Introduction
Wolfram syndrome (WFS) is an autosomal recessive
neurodegenerative disorder characterized by early onset diabetes
mellitus and progressive optic atrophy in children. Patients
typically suffer from diabetes insipidus and deafness. Despite a
number of studies reporting the incidence of WFS as extremely rare
(1,2), several cases have been reported
previously (1–3). It is possible that the incidence of
WFS has been underestimated.
Neuroophthalmologists encounter WFS patients who
present with optical nerve atrophy. Understanding the clinical
characteristics of the disease is important to prevent
misdiagnosis. The prognosis of individuals with WFS is poor and
~70% of patients have an estimated life span of <35 years
(4). Therefore, early diagnosis is
beneficial to the patient and their family. We have found an
extremely limited number of ophthalmologists who are familiar with
this disease and there are also relatively few studies published in
ocular journals.
The WFS1 and CISD2 (also known as WFS2) genes have
been identified in association with WFS. The WFS1 gene is on the
short arm of chromosome 4. WFS1-related disorders vary between WFS
and WFS1-related low-frequency sensory hearing loss. The WFS1
protein (wolframin) is an integral, endoglycosidase H-sensitive
membrane glycoprotein that localizes primarily in the endoplasmic
reticulum. Numerous mutations have been found in WFS1, largely in
exon 8, including missense, insertion, deletion and splice
mutations. Compound heterozygote for 2 missense mutations has also
been reported and leads to a relatively mild phenotype (5–7).
There is no evidence that deletion, splice and insertion mutations
cause more severe symptoms than missense mutations (8). A specific missense mutation is known
to cause WFS in homozygotes but only low-frequency sensorineural
