Introduction
Acute viral myocarditis (AVMC) is characterized by
virus-triggered myocardial inflammation, followed by an autoimmune
response, and leads to a significant minority of dilated
cardiomyopathy (DCM) cases (1).
Myocarditis causes 4–20% of sudden cardiovascular-associated deaths
among young adults, the military and athletes (2).Viral infections, including
coxsackievirus B3 (CVB3), are the most commonly identified cause of
myocarditis in developed countries and are able to induce
myocarditis and DCM in animal models (2,3). An
excessively activated immune response triggered by the virus was
considered to be the dominant cause of myocyte injury in AVMC,
particularly T-cell activation and the secretion of numerous
proinflammatory/protective cytokines. Cytokine-based therapy has
been considered an important strategy in the treatment of AVMC
(4–6). Recently, identification of the
interleukin (IL)-23/Th17 axis in the pathophysiology of AVMC has
shifted the cytokine paradigm from Th1 to Th17 cytokines and is
mainly focused on IL-17A (5,6).
IL-17A, a cytokine predominantly produced by Th17
cells, was identified as important in the pathogenesis of AVMC. The
critical proinflammatory role of this cytokine and its contribution
to CVB3 replication in AVMC is well documented (5,6).
IL-22, a member of the IL-10 family of cytokines, has received
increasing amounts of attention and is currently widely
investigated. IL-22 is an important cytokine, derived from multiple
cells of the innate and adaptive immune system, including but not
limited to Th22, Th1 and Th17 and CD8+T, γδT and NK
cells (7,8). IL-22 stimulation of IL-22
receptor-expressing cells results in the activation of its
downstream signals, including signal transducers and activator of
transcription 3 (STAT3) signaling pathways, namely the IL-22/STAT3
pathway (8–10). A large number of murine and human
studies have demonstrated that IL-22 provides a unique contribution
to tissue inflammation, immune responses and viral infection and
has proinflammatory and tissue-protective properties, depending on
the context in which it is expressed (8,11–13).
In different stages of disease development, IL-22 has shown
opposing short- and long-term effects (14,15).
The biological activities of IL-22 are complex and this has caused
it to be described as ‘a sheep in wolf’s clothing’ (16). Furthermore, IL-22 is involved in
IL-17-mediated diseases and the signaling pathways of these two
cytokines may synergize to induce the expression of
anti-inflammatory factors (8–16).
More specifically, the proinflammatory properties and
tissue-protective functions of IL-22 were regulated by IL-17A in
airway damage and inflammation (17). These data indicate that the
differential temporal and spatial co-expression of IL-17A and IL-22
may underlie the conflicting results for the biological effects of
IL-22 in distinct disease models. This may offer selective
therapeutic potential in the treatment of IL-17A and
IL-22-associated inflammatory diseases.
IL-22 has been considered an ideal therapeutic
candidate since it specifically affects tissue responses without
having direct effects on the immune response (8). Our previous study conducted on
wild-type (WT) mice demonstrated the existence of a specific
pattern of IL-22 cytokines in CVB3-induced AVMC (18). Furthermore, neutralization of IL-22
in vivo exacerbated the severity of AVMC and promoted
cardiac viral replication, which highlights the anti-inflammatory
and antiviral roles of IL-22 in the pathogenesis of myocarditis in
the presence of IL-17A. However, whether IL-17A contributes to the
tissue-protective role of IL-22 in the development of AVMC remains
to be elucidated. The present study, in continuation of our
previous investigations, aimed to determine whether IL-22 plays a
distinctly different role in the absence of IL-17A in AVMC by using
IL-17A-deficient mice (IL-17A−/−).
Materials and methods
Mice
Specific pathogen-free male IL-17A-deficient mice on
the BALB/c background (designated in the present study as
IL-17A−/−), aged 6 weeks, were provided by Dr Yoichiro
Iwakura (Center for Experimental Medicine and Systems Biology, the
Institute of Medical Science, the University of Tokyo, Tokyo,
Japan) (19). All animals were
kept in a pathogen-free mouse room in the Experimental Animal
Center (Guangxi Medical University, Nanning, China). Experiments
were carried out in accordance with protocols approved by the
Guangxi Medical University Animal Ethics Committee.
Virus
CVB3 (Nancy strain from the Institute of Immunology
of Guangxi Medical University) was maintained by passage through
HEp-2 cells. A plaque-forming unit (PFU) assay was used to
determine that the virus titer was 1×108. CVB3 was
diluted in PBS (Solarbio Science & Technology Co., Ltd.,
Beijing, China). BALB/c mice were infected by an intraperitoneal
injection (i.p.) of 100 μl PBS containing ~106 PFU CVB3
to establish the AVMC models.
Neutralization of IL-22 in
IL-17A−/− mice
For in vivo IL-22 neutralization, a total of
32 IL-17A−/− mice were randomly divided into four
groups: Mice in the AVMC group were injected with CVB3 and PBS (50
μg per mouse, n=8); in the anti-IL-22 Ab group, mice were
administered with CVB3 and anti-IL-22 Ab (AF582; 50 μg per mouse;
R&D Systems, Inc., Minneapolis, MN, USA; n=8); IgG control
group, mice were injected with CVB3 and normal IgG control
(AB-108-C; 50 μg per mouse; R&D Systems; n=8); and in the
normal group, IL-17A−/− mice received no treatments
(n=8). The day of intraperitoneal injection was defined as day 0.
All surviving animals were sacrificed on day 14 after CVB3
infection. The ratios for heart weight/body weight (HW/BW) were
recorded. Blood was collected and the serum was prepared for
analysis. Hearts and spleens were removed aseptically as fresh
specimens for analysis.
