiting binding to the PD-L1 ligand expressed by neoplastic
cells. The same effect has been observed in Hodgkin's lymphoma
(97,98), bladder cancer (99), and renal cell carcinoma (100) among others. T-cell activation
requires co-stimulatory molecules to induce signaling pathways that
lead to chemokine production and proliferation. PD-1 is a
co-inhibitory surface molecule present on T cells that helps
regulate homeostasis during inflammatory states to prevent
autoimmunity. Activation of PD-1 by its ligands leads to T-cell
anergy, exhaustion, and apoptosis.
Tumor cells, particularly in glioblastoma, express
high levels of the PD-L1 ligand leading to myriad immunosuppressive
effects within the tumor microenvironment, many of which have
proven challenging to overcome, even with ICB.
In this context, HMAs have garnered interest for
their ability in preclinical studies to rejuvenate exhausted
T-cells by reversing the acquisition of genomic methylation
patterns that act to restrict T-cell expansion and diversification
(101). Nie et al
(98) demonstrated the clinical
translation of this concept in a phase 2 study that examined the
effects of adding low dose DAC (10 mg/d x5 days every 3 weeks) to
camrelizumab, an anti-PD-1 monoclonal antibody, in patients with
classic Hodgkin's lymphoma who were ICB-naïve, and another cohort
of patients that had developed resistance to other ICBs. They found
that combination therapy was tolerable and increased complete
response rates significantly in both cohorts, indicating that DAC
might reverse acquired and primary ICB resistance. This effect was
associated with a broadened peripheral T-cell receptor repertoire,
suggesting that increased tumor immunogenicity might be one
responsible mechanism. In follow up in vitro experiments,
the authors also found evidence that DAC prevents loss of JunD
transcription factor expression in CD8+ T-cells, abrogating their
tendency to develop an exhaustion phenotype during ICB therapy
(102). Working with
CD19-targeted chimeric antigen receptor (CAR) T-cells in a mouse
model of non-Hodgkin's lymphoma, Wang et al (103) observed similar anti-exhaustion
effects in vivo when low dose (10 nM) DAC was added to the
CAR T-cell culture for 7 days.
Although checkpoint inhibition therapies have
received FDA approval in the US for use in several solid cancers,
including melanoma, hepatocellular carcinoma, and renal cell
carcinoma (104,105), all randomized clinical trials for
GBM to date have failed to demonstrate a survival benefit over
standard chemoradiation. The CheckMate 143 phase 3 randomized trial
(106) assessed the efficacy of
the PD-1 inhibitor nivolumab compared to bevacizumab alone in
patients with GBM at first recurrence, and showed no significant
improvement in overall survival with nivolumab (mOS was about 10
months in both arms). More recently, in a phase 3 trial randomizing
560 patients (CheckMate 498), Omuro et al (107) compared combined nivolumab and
radiotherapy (RT) to standard-of-care TMZ and RT in newly-diagnosed
MGMT-unmethylated GBM, demonstrating significantly shorter overall
survival in the nivolumab arm (13.4 vs. 14.9 months). Finally, in a
companion phase 3 study that randomized 716 patients (CheckMate
548), the addition of nivolumab to standard radiotherapy plus TMZ
in newly-diagnosed MGMT-methylated or indeterminate GBM patients
did not improve overall or progression-free survival (108). Based on these negative results,
it is becoming increasingly clear that ICB monotherapy as an
addition to radiotherapy, with or without temozolomide, is unable
to overcome T-cell exhaustion and the powerfully immunosuppressive
tumor microenvironment of GBM. However, given the encouraging
preclinical and clinical data emerging in hematologic malignancies,
using HMAs to address the hurdle of ICB resistance seems to be a
promising next tactic that merits exploration by investigators.
Cancer vaccines
Recent studies have shown that the use of
personalized cancer vaccines to boost the host immune response are
feasible even in tumors that are recognized as insensitive to
immunotherapy, such as GBM (109,110) and pancreatic cancer (111). In these clinical trials, patients
may receive vaccines containing peptides that match the amino acid
sequences of their own tumor-specific antigens, known as
‘personalized neoantigen-targeting vaccines’, or a pre-determined
‘off-the-shelf’ panel of one or more public tumor-specific antigens
(112).
Alternatively, antigen-presenting dendritic cells
collected from patients through leukapheresis can be exposed ex
vivo to tumor-specific antigens in the presence of
immunostimulatory adjuvants, such as poly-ICLC or GM-CSF, which
promote antigen delivery and dendritic cell maturation and
activation (113). Once the
dendritic cells are activated, they are reintroduced into the
patient to stimulate the adaptive immune response (114). This was tested in newly-diagnosed
GBM patients in a recent prospective externally controlled cohort
trial where researchers added an autologous tumor lysate-loaded
dendritic cell vaccine (DCVax-L) to standard-of-care
chemoradiation. The trial was initially designed as a randomized
phase 3 trial comparing vaccine to placebo, but due to a high rate
of crossover diluting the control arm, could not be meaningfully
analyzed as such. Compared to patients receiving only
standard-of-care therapy in other GBM trials, who were matched by
known prognostic factors, overall survival after initial surgery
was modestly but significantly longer (19.3 vs. 16.5 months). The
survival advantage was more pronounced when comparing the subset of
patients who received the vaccine only after tumor progression to
matched external control patients with recurrent GBM (13.2 vs. 7.8
months) (115).