hearing loss in heterozygous carriers (9,10).
In the current study, a case of WFS is discussed following detailed
investigation of its biological presentation.
Case report
Patient and clinical data
A 12-year-old Chinese girl presented with gradual
loss of vision in both eyes over ~3 years. The patient did not
report any associated symptoms or history, including pain with eye
movement, ocular injury, encephalitis or premature delivery. The
patient was also diagnosed with Type 1 diabetes mellitus 7 years
ago and inadequately controlled blood sugar levels even with
insulin treatment. Her parents were not consanguineous and were
healthy. No history of diabetes was known in her family. The study
was approved by the ethics committee of PLA general hospital,
Beijing, China, and written informed consent was obtained from the
patient’s family.
Ophthalmological examination was performed and
revealed counting fingers/10 cm in the right eye and hand
motions/10 cm in the left eye. Random amplification of polymorphic
DNA was negative in both eyes. Results of intraocular pressure,
ocular motility and the anterior segment examination were normal.
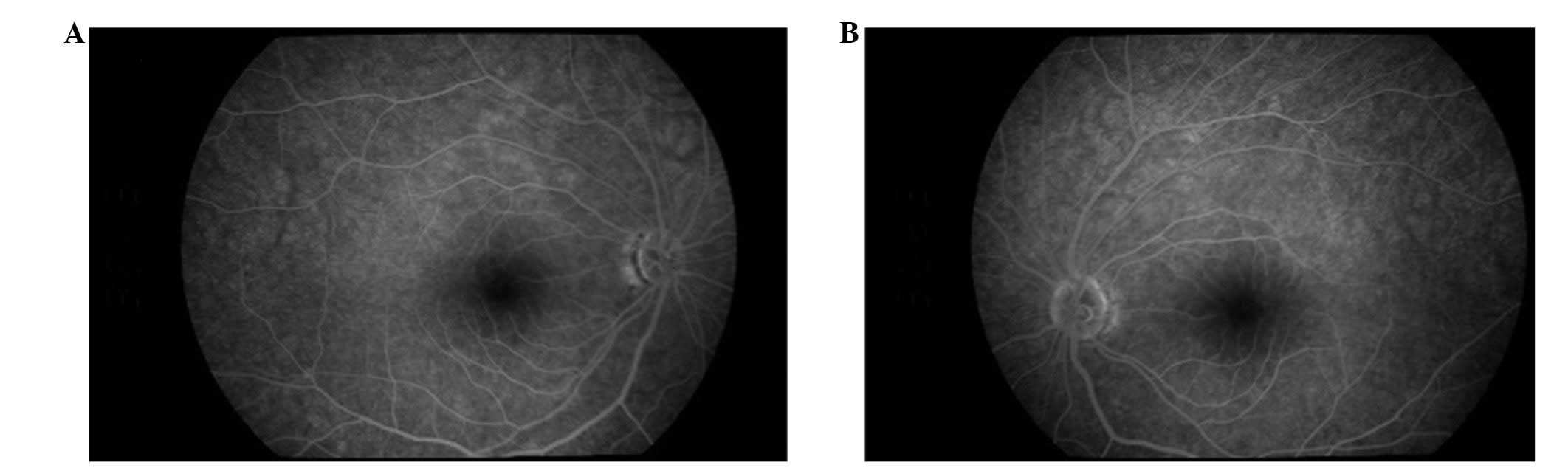
Ophthalmoscopic examination revealed bilateral optic atrophy
without any signs of diabetic retinopathy (Fig. 1). Flash visual-evoked response
revealed that the amplitude of P2 decreased. The vision was too
poor to perform a visual field test. Magnetic resonance imaging of
the brain and spinal cord was normal. Although the patient had no
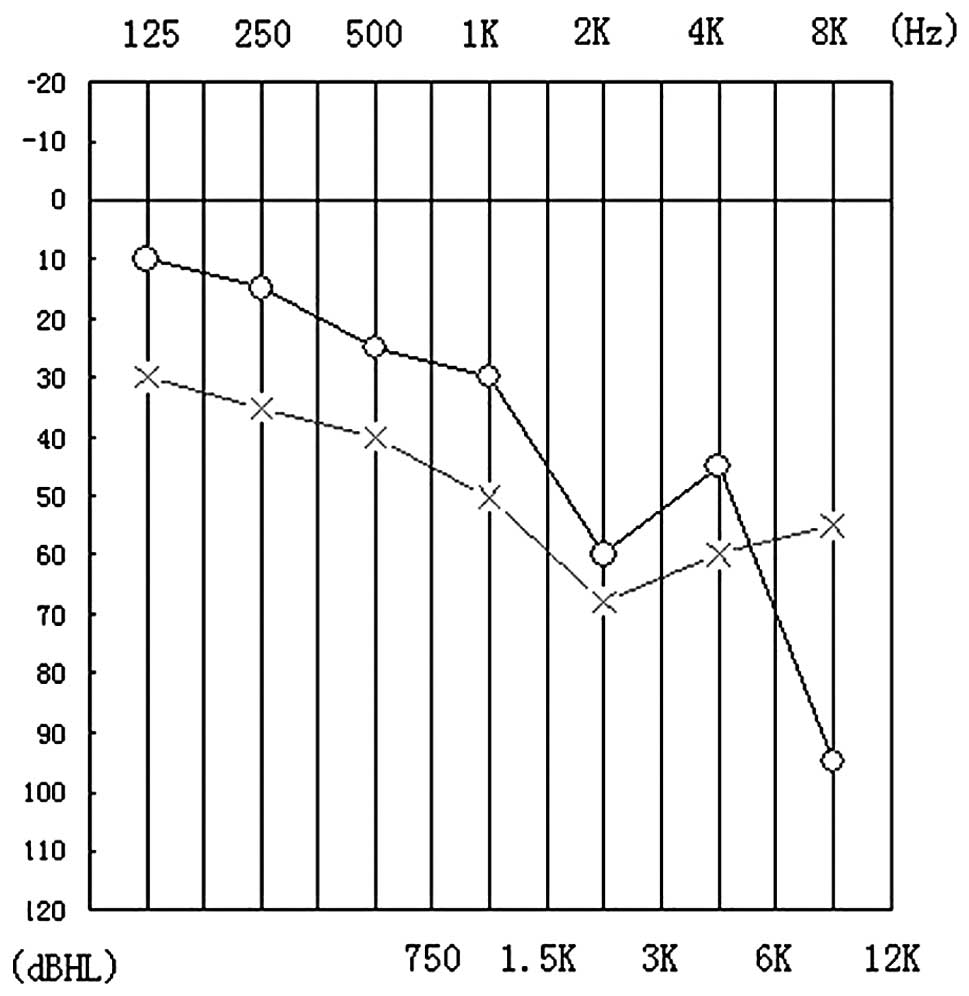
complaint of hearing loss, a hearing test revealed decreased high
frequency hearing ability (Fig.
2). Urinary output (24 h) was >5,000 ml. Despite the high
urine output, the patient had no complaint of polyuria and
considered this output to be normal due to diagnosis of type 1
diabetes mellitus.