Histopathology
The ventricular tissues of the hearts were fixed in
10% formalin and embedded in paraffin. After the entire length of
the heart was sectioned (5 μm) and stained with hematoxylin and
eosin (H&E), histopathological changes were observed using
light microscopy (Nikon Eclipse E800 Microscope, Kawasaki,
Kanagawa, Japan). The pathological scores of heart tissues were
graded as follows: a score of 0 was assigned for no inflammatory
infiltrates, 1 for a small foci of inflammatory cells between
myocytes or inflammatory cells surrounding individual myocytes, 2
for a larger foci of 100 inflammatory cells or involving at least
30 myocytes, 3 when 10% of a myocardial cross-section was involved
or 4 if 30% of a myocardial cross-section was involved (20). The assessment was separately scored
by two independent pathologists in a blinded manner.
Preparation of spleen lymphocytes and
cell cultures
Spleens from AVMC mice were collected aseptically.
After mincing, splenic cells were gently dispersed through a nylon
mesh into a single-cell suspension. The lymphocyte fractions of the
splenic mononuclear cell suspensions were obtained by Ficoll-Paque
(Solarbio Science & Technology Co., Ltd.) gradient
centrifugation and washed twice with PBS. Lymphocytes from each
spleen were divided into 2 groups and cultured in a total volume of
300 μl for 48 h in triplicate wells (5×105 cells/well)
of a 96-well culture plate, in RPMI-1640 supplemented with
penicillin (100 U/ml), streptomycin (100 μg/ml), 10% fetal bovine
serum (Gibco-BRL, Carlsbad, CA, USA) and phytohaemafflutinin (PHA;
10 μg/ml; Sigma-Aldrich, St. Louis, MO, USA) at 37°C and 5%
CO2, in the absence or presence of the recombinant mouse
IL-17 (rIL-17; 25 ng/ml; R&D Systems). Cells were collected and
used in real-time polymerase chain reaction (RT-PCR), while the
culture supernatants were assayed by ELISA.
Plaque-forming assay
Viral titers were determined using a standard plaque
formation assay and results were expressed per organ weight (g). A
section of the heart tissue was weighed and homogenized in 2 ml
PBS. The supernatant was absorbed and sequentially diluted 10-fold
in RPMI-1640 medium after three freeze-thaw cycles and
centrifugation at 500 × g for 10 min. The HeLa cell monolayers were
incubated with the supernatant for 1 h at 37°C and 5%
CO2 in six-well plates, washed in PBS and covered with 2
ml of 0.4% agar, DMEM and 5% FCS. The monolayers were fixed in
paraformaldehyde and stained in crystal violet after cultivation
for 72 h and the number of plaques were counted.
RT-PCR
The total RNA of homogenized heart tissue and
cellular RNA of the cultured lymphocytes was extracted with
TRIzol® reagent (Invitrogen, Carlsbad, CA, USA). Reverse
transcription into cDNA was carried out using a Reverse
Transcription kit (Fermentas, Vilnius, Lithuania), according to the
manufacturer’s instructions. RT-PCR was performed with an ABI 7500
Sequence Detection System using SYBR-Green (Applied Biosystems,
Foster City, CA, USA). An initial denaturation step for 3 min at
94°C, 35 cycles of denaturation at 94°C for 30 sec, annealing at
60°C for 30 sec and extension at 72°C for 60 sec were carried out.
The relative gene expression levels were normalized to the level of
β-actin transcripts and quantified using the ΔΔCT method. Each
reaction was carried out in triplicate. Primers for IL-22, IFN-γ,
TNF-α, IL-6, CVB3, STAT3 and the housekeeping gene β-actin were
designed by Primer Premier 5.0 (Table
I).
 | Table IPrimer sequences for real-time
RT-PCR. |
Table I
Primer sequences for real-time
RT-PCR.
| Molecule | Sequence
(5′-3′) |
|---|
| TNF-α | Sense:
AGTCCGGGCAGGTCTACTTT |
| Anti-sense:
TTGGACCCTGAGCCATAATC |
| IL-6 | Sense:
ACAGAAGGAGTGGCTAAGGACC |
| Anti-sense:
TAGGCATAACGCACTAGGTTT |
| IL-22 | Sense:
CGATTGGGGAACTGGACCTG |
| Anti-sense:
GGACGTTAGCTTCTCACTTT |
| CVB3 | Sense:
CGGTACCTTTGTGCGCCTGT |
| Anti-sense:
CAGGCCGCCAACGCAGCC |
| IFN-γ | Sense:
CTCAAGTGGCATAGATGTGGAAG |
| Anti-sense:
GCTGGACCTGTGGGTTGTTGA |
| STAT3 | Sense:
CCCATATCGTCTGAAACTC |
| Anti-sense:
TTGCTCCCTTCTGCTCT |
| β-actin | Sense:
AATTCCATCATGAAGTGTGA |
| Anti-sense:
ACTCCTGCTTGCTGATCCAC |
Western blot analysis
The total proteins were extracted according to the
manufacturer’s instructions. Protein concentrations were determined
using a BCA protein assay kit (Beyotime Institute of Biotechnology,
Shanghai, China). Equal amounts of denatured sample protein (50 μg)
were separated in 10% SDS-PAGE gel and transferred onto a
nitrocellulose membrane. After blocking with 6% non-fat dry milk in
Tris-buffered saline containing 0.1% Tween-20, the membranes were
treated with anti-phospho-STAT3 (p-Y705-STAT3; 1:12,000; Abcam,
Cambridge, MA, USA) and glyceraldehyde phosphate dehydrogenase
(GAPDH; 1:10,000; Kang Chen Bio-tech, Shanghai, China) antibodies
overnight at 4°C and exposed to the corresponding HRP-conjugated
goat anti-rabbit IgG (1:5,000; Santa Cruz Biotechnology, Inc.,
Santa Cruz, CA, USA) for 1 h at room temperature. Labeled bands
were detected using an enhanced chemiluminescence detection kit
(BestBio Inc., Shanghai, China) and analyzed using the Image Lab
2.0 analysis system (Bio-Rad Laboratory, Hercules, CA, USA). The
intensity of phosphor-STAT3 (p-STAT3) bands was normalized to the
levels of GAPDH.