As discussed above, HMAs enhance the expression of
neoantigens by tumor cells for targeting by antigen-specific
cytotoxic T-cells, presenting an opportunity for synergistic
effects when combined with tumor vaccines. Although no trials using
this strategy have yet been reported in GBM, a phase 1 study
employed standard dose DAC to increase tumor-associated antigen
expression in high-risk MDS patients receiving the CDX-1401
vaccine. CDX-1401 targets the cancer testis antigen NY-ESO-1, which
is aberrantly expressed in a variety of solid and hematologic
cancers. After the vaccine was administered every four weeks,
alternating with cycles of DAC, the investigators observed
increased NY-ESO-1 expression in myeloid cells and
NY-ESO-1-specific CD4+ and CD8+ T-cell responses in a majority of
the patients. In a separate preclinical study, DAC significantly
increased the expression of NY-ESO-1 in cultured GBM cells and
intracranial xenografts in mice after three 10 mg/kg doses.
Adoptively transferred NY-ESO-1 TCR-transduced lymphocytes were
then able to traffic from an injection site in the contralateral
cerebral hemisphere to the xenograft, extending mouse survival
significantly (116). Together,
these studies provide a strong justification for testing the
efficacy of CDX-1401 and other similar vaccines in combination with
DAC in GBM.
Innate immune system
Although most studies to date have focused on
harnessing and enhancing adaptive immune responses against
malignant gliomas with HMAs, evidence also points to the potential
for enhancement of the innate immune system. As a first-line
defense of the immune system, natural killer (NK) cells are
activated by stress-induced ligands and can distinguish ‘self’ from
‘missing-self’ by detecting the absence of the MHC class I molecule
on cell membranes. Depending on the balance of activating and
inhibitory signals from receptors at the cell surface, NK cells
will be cytotoxic or tolerant.
Zhang et al (117) discovered that expression of NK
cell-activating NKG2D ligands is suppressed in IDH-mutant gliomas
due to their hypermethylated phenotype. DAC (1 µM) treatment of
IDH-mutant glioma cells was able to restore NKG2D ligand
expression, with the increase lasting up to 7 days after washout of
DAC from the culture medium. In IDH-mutant glioma xenografts
established in the flanks of athymic nude mice, DAC (10 mg/kg)
increased NKG2D ligand expression, enabling NK cells to recognize
glioma cells, triggering an NK cell-mediated anti-tumor response
and slowing tumor growth (118).
Although no such reversible deficit of NK cell-activating ligand
expression has yet been demonstrated in GBM, which are
IDH-wildtype, these findings raise the possibility that HMAs might
be harnessed as a potent enhancer of immunotherapies based on the
NK cell platform.
7. Conclusion
In conclusion, GBM is an aggressive tumor with poor
prognosis, which currently lacks therapeutic options that can
reliably extend survival beyond two years. Based on successes seen
in hematologic and other solid malignancies, the use of HMAs in GBM
holds promise as a versatile means of enhancing established
treatment paradigms, but much work still needs to be done. With
strong evidence supporting its clinical use in MDS and AML, DAC is
perhaps the most well studied HMA that could also have epigenetic
activity in the CNS with an acceptable toxicity profile. This
review has examined how HMAs exert their effects by modifying the
expression of tumor suppressor genes, oncogenes, tumor-associated
antigens and neoantigens, and genes supporting T-cell
diversification. A growing understanding of these mechanisms has
led investigators to explore the synergies HMAs may have with
alkylating chemotherapy, immune checkpoint blockade, cancer
vaccines, and other forms of immunotherapy. Future research in GBM
should focus on combining HMAs with temozolomide to overcome
resistance mechanisms; exploring the utility HMAs might have in
changing the immunosuppressive tumor microenvironment in GBM to a
more favorable one for tumor-infiltrating lymphocytes; and
improving the expression of GBM neoantigens for detection by the
immune system. A more complete understanding of the varied
mechanisms by which HMAs exert their effects will lead to the
identification of biomarkers that will enable clinicians to select
the patients most likely to benefit from epigenetic therapy, based
on immune system and molecular tumor profiling. Personalized
approaches that combine HMAs with rationally chosen,
mechanistically complementary therapies offer hope for improving
outcomes for GBM patients in the future.
Acknowledgements
Not applicable.
Funding
Funding: Funding for this work was provided by the National
Institute of Neurological Disorders and Stroke (Bethesda, Maryland;
grant no. R03NS112572).
Availability of data and materials
Not applicable.
Authors' contributions
TJSH drafted the manuscript. JFI contributed to
drafting the manuscript and critically edited the manuscript. RLY
conceived the study and critically edited the manuscript. All
authors read and approved the final manuscript. Data authentication
is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Di Croce L, Raker VA, Corsaro M, Fazi F,
Fanelli M, Faretta M, Fuks F, Lo Coco F, Kouzarides T, Nervi C, et
al: Methyltransferase recruitment and DNA hypermethylation of
target promoters by an oncogenic transcription factor. Science.
295:1079–1082. 2002.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Turcan S, Rohle D, Goenka A, Walsh LA,
Fang F, Yilmaz E, Campos C, Fabius AW, Lu C, Ward PS, et al: IDH1
mutation is sufficient to establish the glioma hypermethylator
phenotype. Nature. 483:479–483. 2012.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Lu C, Ward PS, Kapoor GS, Rohle D, Turcan
S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, et
al: IDH mutation impairs histone demethylation and results in a
block to cell differentiation. Nature. 483:474–478. 2012.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Louis DN, Perry A, Wesseling P, Brat DJ,
Cree IA, Figarella-Branger D, Hawkins C, Ng HK, Pfister SM,
Reifenberger G, et al: The 2021 WHO classification of tumors of the
central nervous system: A summary. Neuro Oncol. 23:1231–1251.