Identification of mutations in the WFS1
gene
To detect the presence of WFS1 gene mutations,
direct DNA sequencing was performed to screen the entire coding
region of the gene in the patient’s family (Invitrogen Life
Technologies, Beijing, China), including the father, mother and
brother. A base substitution at c.2411T>C (Leu804Pro) was
identified in exon 8 which was homozygous with the patient and
heterozygous with the healthy parents and brother of the patient
(Fig. 3 and Table I).
 | Table IVariations in the WFS1 gene of the
family. |
Table I
Variations in the WFS1 gene of the
family.
| Type of
variation | Exon | Nucleotide
change | Amino acid
change | Status |
|---|
| SNP | 6 | c.684C>G | - | - |
| SNP | 8 | c.997G>A | V333I | - |
| SNP | 8 | c.1185C>T | - | - |
| SNP | 8 | c.1500C>T | - | - |
| SNP | 8 | c.1832G>A | R611H | - |
| Missense | 8 | c.2411T>C | L804P | I:1 heterozygous |
| | | | I:2 heterozygous |
| | | | II:1 homozygous |
| | | | II:2
heterozygous |
| SNP | 8 | c.2433G>A | - | - |
| SNP | 8 | c.2565A>G | - | - |
Discussion
Wolfram syndrome (WFS) often begins with type 1
diabetes. As there are no other early symptoms, diagnosis is
extremely difficult for endocrinologists. As the disease develops,
the patient may become aware of worsening vision 2–3 years
following diagnosis, due to atrophy of the optic nerve. Patients
are often referred to a neuroophthalmologist whose role is crucial
to the early diagnosis of this disease.
WFS patients often present with 4 common symptoms.
The first is insulin-dependent diabetes mellitus, which is often
the first sign of the disease, occuring primarily in childhood. The
mean age of onset is ~5–7 years old (5,6). The
current patient was diagnosed with insulin-dependent diabetes
mellitus at 5 years old. The second symptom is ocular pathology
which often occurs 2–3 years following diagnosis of diabetes.
Almost 100% of WFS individuals present with apparent optic nerve
atrophy and additional eye symptoms include visual loss or field
defects which may occur earlier than optic nerve atrophy. The
patient in this case suffered from considerable optic nerve atrophy
with a history of worsening vision loss over 3 years. In addition,
diabetes insipidus of hypothalamic origin may occur in the third
decade of life in up to 75% of cases (7). Urinary output (24 h) is often 3–12
liters and may be reduced by administration of an anti-diuretic
hormone. Diagnosis of pituitary diabetes insipidus is performed
upon additional signs of elevated urine-specific gravity and
osmotic pressure. Since individuals with diabetes mellitus commonly
suffer from polyuria, WFS patients often consider this output to be
normal, however, the high urine volume is actually caused by
diabetes insipidus. In the current case, patient 24-h urinary
output exceeded 5,000 ml, however, the individual had no complaint
of polyuria. Finally, hearing impairment is commonly noted at early
stages of WFS. Since the impairment is commonly at high frequency
levels of hearing and the frequency range of sounds in everyday
life is between 500 and 2,000 Hz, patients often have no complaints
of a hearing defect. This was the case in the present WFS
individual. The common characteristics of this syndrome demonstrate
the importance of performing examination of urine output and a
hearing test in patients presenting with insulin-dependent diabetes
mellitus and optic nerve atrophy.
It is important for neuroophthalmologists to be able
to differentiate WFS from Leber’s hereditary optic atrophy and
diabetic optic neuropathy, as the prognosis is significantly
different. In the case of WFS, ~70% of patients are likely to
succumb to the disease prior to 35 years old (4,11).
Additional methods, including genetic tests may also aid
diagnosis.
Previously, Amr et al(12) revealed that the CISD2 gene, which
maps to chromosome 4q22-q25, was localized to the endoplasmic
reticulum, colocalizing with calnexin. The authors identified a
mutation in the CISD2 gene in 3 consanguineous families of
Jordanian descent with WFS. The patient developed renal failure,
diabetes mellitus, optic atrophy and high-frequency sensorineural
hearing loss.
A genetic mutation associated with the mitochondria
and WFS has been reported in a single study. Rotig et
al(13) reported a patient who
was diagnosed with type 1 diabetes mellitus at 1 year old. During
her sixth year the individual gradually developed failure to
thrive, cerebellar ataxia, night blindness, progressive external
ophthalmoplegia, extrapyramidal syndrome and mental retardation
with elevated protein levels in the cerebral spinal fluid (1–1.5
g/l; normal, <0.30) and low-density areas in peduncles and
putamen upon nuclear magnetic resonance examination of the brain.
The authors identified a 7.6-kb heteroplasmic deletion of
mitochondrial DNA in the patient. By contrast, in a previous study
of 6 Spanish families with a total of 7 WFS patients, Domenech
et al(14) reported no
mitochondrial DNA abnormalities (15). Genetic defects in mitochondrial DNA
commonly present with several clinical symptoms or a rapidly
progressive course with unexplained association of symptoms
(12,16).