Cytokine assay
ELISA was performed to determine the cytokine
content in serum or cell culture supernatants. The Quantikine Mouse
IL-22 Immunoassay (Cat. No. 436307, Biolegend, San Diego, CA, USA)
was used to detect the levels of IL-22. The minimal detectable
concentration was 5 pg/ml. No cross-reactivity was observed during
detection. Samples were measured in triplicate.
Statistical analysis
Data were expressed as the mean ± standard deviation
(SD). For statistical analysis, differences between the mean values
were tested by Student’s t-test or ANOVA, using SPSS 17.0.
Correlations were determined by Spearman’s rank correlation
coefficients. The Mann-Whitney U test was carried out for the
differences between pathological scores. P<0.05 was considered
to indicate a statistically significant difference.
Results
Neutralization of IL-22 in
IL-17A−/− mice alleviated the severity of
myocarditis
No mice died in the normal, AVMC, anti-IL-22 Ab or
IgG control groups until day 14. With the exception of the normal
group, inflammatory cell infiltration or necrotic lesions and
visible clinical signs of myocarditis were observed in the AVMC,
anti-IL-22 Ab and IgG control groups (Fig. 1A and B). Data showed that injection
of IL-22 neutralizing antibodies alleviated the severity of
myocarditis. The pathological scores of heart sections from mice
receiving anti-IL-22 Ab (1.48±0.18) were lower than those in mice
in the AVMC (2.12±0.37) and IgG control (2.08±0.29) groups (all
P<0.01). Compared with the AVMC and IgG control groups,
significantly decreased values for HW/BW were observed in the
anti-IL-22 Ab group (Fig. 1C; all
P<0.01). With regard to the pathological scores and ratios of
HW/BW, no significant difference was observed between the AVMC and
IgG control groups (P>0.05).
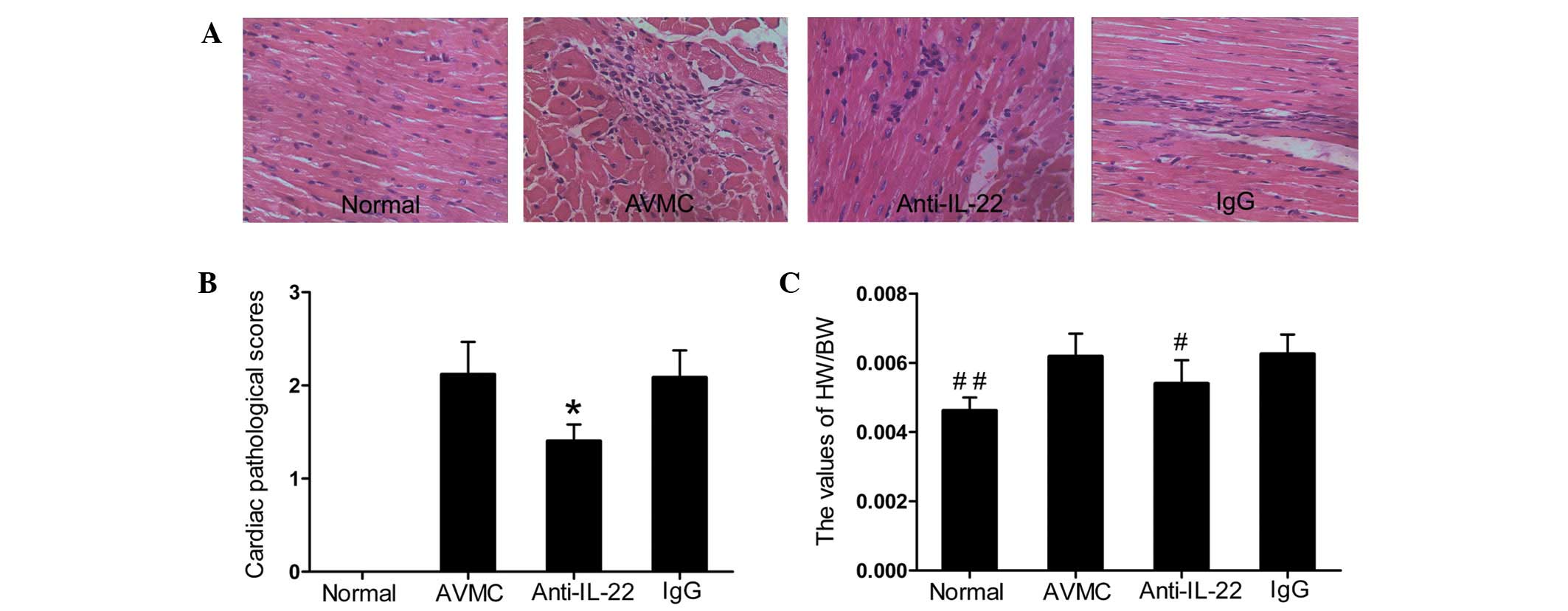 | Figure 1Neutralization of IL-22 in
IL-17A−/− mice alleviated the severity of myocarditis.
(A) Representative histopathological images in heart tissues
(H&E, original magnification ×400). (B) The pathological scores
in the normal, AVMC, anti-IL-22 Ab and IgG control groups.