2021.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Ostrom QT, Price M, Neff C, Cioffi G,
Waite KA, Kruchko C and Barnholtz-Sloan JS: CBTRUS statistical
report: primary brain and other central nervous system tumors
diagnosed in the United States in 2016-2020. Neuro Oncol. 25 (12
Suppl 2):iv1–iv99. 2023.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Alnahhas I, Alsawas M, Rayi A, Palmer JD,
Raval R, Ong S, Giglio P, Murad MH and Puduvalli V: Characterizing
benefit from temozolomide in MGMT promoter unmethylated and
methylated glioblastoma: A systematic review and meta-analysis.
Neurooncol Adv. 2(vdaa082)2020.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Szklener K, Mazurek M, Wieteska M,
Wacławska M, Bilski M and Mańdziuk S: New directions in the therapy
of glioblastoma. Cancers (Basel). 14(5377)2022.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Handy DE, Castro R and Loscalzo J:
Epigenetic modifications: Basic mechanisms and role in
cardiovascular disease. Circulation. 123:2145–2156. 2011.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Ivanova E, Canovas S, Garcia-Martínez S,
Romar R, Lopes JS, Rizos D, Sanchez-Calabuig MJ, Krueger F, Andrews
S, Perez-Sanz F, et al: DNA methylation changes during
preimplantation development reveal inter-species differences and
reprogramming events at imprinted genes. Clin Epigenetics.
12(64)2020.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Duncan CG, Grimm SA, Morgan DL, Bushel PR
and Bennett BD: NISC Comparative Sequencing Program. Roberts JD,
Tyson FL, Merrick BA and Wade PA: Dosage compensation and DNA
methylation landscape of the X chromosome in mouse liver. Sci Rep.
8(10138)2018.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Brabson JP, Leesang T, Mohammad S and
Cimmino L: Epigenetic regulation of genomic stability by vitamin C.
Front Genet. 12(675780)2021.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Dhar GA, Saha S, Mitra P and Nag Chaudhuri
R: DNA methylation and regulation of gene expression: Guardian of
our health. Nucleus (Calcutta). 64:259–270. 2021.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Compere SJ and Palmiter RD: DNA
methylation controls the inducibility of the mouse
metallothionein-I gene lymphoid cells. Cell. 25:233–240.
1981.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Moore LD, Le T and Fan G: DNA methylation
and its basic function. Neuropsychopharmacology. 38:23–38.
2013.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Mortusewicz O, Schermelleh L, Walter J,
Cardoso MC and Leonhardt H: Recruitment of DNA methyltransferase I
to DNA repair sites. Proc Natl Acad Sci USA. 102:8905–8909.
2005.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Kaneda M, Okano M, Hata K, Sado T,
Tsujimoto N, Li E and Sasaki H: Essential role for de novo DNA
methyltransferase Dnmt3a in paternal and maternal imprinting.
Nature. 429:900–903. 2004.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Aapola U, Kawasaki K, Scott HS, Ollila J,
Vihinen M, Heino M, Shintani A, Kawasaki K, Minoshima S, Krohn K,
et al: Isolation and initial characterization of a novel zinc
finger gene, DNMT3L, on 21q22.3, related to the
cytosine-5-methyltransferase 3 gene family. Genomics. 65:293–298.
2000.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Jin B, Li Y and Robertson KD: DNA
methylation: Superior or subordinate in the epigenetic hierarchy?
Genes Cancer. 2:607–617. 2011.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Rideout WM III, Coetzee GA, Olumi AF and
Jones PA: 5-Methylcytosine as an endogenous mutagen in the human
LDL receptor and p53 genes. Science. 249:1288–1290. 1990.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Ramirez-Carrozzi VR, Braas D, Bhatt DM,
Cheng CS, Hong C, Doty KR, Black JC, Hoffmann A, Carey M and Smale
ST: A unifying model for the selective regulation of inducible
transcription by CpG islands and nucleosome remodeling. Cell.
138:114–128. 2009.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Mikkelsen TS, Ku M, Jaffe DB, Issac B,
Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP,
et al: Genome-wide maps of chromatin state in pluripotent and
lineage-committed cells. Nature. 448:553–560. 2007.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Brenet F, Moh M, Funk P, Feierstein E,
Viale AJ, Socci ND and Scandura JM: DNA methylation of the first
exon is tightly linked to transcriptional silencing. PLoS One.
6(e14524)2011.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Hellman A and Chess A: Gene body-specific
methylation on the active X chromosome. Science. 315:1141–1143.
2007.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Bogdanović O and Veenstra GJ: DNA
methylation and methyl-CpG binding proteins: Developmental
requirements and function. Chromosoma. 118:549–565. 2009.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Li Y, Zheng H, Wang Q, Zhou C, Wei L, Liu
X, Zhang W, Zhang Y, Du Z, Wang X and Xie W: Genome-wide analyses
reveal a role of polycomb in promoting hypomethylation of DNA
methylation valleys. Genome Biol. 19(18)2018.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Mohn F, Weber M, Rebhan M, Roloff TC,
Richter J, Stadler MB, Bibel M and Schübeler D: Lineage-specific
polycomb targets and de novo DNA methylation define restriction and
potential of neuronal progenitors. Mol Cell. 30:755–766.