In the family of the current patient, the parents
and brother were heterozygous at c.2411 and did not present with
any symptoms of WFS, however, the patient was homozygous and
revealed a complete phenotype.
The aim of this case study is to draw attention to
the rare disease WFS and highlight the importance of performing a
hearing test and analysis of 24-h urinary output when encountering
juvenile diabetes mellitus patients with optic atrophy. Genetic
tests to identify mutations in WFS1 may also aid diagnosis. In
addition, early diagnosis of WFS is likely to aid rapid prognosis
prediction. Finally, the differentiation of WFS from diabetic
papillopathy and additional diseases caused by trauma,
inflammation, tumor or metastasis is extremely important.
References
|
1
|
Barrett TG, Bundey SE and Macleod AF:
Neurodegeneration and diabetes: UK nationwide study of Wolfram
(DIDMOAD) syndrome. Lancet. 346:1458–1463. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Inukai K, Awata T, Inoue K, et al:
Identification of a novel WFS1 mutation (AFF344-345ins) in Japanese
patients with Wolfram syndrome. Diabetes Res Clin Pract.
69:136–141. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Naderian G, Ashtari F, Nouri-Mahdavi K and
Sajjadi V: A case of wolfram syndrome. J Ophthalmic Vis Res.
5:53–56. 2010.PubMed/NCBI
|
|
4
|
Fabbri LP, Nucera M, Grippo A, et al:
Wolfram syndrome. How much could knowledge challenge the fate? A
case report. Med Sci Monit. 11:CS40–44. 2005.PubMed/NCBI
|
|
5
|
Simsek E, Simsek T, Tekgul S, Hosal S,
Seyrantepe V and Aktan G: Wolfram (DIDMOAD) syndrome: a
multidisciplinary clinical study in nine Turkish patients and
review of the literature. Acta Paediatr. 92:55–61. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Medlej R, Wasson J, Baz P, et al: Diabetes
mellitus and optic atrophy: a study of Wolfram syndrome in the
Lebanese population. J Clin Endocrinol Metab. 89:1656–1661. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Piccoli GB, Mezza E, Jeantet A and
Segoloni GP: An uncommon genetic syndrome with acute renal failure
in a 30-year-old diabetic patient. Nephrol Dial Transplant.
18:206–208. 2003.PubMed/NCBI
|
|
8
|
d’Annunzio G, Minuto N, D’Amato E, et al:
Wolfram syndrome (diabetes insipidus, diabetes, optic atrophy and
deafness): clinical and genetic study. Diabetes Care. 31:1743–1745.
2008.PubMed/NCBI
|
|
9
|
Hardy C, Khanim F, Torres R, et al:
Clinical and molecular genetic analysis of 19 Wolfram syndrome
kindreds demonstrating a wide spectrum of mutations in WFS1. Am J
Hum Genet. 65:1279–1290. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Young TL, Ives E, Lynch E, et al:
Non-syndromic progressive hearing loss DFNA38 is caused by
heterozygous missense mutation in the Wolfram syndrome gene WFS1.
Hum Mol Genet. 10:2509–2514. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cryns K, Sivakumaran TA, Van den Ouweland
JM, et al: Mutational spectrum of the WFS1 gene in Wolfram
syndrome, nonsyndromic hearing impairment, diabetes mellitus and
psychiatric disease. Hum Mutat. 22:275–287. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Amr S, Heisey C, Zhang M, et al: A
homozygous mutation in a novel zinc-finger protein, ERIS, is
responsible for Wolfram syndrome 2. Am J Hum Genet. 81:673–683.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rotig A, Cormier V, Chatelain P, et al:
Deletion of mitochondrial DNA in a case of early-onset diabetes
mellitus, optic atrophy and deafness (Wolfram syndrome, MIM
222300). J Clin Invest. 91:1095–1098. 1993. View Article : Google Scholar
|
|
14
|
Domenech E, Gomez-Zaera M and Nunes V:
Study of the WFS1 gene and mitochondrial DNA in Spanish Wolfram
syndrome families. Clin Genet. 65:463–469. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chaussenot A, Bannwarth S, Rouzier C, et
al: Neurologic features and genotype-phenotype correlation in
Wolfram syndrome. Ann Neurol. 69:501–508. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zalloua PA, Azar ST, Delepine M, et al:
WFS1 mutations are frequent monogenic causes of juvenile-onset
diabetes mellitus in Lebanon. Hum Mol Genet. 17:4012–4021. 2008.
View Article : Google Scholar : PubMed/NCBI
|