*P<0.01, versus normal, AVMC and IgG groups. (C) The
ratio of HW/BW in different groups. Eight mice in each group were
analyzed. #P<0.05 versus the normal, AVMC and IgG
groups. ##P<0.05 versus the AVMC, anti-IL-22 and IgG
groups. IL, interleukin; AVMC, acute viral myocarditis; H&E,
hematoxylin and eosin; HW/BW, heart weight/body weight. |
Neutralization of IL-22 in
IL-17A−/− mice promoted viral replication
On day 14, the levels of cardiac CVB3 titers (values
expressed in PFU/g) were 0 in the normal,
(9.17±2.22)x103 in the AVMC, (1.08±0.15)x105
in the anti-IL-22 Ab and (9.13±2.30)x103 in the IgG
control groups. Data revealed that the levels of cardiac viral
titers were elevated significantly in the anti-IL-22 Ab group
compared with the normal, AVMC and IgG control groups (all
P<0.01). At the same time-point, the relative expression levels
of cardiac CVB3 RNA in the anti-IL-22 Ab group were higher than
those in the normal, AVMC and IgG control groups (all P<0.01).
Cardiac CVB3 RNA was not detected in the normal group. Differences
in CVB3 titers and cardiac CVB3 RNA expression levels between the
AVMC and IgG control groups were not statistically significant
(Fig. 2A and B).
 | Figure 2Neutralization of IL-22 in
IL-17A−/− mice promoted viral replication and decreased
the production of TNF-α, IL-6 and IFN-γ. (A) The levels of cardiac
CVB3 titers on day 14. Data show the mean values of CVB3 PFU/g of
heart. (B) The relative expression levels of cardiac CVB3 RNA in
different groups. (C-E) The result of statistical analysis for the
alterations in cardiac TNF-α, IL-6 and IFN-γ mRNA, as measured by
RT-PCR. (F) The correlation analysis of cardiac CVB3 RNA, cardiac
IL-22 mRNA and IFN-γ mRNA. Each point represents an individual
mouse. *P<0.01 and #P<0.05 versus the
normal, AVMC and IgG groups. **P<0.01 and
##P<0.05 versus the AVMC, anti-IL-22 and IgG groups.
Values are presented as the mean ± SD. IL, interleukin; CVB3,
coxsackievirus B3; RT-PCR, real-time polymerase chain reaction;
AVMC, acute viral myocarditis; SD, standard deviation. |
Neutralization of IL-22 in
IL-17A−/− mice decreased the production of TNF-α, IL-6
and IFN-γ
Compared with the normal group, an increased
production of TNF-α, IL-6 and IFN-γ was observed in the AVMC, IgG
control and anti-IL-22 Ab groups (Fig.
2C-E). Compared with the AVMC and IgG control groups, lower
expression levels of cardiac TNF-α, IL-6 and IFN-γ mRNA were