2008.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Ghadiri Moghaddam F, Farajnia S,
Karbalaei-Mahdi M and Monir L: Epigenetic insights in the
diagnosis, prognosis, and treatment selection in CRC, an updated
review. Mol Biol Rep. 49:10013–10022. 2022.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Collings CK and Anderson JN: Links between
DNA methylation and nucleosome occupancy in the human genome.
Epigenetics Chromatin. 10(18)2017.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Huang KK, Ramnarayanan K, Zhu F,
Srivastava S, Xu C, Tan ALK, Lee M, Tay S, Das K, Xing M, et al:
Genomic and epigenomic profiling of high-risk intestinal metaplasia
reveals molecular determinants of progression to gastric cancer.
Cancer Cell. 33:137–150.e5. 2018.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Wang Q, Xiong F, Wu G, Liu W, Chen J, Wang
B and Chen Y: Gene body methylation in cancer: Molecular mechanisms
and clinical applications. Clin Epigenetics. 14(154)2022.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Santini V and Ossenkoppele GJ:
Hypomethylating agents in the treatment of acute myeloid leukemia:
A guide to optimal use. Crit Rev Oncol Hematol. 140:1–7.
2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Holliday R and Ho T: DNA methylation and
epigenetic inheritance. Methods. 27:179–183. 2002.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Jabbour E, Issa JP, Garcia-Manero G and
Kantarjian H: Evolution of decitabine development: Accomplishments,
ongoing investigations, and future strategies. Cancer.
112:2341–2351. 2008.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Sorm F and Veselý J: Effect of
5-aza-2'-deoxycytidine against leukemic and hemopoietic tissues in
AKR mice. Neoplasma. 15:339–343. 1968.PubMed/NCBI
|
|
35
|
Xu K and Hansen E: Novel agents for
myelodysplastic syndromes. J Oncol Pharm Pract. 27:1982–1992.
2021.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Kordella C, Lamprianidou E and Kotsianidis
I: Mechanisms of action of hypomethylating agents: Endogenous
retroelements at the epicenter. Front Oncol.
11(650473)2021.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Quintás-Cardama A, Santos FP and
Garcia-Manero G: Therapy with azanucleosides for myelodysplastic
syndromes. Nat Rev Clin Oncol. 7:433–444. 2010.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Hollenbach PW, Nguyen AN, Brady H,
Williams M, Ning Y, Richard N, Krushel L, Aukerman SL, Heise C and
MacBeth KJ: A comparison of azacitidine and decitabine activities
in acute myeloid leukemia cell lines. PLoS One.
5(e9001)2010.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Seelan RS, Mukhopadhyay P, Pisano MM and
Greene RM: Effects of 5-Aza-2'-deoxycytidine (decitabine) on gene
expression. Drug Metab Rev. 50:193–207. 2018.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Cashen AF, Shah AK, Todt L, Fisher N and
DiPersio J: Pharmacokinetics of decitabine administered as a 3-h
infusion to patients with acute myeloid leukemia (AML) or
myelodysplastic syndrome (MDS). Cancer Chemother Pharmacol.
61:759–766. 2008.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Chabot GG, Rivard GE and Momparler RL:
Plasma and cerebrospinal fluid pharmacokinetics of
5-Aza-2'-deoxycytidine in rabbits and dogs. Cancer Res. 43:592–597.
1983.PubMed/NCBI
|
|
42
|
Kim N, Norsworthy KJ, Subramaniam S, Chen
H, Manning ML, Kitabi E, Earp J, Ehrlich LA, Okusanya OO, Vallejo
J, et al: FDA approval summary: Decitabine and cedazuridine tablets
for myelodysplastic syndromes. Clin Cancer Res. 28:3411–3416.
2022.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Kantarjian H, Oki Y, Garcia-Manero G,
Huang X, O'Brien S, Cortes J, Faderl S, Bueso-Ramos C, Ravandi F,
Estrov Z, et al: Results of a randomized study of 3 schedules of
low-dose decitabine in higher-risk myelodysplastic syndrome and
chronic myelomonocytic leukemia. Blood. 109:52–57. 2007.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Kantarjian H, Issa JP, Rosenfeld CS,
Bennett JM, Albitar M, DiPersio J, Klimek V, Slack J, de Castro C,
Ravandi F, et al: Decitabine improves patient outcomes in
myelodysplastic syndromes: Results of a phase III randomized study.
Cancer. 106:1794–1803. 2006.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Müller A and Florek M:
5-Azacytidine/azacitidine. Recent Results Cancer Res. 184:159–170.
2010.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Krawczyk J, Keane N, Freeman CL, Swords R,
O'Dwyer M and Giles FJ: 5-Azacytidine for the treatment of
myelodysplastic syndromes. Expert Opin Pharmacother. 14:1255–1268.
2013.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Glover AB, Leyland-Jones BR, Chun HG,
Davies B and Hoth DF: Azacitidine: 10 Years later. Cancer Treat
Rep. 71:737–746. 1987.PubMed/NCBI
|
|
48
|
Kaminskas E, Farrell AT, Wang YC, Sridhara
R and Pazdur R: FDA drug approval summary: Azacitidine
(5-azacytidine, Vidaza) for injectable suspension. Oncologist.