observed in the anti-IL-22 Ab group (all P<0.05) on day 14
postinjection. With regard to the levels of TNF-α, IL-6 and IFN-γ
genes, no significant changes were observed between the AVMC and
IgG control groups (all P>0.05). It was determined that the
cardiac IL-22 mRNA was correlated negatively with the CVB3 RNA
(R=−0.805, P<0.01; Fig.
2F).
Neutralization of IL-22 in
IL-17A−/− mice decreased the levels of IL-22 and
STAT3
Compared with the normal group, an increased
production of serum IL-22 and cardiac IL-22 mRNA was observed in
the AVMC, anti-IL-22 Ab and IgG control groups (all P<0.01;
Fig. 3A and B). However, compared
with the AVMC and IgG control groups, anti-IL-22 Ab markedly
decreased the expression levels of circulating IL-22 and cardiac
IL-22 (all P<0.05) on day 14 postinjection. A negative
correlation was established between cardiac IL-22 mRNA and CVB3 RNA
(R=−0.825, P<0.01; Fig. 3C).
Furthermore, decreased expression levels of cardiac STAT3 mRNA and
p-STAT3, analyzed by RT-PCR and western blot analysis,
respectively, were observed in the anti-IL-22 Ab group (all
P<0.05; Fig. 3D-F). For IL-22
and STAT3, no significant difference was observed between the AVMC
and IgG control groups.
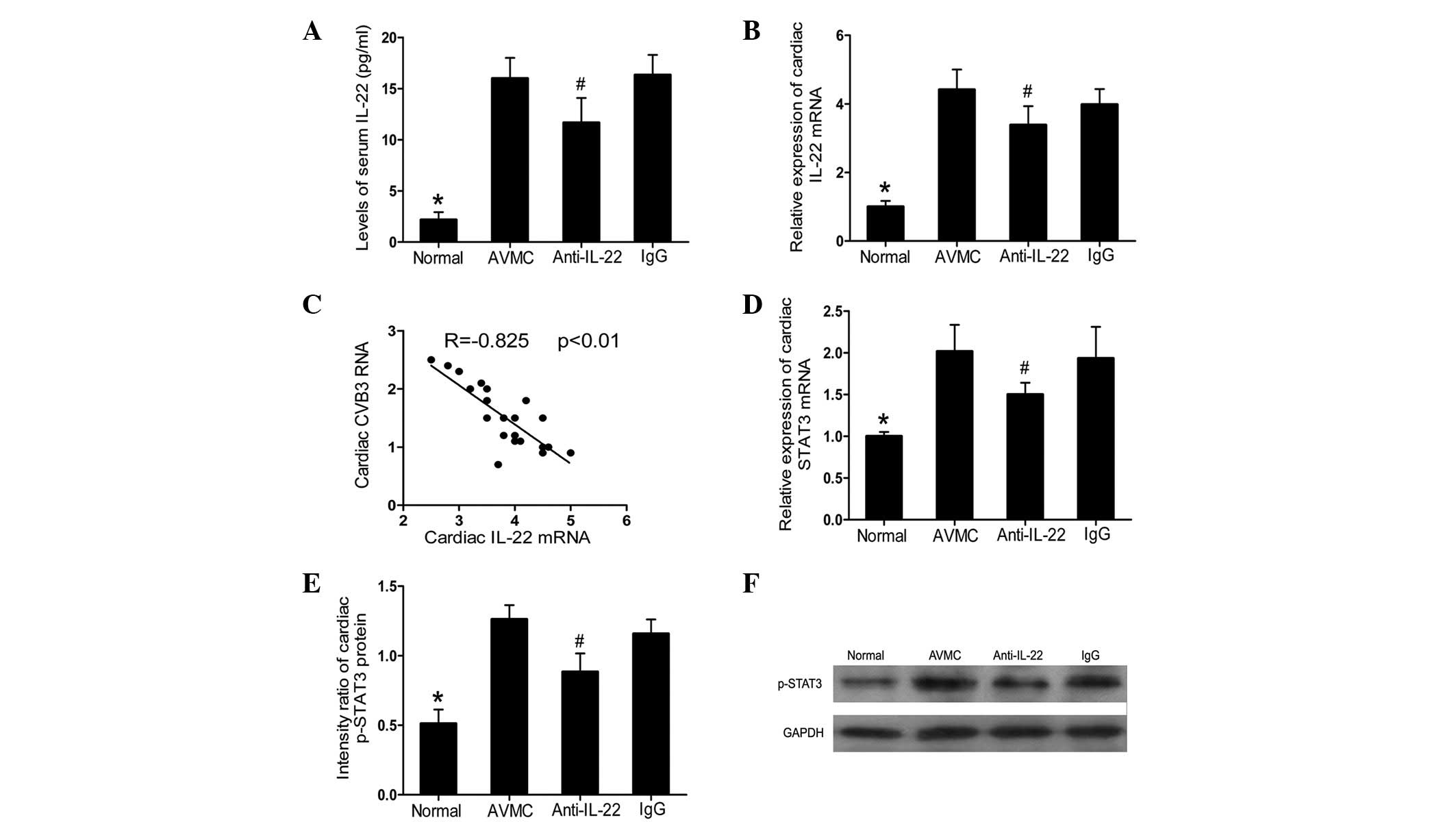 | Figure 3Neutralization of IL-22 in
IL-17A−/− mice decreased the levels of IL-22 and STAT3.
(A) Results of the statistical analysis for the alteration of serum
IL-22 protein levels, as investigated by ELISA. (B) The relative
cardiac expression levels of IL-22, as analyzed by RT-PCR. (C) The
correlation analysis of cardiac CVB3 RNA and cardiac IL-22 mRNA. (D
and E) Results of the statistical analysis for the levels of
cardiac STAT3 mRNA and STAT3 proteins, as measured by RT-PCR and
western blot analysis, respectively. (F) Representative images for
the levels of cardiac STAT3 proteins from each treated group.
#P<0.05 versus the normal, AVMC and IgG groups.
*P<0.01 versus the AVMC, anti-IL-22 and IgG groups.
Values are presented as the mean ± SD. IL, interleukin; STAT3,
activator of transcription 3; RT-PCR, real-time polymerase chain
reaction; CVB3, coxsackievirus B3; SD, standard deviation; GAPDH,
glyceraldehyde phosphate dehydrogenase. |
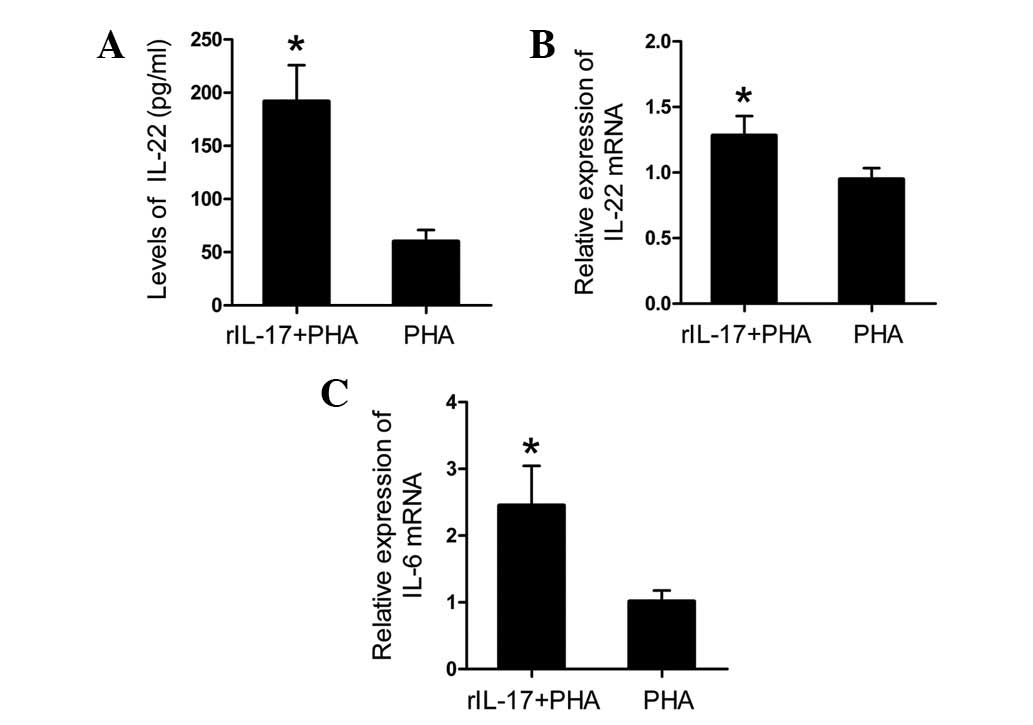
rIL-17 increased the production of IL-22
and IL-6
To assess the effect of IL-17 on IL-22 production,
spleen lymphocytes of AVMC from IL-17A−/− mice were
incubated for 24 h with PHA and 25 ng/ml rIL-17 (R&D Systems).