10:176–182. 2005.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Marcucci G, Silverman L, Eller M, Lintz L
and Beach CL: Bioavailability of azacitidine subcutaneous versus
intravenous in patients with the myelodysplastic syndromes. J Clin
Pharmacol. 45:597–602. 2005.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Garcia-Manero G, Roboz G, Walsh K,
Kantarjian H, Ritchie E, Kropf P, O'Connell C, Tibes R, Lunin S,
Rosenblat T, et al: Guadecitabine (SGI-110) in patients with
intermediate or high-risk myelodysplastic syndromes: phase 2
results from a multicentre, open-label, randomised, phase 1/2
trial. Lancet Haematol. 6:e317–e327. 2019.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Chuang JC, Warner SL, Vollmer D,
Vankayalapati H, Redkar S, Bearss DJ, Qiu X, Yoo CB and Jones PA:
S110, a 5-Aza-2'-deoxycytidine-containing dinucleotide, is an
effective DNA methylation inhibitor in vivo and can reduce tumor
growth. Mol Cancer Ther. 9:1443–1450. 2010.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Issa JJ, Roboz G, Rizzieri D, Jabbour E,
Stock W, O'Connell C, Yee K, Tibes R, Griffiths EA, Walsh K, et al:
Safety and tolerability of guadecitabine (SGI-110) in patients with
myelodysplastic syndrome and acute myeloid leukaemia: A
multicentre, randomised, dose-escalation phase 1 study. Lancet
Oncol. 16:1099–1110. 2015.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Ramakrishnan S, Hu Q, Krishnan N, Wang D,
Smit E, Granger V, Rak M, Attwood K, Johnson C, Morrison C, et al:
Decitabine, a DNA-demethylating agent, promotes differentiation via
NOTCH1 signaling and alters immune-related pathways in
muscle-invasive bladder cancer. Cell Death Dis.
8(3217)2017.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Li M and Zhang D: DNA methyltransferase-1
in acute myeloid leukaemia: Beyond the maintenance of DNA
methylation. Ann Med. 54:2011–2023. 2022.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Pappalardi MB, Keenan K, Cockerill M,
Kellner WA, Stowell A, Sherk C, Wong K, Pathuri S, Briand J,
Steidel M, et al: Discovery of a first-in-class reversible
DNMT1-selective inhibitor with improved tolerability and efficacy
in acute myeloid leukemia. Nat Cancer. 2:1002–1017. 2021.PubMed/NCBI
|
|
56
|
Quesnel B and Fenaux P: P15INK4b gene
methylation and myelodysplastic syndromes. Leuk Lymphoma.
35:437–443. 1999.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Daver NG, Maiti A, Kadia TM, Vyas P,
Majeti R, Wei AH, Garcia-Manero G, Craddock C, Sallman DA and
Kantarjian HM: TP53-mutated myelodysplastic syndrome and acute
myeloid leukemia: Biology, current therapy, and future directions.
Cancer Discov. 12:2516–2529. 2022.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Claus R, Hackanson B, Poetsch AR, Zucknick
M, Sonnet M, Blagitko-Dorfs N, Hiller J, Wilop S, Brümmendorf TH,
Galm O, et al: Quantitative analyses of DAPK1 methylation in AML
and MDS. Int J Cancer. 131:E138–E142. 2012.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Flotho C, Claus R, Batz C, Schneider M,
Sandrock I, Ihde S, Plass C, Niemeyer CM and Lübbert M: The DNA
methyltransferase inhibitors azacitidine, decitabine and zebularine
exert differential effects on cancer gene expression in acute
myeloid leukemia cells. Leukemia. 23:1019–1028. 2009.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Xie B, Peng F, He F, Cheng Y, Cheng J,
Zhou Z and Mao W: DNA methylation influences the CTCF-modulated
transcription of RASSF1A in lung cancer cells. Cell Biol Int.
46:1900–1914. 2022.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Tang Q, Cheng J, Cao X, Surowy H and
Burwinkel B: Blood-based DNA methylation as biomarker for breast
cancer: A systematic review. Clin Epigenetics.
8(115)2016.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Cheng W, Jiang Y, Liu C, Shen O, Tang W
and Wang X: Identification of aberrant promoter hypomethylation of
HOXA10 in ovarian cancer. J Cancer Res Clin Oncol. 136:1221–1227.
2010.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Ranjan N, Pandey V, Panigrahi MK, Klumpp
L, Naumann U and Babu PP: The tumor suppressor MTUS1/ATIP1
modulates tumor promotion in glioma: Association with epigenetics
and DNA repair. Cancers (Basel). 13(1245)2021.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Götze S, Feldhaus V, Traska T, Wolter M,
Reifenberger G, Tannapfel A, Kuhnen C, Martin D, Müller O and
Sievers S: ECRG4 is a candidate tumor suppressor gene frequently
hypermethylated in colorectal carcinoma and glioma. BMC Cancer.
9(447)2009.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Alaminos M, Dávalos V, Ropero S, Setién F,
Paz MF, Herranz M, Fraga MF, Mora J, Cheung NK, Gerald WL and
Esteller M: EMP3, a myelin-related gene located in the critical
19q13.3 region, is epigenetically silenced and exhibits features of
a candidate tumor suppressor in glioma and neuroblastoma. Cancer
Res. 65:2565–2571. 2005.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Sanaei M and Kavoosi F: The effect of
5-aza,2'-deoxyCytidine (5 AZA CdR or decitabine) on extrinsic,
intrinsic, and JAK/STAT pathways in neuroblastoma and glioblastoma
cells lines. Asian Pac J Cancer Prev. 24:1841–1854. 2023.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Yang X, Han H, De Carvalho DD, Lay FD,
Jones PA and Liang G: Gene body methylation can alter gene
expression and is a therapeutic target in cancer. Cancer Cell.
26:577–590. 2014.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Ochs K and Kaina B: Apoptosis induced by
DNA damage O6-methylguanine is Bcl-2 and caspase-9/3 regulated and
Fas/caspase-8 independent. Cancer Res. 60:5815–5824.
2000.PubMed/NCBI
|
|
69
|
Tabu K, Sasai K, Kimura T, Wang L,
Aoyanagi E, Kohsaka S, Tanino M, Nishihara H and Tanaka S: Promoter
hypomethylation regulates CD133 expression in human gliomas. Cell
Res. 18:1037–1046. 2008.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Yi JM, Tsai HC, Glöckner SC, Lin S, Ohm
JE, Easwaran H, James CD, Costello JF, Riggins G, Eberhart CG, et
al: Abnormal DNA methylation of CD133 in colorectal and
glioblastoma tumors. Cancer Res. 68:8094–8103. 2008.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Federici L, Capelle L, Annereau M, Bielle
F, Willekens C, Dehais C, Laigle-Donadey F, Hoang-Xuan K, Delattre
JY, Idbaih A, et al: 5-Azacitidine in patients with IDH1/2-mutant
recurrent glioma. Neuro Oncol. 22:1226–1228. 2020.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Sato T, Issa JJ and Kropf P: DNA
Hypomethylating drugs in cancer therapy. Cold Spring Harb Perspect
Med. 7(a026948)2017.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Matei D, Fang F, Shen C, Schilder J,
Arnold A, Zeng Y, Berry WA, Huang T and Nephew KP: Epigenetic
resensitization to platinum in ovarian cancer. Cancer Res.
72:2197–2205. 2012.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Glaysher S, Gabriel FG, Johnson P, Polak
M, Knight LA, Parker K, Poole M, Narayanan A and Cree IA: NHS
Collaborative Research Programme for Predictive Oncology. Molecular
basis of chemosensitivity of platinum pre-treated ovarian cancer to
chemotherapy. Br J Cancer. 103:656–662. 2010.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Hannon CE and Eisen MB: Intrinsic protein
disorder is insufficient to drive subnuclear clustering in
embryonic transcription factors. Elife. 12(RP88221)2024.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Moen EL, Stark AL, Zhang W, Dolan ME and
Godley LA: The role of gene body cytosine modifications in MGMT
expression and sensitivity to temozolomide. Mol Cancer Ther.
13:1334–1344. 2014.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Plumb JA, Strathdee G, Sludden J, Kaye SB
and Brown R: Reversal of drug resistance in human tumor xenografts
by 2'-deoxy-5-azacytidine-induced demethylation of the hMLH1 gene
promoter. Cancer Res. 60:6039–6044. 2000.PubMed/NCBI
|
|
78
|
Tawbi HA, Beumer JH, Tarhini AA, Moschos
S, Buch SC, Egorin MJ, Lin Y, Christner S and Kirkwood JM: Safety
and efficacy of decitabine in combination with temozolomide in
metastatic melanoma: A phase I/II study and pharmacokinetic
analysis. Ann Oncol. 24:1112–1119. 2013.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Skiriutė D, Vaitkienė P, Ašmonienė V,
Steponaitis G, Deltuva VP and Tamašauskas A: Promoter methylation
of AREG, HOXA11, hMLH1, NDRG2, NPTX2 and Tes genes in glioblastoma.
J Neurooncol. 113:441–449. 2013.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Rodríguez-Hernández I, Garcia JL,
Santos-Briz A, Hernández-Laín A, González-Valero JM, Gómez-Moreta
JA, Toldos-González O, Cruz JJ, Martin-Vallejo J and
González-Sarmiento R: Integrated analysis of mismatch repair system
in malignant astrocytomas. PLoS One. 8(e76401)2013.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Fukushima T, Katayama Y, Watanabe T,
Yoshino A, Ogino A, Ohta T and Komine C: Promoter hypermethylation
of mismatch repair gene hMLH1 predicts the clinical response of
malignant astrocytomas to nitrosourea. Clin Cancer Res.
11:1539–1544. 2005.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Gallitto M, Cheng He R, Inocencio JF, Wang
H, Zhang Y, Deikus G, Wasserman I, Strahl M, Smith M, Sebra R and
Yong RL: Epigenetic preconditioning with decitabine sensitizes
glioblastoma to temozolomide via induction of MLH1. J Neurooncol.
147:557–566. 2020.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Mehnert JM, Panda A, Zhong H, Hirshfield
K, Damare S, Lane K, Sokol L, Stein MN, Rodriguez-Rodriquez L,
Kaufman HL, et al: Immune activation and response to pembrolizumab
in POLE-mutant endometrial cancer. J Clin Invest. 126:2334–2340.
2016.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Panda A, Betigeri A, Subramanian K, Ross
JS, Pavlick DC, Ali S, Markowski P, Silk A, Kaufman HL, Lattime E,
et al: Identifying a clinically applicable mutational burden
threshold as a potential biomarker of response to immune checkpoint
therapy in solid tumors. JCO Precis Oncol.
2017(PO.17.00146)2017.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Rizvi NA, Hellmann MD, Snyder A, Kvistborg
P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, et al: Cancer
immunology. Mutational landscape determines sensitivity to PD-1
blockade in non-small cell lung cancer. Science. 348:124–128.