Compared with those cultured with PHA only, data showed that the
addition of rIL-17A to spleen lymphocyte cultures resulted in a
significantly increased level of the IL-22 protein in culture
supernatants and an upregulation of IL-22 mRNA in spleen
lymphocytes (Fig. 4A and B; all
P<0.01). Furthermore, a significant increase in the mRNA
expression levels of IL-6 was observed in the rIL-17A + PHA group
in comparison with the PHA only group (Fig. 4C; P<0.01).
Discussion
The secretion of numerous proinflammatory or
protective cytokines is important in the pathogenesis of AVMC
(4–6). Our previous study demonstrated that
IL-17A and IL-22 responses were developed in CVB3-induced AVMC and
characterized the myocardium-protective and antiviral roles of
IL-22 in the presence of IL-17A (18). Enhanced co-expression of these two
cytokines suggested that they may have a functional interaction.
Studies conducted on the mouse model of intestinal intracellular
parasites showed that the concurrent neutralization of IL-17A and
IL-22 resulted in a reduction in infection-induced body weight
loss, while the neutralization of IL-22 alone had no effect on body
weight loss (21). Furthermore,
Liang et al and Aujla et al observed synergy between
IL-22 and IL-17A after infection with the pulmonary pathogen K.
pneumonia, which promoted the production of inflammatory
mediators and antimicrobial peptides (22) and contributed to the host defense
after pulmonary infection (23).
However, administration of exogenous IL-22 alone was not enough to
promote neutrophil recruitment to the airway (24). On the basis of these results, it is
possible that IL-17A contributes to the tissue-protective and
antiviral properties of IL-22 in AVMC, therefore, we carried out
the present study using IL-17A knockout mice.
Notably, in the present study, neutralization of
IL-22 in the absence of IL-17A improved measures of the severity of
AVMC, which was verified by lower values of HW/BW and pathological
scores of heart sections and the decreased production of cardiac
proinflammatory cytokines IL-6 and TNF-α. These results and those
from our previous studies (18)
indicate that IL-22 appears to confer anti-inflammatory properties
in a murine model of AVMC in the presence of IL-17A, whereas in the
absence of IL-17A, IL-22 was no longer protective and instead
conferred a proinflammatory and pathological outcome. By contrast,
previous observations have shown that bleomycin induces acute
tissue damage and airway inflammation (17), which indicates that IL-17A and
IL-22 acted synergistically to promote inflammation, while IL-22
had tissue-protective functions in the absence of IL-17A. Our
experiment demonstrates opposite data, where IL-17A prevented the
proinflammatory properties and promoted the tissue-protective
functions of IL-22, adding another layer of complexity to these
cytokines. The first possible explanation for these observations
may be that IL-17R and IL-22R expression are not overlapped. IL-17R
is expressed in the tissues and cells of immune system (25,26)
and IL-17 activates T cells and other immune cells to produce a
variety of cytokines, chemokines and cell adhesion molecules
(27). By contrast, IL-22R is
expressed in the tissues but is absent from cells of a hematopoetic
origin. Thus, IL-22 acts as a middle-man between the immune system
and its environment (8). The
presence or absence of direct immune response activation by IL-17
may determine whether IL-22 alleviates or exacerbates the severity
of AVMC. Secondly, STAT3, the important downstream signal protein
of IL-22 (8–10), regulates a wide range of biological
processes (28). In addition to
our published data (6), the
detection of enhanced levels of IL-22 and STAT3 in mice infected
with CVB3 and the blockade of IL-22 causing a decrease in the
amount of STAT3 in the present study suggests the importance of the
IL-22/STAT3 pathway in the pathogenesis of AVMC. A number of
studies have demonstrated the complex interactions between STAT3
and the downstream signal molecules of IL-17, including NF-κB
(29). This interplay between
IL-17A and IL-22 signaling pathways may regulate the production of
inflammatory cytokines, including IL-6 and TNF-α, to determine the
balance between pathological versus tissue-protective outcomes.
Therefore, although further investigation is required, cytokine
receptors and the signaling pathways of IL-17A and IL-22 may affect
the functional properties of IL-22 and provide possible
explanations for the distinct roles of IL-22 in murine models of
AVMC.
Notably, cardiac CVB3 RNA was correlated negatively
with the cardiac IFN-γ and IL-22 mRNA and the upregulation of viral
replication was observed in the anti-IL-22 Ab group. Additionally,
we identified a lower amount of cardiac IFN-γ, the vital Th1 cell
cytokine, in the anti-IL-22 Ab group, which is consistent with our
earlier observation that the number of IL-22-producing Th22 cells
was correlated positively with the number of IFN-γ-producing Th1
cells in mice infected with CVB3 (18). Th1 responses have been demonstrated
to protect against viral replication and prevent chronic
myocarditis/DCM and increase acute inflammation, particularly in
males (30). Thus, the decreased
production of the IFN-γ gene, which may be modulated by the
IL-22/STAT3 pathway, is a possible explanation for our observation
that the enhancement of viral replication is followed by a decrease
in acute cardiac inflammation, rather than an increase in
myocarditis. These results also suggest that cardiac inflammation
is not positively correlated with viral replication, since either
virus-specific or immune responses also contribute to myocarditis.
For instance, T-cell-deficient mice develop minimal cardiac injury
despite high virus titers in the heart (30,31).
Together with our earlier studies, results of the present study
indicate that IL-22 has an important antiviral function in AVMC
microenvironmental conditions, independent of IL-17A.