2015.PubMed/NCBI View Article : Google Scholar
|
|
86
|
DeCordova S, Shastri A, Tsolaki AG, Yasmin
H, Klein L, Singh SK and Kishore U: Molecular heterogeneity and
immunosuppressive microenvironment in glioblastoma. Front Immunol.
11(1402)2020.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Zaidi N and Jaffee EM: Immune cells track
hard-to-target brain tumours. Nature. 565:170–171. 2019.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Zhong J, Yang X, Chen J, He K, Gao X, Wu
X, Zhang M, Zhou H, Xiao F, An L, et al: Circular EZH2-encoded
EZH2-92aa mediates immune evasion in glioblastoma via inhibition of
surface NKG2D ligands. Nat Commun. 13(4795)2022.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Long S, Huang G, Ouyang M, Xiao K, Zhou H,
Hou A, Li Z, Zhong Z, Zhong D, Wang Q, et al: Epigenetically
modified AP-2α by DNA methyltransferase facilitates glioma immune
evasion by upregulating PD-L1 expression. Cell Death Dis.
14(365)2023.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Tompa M, Kraboth Z, Galik B, Kajtar B,
Gyenesei A and Kalman B: Epigenetic suppression of the IL-7 pathway
in progressive glioblastoma. Biomedicines. 10(2174)2022.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Héninger E, Krueger TE and Lang JM:
Augmenting antitumor immune responses with epigenetic modifying
agents. Front Immunol. 6(29)2015.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Malekzadeh P, Pasetto A, Robbins PF,
Parkhurst MR, Paria BC, Jia L, Gartner JJ, Hill V, Yu Z, Restifo
NP, et al: Neoantigen screening identifies broad TP53 mutant
immunogenicity in patients with epithelial cancers. J Clin Invest.
129:1109–1114. 2019.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Tran E, Robbins PF, Lu YC, Prickett TD,
Gartner JJ, Jia L, Pasetto A, Zheng Z, Ray S, Groh EM, et al:
T-cell transfer therapy targeting mutant KRAS in cancer. N Engl J
Med. 375:2255–2262. 2016.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Ma R, Rei M, Woodhouse I, Ferris K,
Kirschner S, Chandran A, Gileadi U, Chen JL, Pereira Pinho M,
Ariosa-Morejon Y, et al: Decitabine increases neoantigen and cancer
testis antigen expression to enhance T-cell-mediated toxicity
against glioblastoma. Neuro Oncol. 24:2093–2106. 2022.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Okemoto K, Kasai K, Wagner B, Haseley A,
Meisen H, Bolyard C, Mo X, Wehr A, Lehman A, Fernandez S, et al:
DNA demethylating agents synergize with oncolytic HSV1 against
malignant gliomas. Clin Cancer Res. 19:5952–5959. 2013.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Nebhan CA and Johnson DB: Pembrolizumab in
the adjuvant treatment of melanoma: Efficacy and safety. Expert Rev
Anticancer Ther. 21:583–590. 2021.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Allen PB, Savas H, Evens AM, Advani RH,
Palmer B, Pro B, Karmali R, Mou E, Bearden J, Dillehay G, et al:
Pembrolizumab followed by AVD in untreated early unfavorable and
advanced-stage classical Hodgkin lymphoma. Blood. 137:1318–1326.
2021.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Nie J, Wang C, Liu Y, Yang Q, Mei Q, Dong
L, Li X, Liu J, Ku W, Zhang Y, et al: Addition of low-dose
decitabine to anti-PD-1 antibody camrelizumab in
relapsed/refractory classical hodgkin lymphoma. J Clin Oncol.
37:1479–1489. 2019.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Merseburger AS, Apolo AB, Chowdhury S,
Hahn NM, Galsky MD, Milowsky MI, Petrylak D, Powles T, Quinn DI,
Rosenberg JE, et al: SIU-ICUD recommendations on bladder cancer:
Systemic therapy for metastatic bladder cancer. World J Urol.
37:95–105. 2019.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Chowdhury S, Infante JR, Hawkins R, Voss
MH, Perini R, Arkenau T, Voskoboynik M, Aimone P, Naeije I, Reising
A and McDermott DF: A phase I/II study to assess the safety and
efficacy of pazopanib and pembrolizumab combination therapy in
patients with advanced renal cell carcinoma. Clin Genitourin
Cancer. 19:434–446. 2021.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Ghoneim HE, Fan Y, Moustaki A, Abdelsamed
HA, Dash P, Dogra P, Carter R, Awad W, Neale G, Thomas PG and
Youngblood B: De novo epigenetic programs inhibit PD-1
blockade-mediated T cell rejuvenation. Cell. 170:142–157.e19.
2017.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Li X, Li Y, Dong L, Chang Y, Zhang X, Wang
C, Chen M, Bo X, Chen H, Han W and Nie J: Decitabine priming
increases anti-PD-1 antitumor efficacy by promoting CD8+ progenitor
exhausted T cell expansion in tumor models. J Clin Invest.
133(e165673)2023.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Wang Y, Tong C, Dai H, Wu Z, Han X, Guo Y,
Chen D, Wei J, Ti D, Liu Z, et al: Low-dose decitabine priming
endows CAR T cells with enhanced and persistent antitumour
potential via epigenetic reprogramming. Nat Commun.
12(409)2021.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Papadatos-Pastos D, Yuan W, Pal A, Crespo
M, Ferreira A, Gurel B, Prout T, Ameratunga M, Chénard-Poirier M,
Curcean A, et al: Phase 1, dose-escalation study of guadecitabine
(SGI-110) in combination with pembrolizumab in patients with solid
tumors. J Immunother Cancer. 10(e004495)2022.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Wei SC, Duffy CR and Allison JP:
Fundamental mechanisms of immune checkpoint blockade therapy.
Cancer Discov. 8:1069–1086. 2018.PubMed/NCBI View Article : Google Scholar
|
|
106
|
Reardon DA, Brandes AA, Omuro A,
Mulholland P, Lim M, Wick A, Baehring J, Ahluwalia MS, Roth P, Bähr
O, et al: Effect of nivolumab vs bevacizumab in patients with
recurrent glioblastoma: The CheckMate 143 phase 3 randomized
clinical trial. JAMA Oncol. 6:1003–1010. 2020.PubMed/NCBI View Article : Google Scholar
|
|
107
|
Omuro A, Brandes AA, Carpentier AF, Idbaih
A, Reardon DA, Cloughesy T, Sumrall A, Baehring J, van den Bent M,
Bähr O, et al: Radiotherapy combined with nivolumab or temozolomide
for newly diagnosed glioblastoma with unmethylated MGMT promoter:
An international randomized phase III trial. Neuro Oncol.
25:123–134. 2023.PubMed/NCBI View Article : Google Scholar
|
|
108
|
Lim M, Weller M, Idbaih A, Steinbach J,
Finocchiaro G, Raval RR, Ansstas G, Baehring J, Taylor JW, Honnorat
J, et al: Phase III trial of chemoradiotherapy with temozolomide
plus nivolumab or placebo for newly diagnosed glioblastoma with
methylated MGMT promoter. Neuro Oncol. 24:1935–1949.
2022.PubMed/NCBI View Article : Google Scholar
|
|
109
|
Hilf N, Kuttruff-Coqui S, Frenzel K, Bukur
V, Stevanović S, Gouttefangeas C, Platten M, Tabatabai G, Dutoit V,
van der Burg SH, et al: Actively personalized vaccination trial for
newly diagnosed glioblastoma. Nature. 565:240–245. 2019.PubMed/NCBI View Article : Google Scholar
|
|
110
|
Keskin DB, Anandappa AJ, Sun J, Tirosh I,
Mathewson ND, Li S, Oliveira G, Giobbie-Hurder A, Felt K, Gjini E,
et al: Neoantigen vaccine generates intratumoral T cell responses
in phase Ib glioblastoma trial. Nature. 565:234–239.
2019.PubMed/NCBI View Article : Google Scholar
|
|
111
|
Rojas LA, Sethna Z, Soares KC, Olcese C,
Pang N, Patterson E, Lihm J, Ceglia N, Guasp P, Chu A, et al:
Personalized RNA neoantigen vaccines stimulate T cells in
pancreatic cancer. Nature. 618:144–150. 2023.PubMed/NCBI View Article : Google Scholar
|
|
112
|
Weller M, Butowski N, Tran DD, Recht LD,
Lim M, Hirte H, Ashby L, Mechtler L, Goldlust SA, Iwamoto F, et al:
Rindopepimut with temozolomide for patients with newly diagnosed,
EGFRvIII-expressing glioblastoma (ACT IV): A randomised,
double-blind, international phase 3 trial. Lancet Oncol.
18:1373–1385. 2017.PubMed/NCBI View Article : Google Scholar
|
|
113
|
Zhao T, Cai Y, Jiang Y, He X, Wei Y, Yu Y
and Tian X: Vaccine adjuvants: Mechanisms and platforms. Signal
Transduct Target Ther. 8(283)2023.PubMed/NCBI View Article : Google Scholar
|
|
114
|
Palucka K and Banchereau J: Cancer
immunotherapy via dendritic cells. Nat Rev Cancer. 12:265–277.
2012.PubMed/NCBI View Article : Google Scholar
|
|
115
|
Liau LM, Ashkan K, Brem S, Campian JL,
Trusheim JE, Iwamoto FM, Tran DD, Ansstas G, Cobbs CS, Heth JA, et
al: Association of autologous tumor lysate-loaded dendritic cell
vaccination with extension of survival among patients with newly
diagnosed and recurrent glioblastoma: A phase 3 prospective
externally controlled cohort trial. JAMA Oncol. 9:112–121.
2023.PubMed/NCBI View Article : Google Scholar
|
|
116
|
Everson RG, Antonios JP, Lisiero DN, Soto
H, Scharnweber R, Garrett MC, Yong WH, Li N, Li G, Kruse CA, et al:
Efficacy of systemic adoptive transfer immunotherapy targeting
NY-ESO-1 for glioblastoma. Neuro Oncol. 18:368–378. 2016.PubMed/NCBI View Article : Google Scholar
|
|
117
|
Zhang X, Rao A, Sette P, Deibert C,
Pomerantz A, Kim WJ, Kohanbash G, Chang Y, Park Y, Engh J, et al:
IDH mutant gliomas escape natural killer cell immune surveillance
by downregulation of NKG2D ligand expression. Neuro Oncol.
18:1402–1412. 2016.PubMed/NCBI View Article : Google Scholar
|
|
118
|
Zhang X, Kim WJ, Rao AV, Jaman E, Deibert
CP, Sandlesh P, Krueger K, Allen JC and Amankulor NM: In vivo
efficacy of decitabine as a natural killer cell-mediated
immunotherapy against isocitrate dehydrogenase mutant gliomas.
Neurosurg Focus. 52(E3)2022.PubMed/NCBI View Article : Google Scholar
|