Our published studies have demonstrated a positive
correlation between IL-22-producing Th22 and IL-17-producing Th17
cells in WT mice (18). With
regard to the serum levels of IL-22 in mice infected with CVB3, we
observed lower amounts of IL-22 in IL-17A−/− mice
compared with WT mice (18). Data
indicated that IL-17 may contribute to the production of IL-22 in
AVMC in vivo. Therefore, we investigated the contribution of
IL-17 to IL-22 in vitro and demonstrated that rIL-17 was
able to enhance the mRNA expression and protein secretion of IL-22
in spleen lymphocytes, in addition to IL-6. The main explanation
for this observation was that IL-17 induces the expression of IL-6,
which is important for the differentiation and proliferation of
IL-22-producing Th22 cells (32).
Pertinent to our data, previous studies have observed increased
IL-22 mRNA levels in the absence of IL-17A in mouse models of
colitis (33) or decreased IL-22
mRNA levels in splenocyte cultures with the addition of exogenous
IL-17A (34,35). This discrepancy may be explained by
a disease-specific difference in the role of IL-17A in regulating
the production of IL-22.
A number of limitations should be noted in the
present study. Firstly, the biological activity, affinity and/or
potency of IL-22 antibodies in vivo are uncertain. The
administered dose of anti-IL-22 Ab may not be able to completely
antagonize the circulating IL-22 in vivo. Secondly, the
exact mechanisms by which IL-17A regulates the proinflammatory or
anti-inflammatory properties of IL-22 in AVMC remain unclear.
Further studies with regard to this may be conducted in the future
to clarify the complicated interactions among their downstream
signaling pathways.
In conclusion, the present study furthers our
previous findings and provides preliminary evidence for
demonstrating the critical pathological and antiviral roles of
IL-22 in AVMC, in the absence of IL-17A. To the best of our
knowledge, in combination with our previous studies, our data
provide the first demonstration that IL-17A is unable to govern the
antiviral role of IL-22, although it is able to regulate the
proinflammatory or tissue-protective properties and expression
levels of IL-22, thereby determining the functional consequences of
IL-22 in the pathogenesis of CVB3-induced AVMC. Our observation
indicates that elucidation of the roles of IL-22 and the mechanisms
of local cytokine dependency, including IL-17A, is likely to aid
the understanding of the pathophysiology of IL-22-associated
infections, inflammation and immune responses.
Acknowledgements
The study described in this manuscript was supported
by grants from the National Natural Science Foundations of China
(nos. 30960129 and 81260045). We would like to thank Dr Jiao Lan,
Zoujiu Liang and Qiguang Huang for their technical assistance.
References
|
1
|
Dennert R, Crijns HJ and Heymans S: Acute
viral myocarditis. Eur Heart J. 29:2073–2082. 2008. View Article : Google Scholar
|
|
2
|
Gupta S, Markham DW, Drazner MH and Mammen
PP: Fulminant myocarditis. Nat Clin Pract Cardiovasc Med.
5:693–706. 2008. View Article : Google Scholar
|
|
3
|
Cooper LT Jr: Myocarditis. N Engl J Med.
360:1526–1538. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fairweather D and Rose NR:
Coxsackievirus-induced myocarditis in mice: a model of autoimmune
disease for studying immunotoxicity. Methods. 41:118–122. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yuan J, Yu M, Lin QW, Cao AL, Yu X, Dong
JH, Wang JP, Zhang JH, Wang M, Guo HP, Cheng X and Liao YH: Th17
cells contribute to viral replication in coxsackievirus B3-induced
acute viral myocarditis. J Immunol. 185:4004–4010. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yang F, Wu WF, Yan YL, Pang Y, Kong Q and
Huang YL: Expression of IL-23/Th17 pathway in a murine model of
Coxsackie virus B3-induced viral myocarditis. Virol J. 8:3012011.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Trifari S, Kaplan CD, Tran EH, Crellin NK
and Spits H: Identification of a human helper T cell population
that has abundant production of interleukin 22 and is distinct from
T(H)-17, T(H)1 and T(H)2 cells. Nat Immunol. 10:864–871. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sonnenberg GF, Fouser LA and Artis D:
Border patrol: regulation of immunity, inflammation and tissue
homeostasis at barrier surfaces by IL-22. Nat Immunol. 12:383–390.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Radaeva S, Sun R, Pan HN, Hong F and Gao
B: Interleukin 22 (IL-22) plays a protective role in T
cell-mediated murine hepatitis: IL-22 is a survival factor for
hepatocytes via STAT3 activation. Hepatology. 39:1332–1342. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ziesché E, Bachmann M, Kleinert H,
Pfeilschifter J and Mühl H: The interleukin-22/STAT3 pathway
potentiates expression of inducible nitric-oxide synthase in human
colon carcinoma cells. J Biol Chem. 282:16006–16015.
2007.PubMed/NCBI
|
|
11
|
Takahashi K, Hirose K, Kawashima S, Niwa
Y, Wakashin H, Iwata A, Tokoyoda K, Renauld JC, Iwamoto I, Nakayama
T and Nakajima H: IL-22 attenuates IL-25 production by lung
epithelial cells and inhibits antigen-induced eosinophilic airway
inflammation. J Allergy Clin Immunol. 128:1067–1076. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang P, Bai F, Zenewicz LA, Dai J, Gate D,
Cheng G, Yang L, Qian F, Yuan X, Montgomery RR, Flavell RA, Town T
and Fikrig E: IL-22 signaling contributes to West Nile encephalitis
pathogenesis. PLoS One. 7:e441532012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Missé D, Yssel H, Trabattoni D, Oblet C,
Lo Caputo S, Mazzotta F, Pène J, Gonzalez JP, Clerici M and Veas F:
IL-22 participates in an innate anti-HIV-1 host-resistance network
through acute-phase protein induction. J Immunol. 178:407–415.
2007.PubMed/NCBI
|
|
14
|
Zenewicz LA, Yancopoulos GD, Valenzuela
DM, Murphy AJ, Karow M and Flavell RA: Interleukin-22 but not
interleukin-17 provides protection to hepatocytes during acute
liver inflammation. Immunity. 27:647–659. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Huber S, Gagliani N, Zenewicz LA, Huber
FJ, Bosurgi L, Hu B, Hedl M, Zhang W, O’Connor W Jr, Murphy AJ,
Valenzuela DM, Yancopoulos GD, Booth CJ, Cho JH, Ouyang W, Abraham
C and Flavell RA: IL-22BP is regulated by the inflammasome and
modulates tumorigenesis in the intestine. Nature. 491:259–263.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Laurence A, O’Shea JJ and Watford WT:
Interleukin-22: a sheep in wolf’s clothing. Nat Med. 14:247–249.
2008.
|
|
17
|
Sonnenberg GF, Nair MG, Kirn TJ, Zaph C,
Fouser LA and Artis D: Pathological versus protective functions of
IL-22 in airway inflammation are regulated by IL-17A. J Exp Med.
207:1293–1305. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kong Q, Wu W, Yang F, Liu Y, Xue Y, Gao M,
Lai W, Pan X, Yan Y, Pang Y and Deng Y: Increased expressions of
IL-22 and Th22 cells in the coxsackievirus B3-Induced mice acute
viral myocarditis. Virol J. 9:2322012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nakae S, Komiyama Y, Nambu A, Sudo K,
Iwase M, Homma I, Sekikawa K, Asano M and Iwakura Y:
Antigen-specific T cell sensitization is impaired in
IL-17-deficient mice, causing suppression of allergic cellular and
humoral responses. Immunity. 17:375–387. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Eriksson U, Ricci R, Hunziker L, Kurrer
MO, Oudit GY, Watts TH, Sonderegger I, Bachmaier K, Kopf M and
Penninger JM: Dendritic cell-induced autoimmune heart failure
requires cooperation between adaptive and innate immunity. Nat Med.
9:1484–1490. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Stange J, Hepworth MR, Rausch S, Zajic L,
Kühl AA, Uyttenhove C, Renauld JC, Hartmann S and Lucius R: IL-22
mediates host defense against an intestinal intracellular parasite
in the absence of IFN-γ at the cost of Th17-driven immunopathology.
J Immunol. 188:2410–2418. 2012.PubMed/NCBI
|
|
22
|
Liang SC, Tan XY, Luxenberg DP, Karim R,
Dunussi-Joannopoulos K, Collins M and Fouser LA: Interleukin
(IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively
enhance expression of antimicrobial peptides. J Exp Med.
203:2271–2279. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Aujla SJ, Chan YR, Zheng M, Fei M, Askew
DJ, Pociask DA, Reinhart TA, McAllister F, Edeal J, Gaus K, Husain
S, Kreindler JL, Dubin PJ, Pilewski JM, Myerburg MM, Mason CA,
Iwakura Y and Kolls JK: IL-22 mediates mucosal host defense against
Gram-negative bacterial pneumonia. Nat Med. 14:275–281. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liang SC, Long AJ, Bennett F, Whitters MJ,
Karim R, Collins M, Goldman SJ, Dunussi-Joannopoulos K, Williams
CM, Wright JF and Fouser LA: An IL-17F/A heterodimer protein is
produced by mouse Th17 cells and induces airway neutrophil
recruitment. J Immunol. 179:7791–7799. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yao Z, Fanslow WC, Seldin MF, Rousseau AM,
Painter SL, Comeau MR, Cohen JI and Spriggs MK: Herpesvirus Saimiri
encodes a new cytokine, IL-17, which binds to a novel cytokine
receptor. Immunity. 3:811–821. 1995. View Article : Google Scholar
|
|
26
|
Gaffen SL: Structure and signalling in the
IL-17 receptor family. Nat Rev Immunol. 9:556–567. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Komiyama Y, Nakae S, Matsuki T, Nambu A,
Ishigame H, Kakuta S, Sudo K and Iwakura Y: IL-17 plays an
important role in the development of experimental autoimmune
encephalomyelitis. J Immunol. 177:566–573. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dauer DJ, Ferraro B, Song L, Yu B, Mora L,
Buettner R, Enkemann S, Jove R and Haura EB: Stat3 regulates genes
common to both wound healing and cancer. Oncogene. 24:3397–3408.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bollrath J and Greten FR: IKK/NF-kappaB
and STAT3 pathways: central signalling hubs in
inflammation-mediated tumour promotion and metastasis. EMBO Rep.
10:1314–1319. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fairweather D, Stafford KA and Sung YK:
Update on coxsackievirus B3 myocarditis. Curr Opin Rheumatol.
24:401–407. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Woodruff JF and Woodruff JJ: Involvement
of T lymphocytes in the pathogenesis of coxsackievirus B3 heart
disease. J Immunol. 113:1726–1734. 1974.PubMed/NCBI
|
|
32
|
Duhen T, Geiger R, Jarrossay D,
Lanzavecchia A and Sallusto F: Production of interleukin 22 but not
interleukin 17 by a subset of human skin-homing memory T cells. Nat
Immunol. 10:857–863. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
O’Connor W Jr, Kamanaka M, Booth CJ, Town
T, Nakae S, Iwakura Y, Kolls JK and Flavell RA: A protective
function for interleukin 17A in T cell-mediated intestinal
inflammation. Nat Immunol. 10:603–609. 2009.PubMed/NCBI
|
|
34
|
Smith E, Stark MA, Zarbock A, Burcin TL,
Bruce AC, Vaswani D, Foley P and Ley K: IL-17A inhibits the
expansion of IL-17A-producing T cells in mice through ‘short-loop’
inhibition via IL-17 receptor. J Immunol. 181:1357–1364. 2008.
|
|
35
|
von Vietinghoff S and Ley K: IL-17A
controls IL-17F production and maintains blood neutrophil counts in
mice. J Immunol. 183:865–873. 2009.PubMed/NCBI
|