Introduction
A chronic imbalance between energy intake and
expenditure leads to obesity, which is characterized by an
increased number and size of adipocytes. As an active endocrine
organ, adipose tissue is part of an innate immune system capable of
producing biologically active proteins referred to as adipokines,
including leptin, tumor necrosis factor-α (TNF-α), interleukin-6
(IL-6), resistin and adiponectin. Adipocyte-derived peptides are
reciprocally reflected by energy homeostasis. Energy balance and
body weight regulation affect the response and activities of
adipose tissue by regulating adipokine production and secretion,
all of which contribute to obesity-associated comorbidities and
mortality (1–4).
The first adipose-derived proinflammatory mediator,
TNF-α, is well established as a major inflammatory adipokine that
has been associated with adiposity, obesity, inflammation and the
development of insulin resistance (4–6). The
deleterious effects of TNF-α on insulin sensitivity result from its
effects on lipolysis (7),
adipocyte metabolism (8) and the
insulin signaling network (9,10).
By contrast to TNF-α, another adipose-secreted protein,
adiponectin, is an established insulin-sensitizing adipokine with
anti-inflammatory and anti-atherogenic properties (11,12).
Circulating levels of adiponectin and its synthesis in adipose
tissue are decreased in patients and animals with obesity and type
2 diabetes (13,14). In terms of the regulation of
adiponectin, in vitro and in vivo studies have
demonstrated the involvement of proinflammatory cytokines (15,16).
TNF-α directly inhibits adiponectin gene expression; however, this
effect is reversed following stimulus removal (17). The mechanisms by which exposure and
removal of obesity-inducing proinflammatory adipokines affect
adiponectin remain to be fully elucidated.
Extensive studies have revealed that obesity is a
state of chronic, low-grade inflammation, which may act as the
potential link between adipose tissue expansion and obesity-induced
health complications (18,19). Proteins produced and secreted from
adipose tissue fluctuate in response to energy balance, including
weight change or adiposity size, which in turn reflects health
outcome (20–22). However, to the best of our
knowledge, the mechanisms by which metabolic stress from chronic
energy imbalance increases proinflammatory cytokines and affects
adiponectin remain elusive. In the present study, the mechanisms
underlying the adverse effects of TNF-α on adiponectin were
examined and the recovery time required to reverse the
TNF-α-induced decrease in adiponectin mRNA expression was
determined.
Materials and methods
Materials
3T3-L1 mouse fibroblast cells were obtained from the
American Type Culture Collection (Manassas, VA, USA). Tissue
culture reagents, including Dulbecco’s modified Eagle’s medium
(DMEM), bovine calf serum (CS) and fetal bovine serum (FBS), were
purchased from Invitrogen Life Technologies (Carlsbad, CA, USA).
Antibodies against adiponectin, phospho-c-Jun N-terminal kinases
(p-JNK; Thr183/Tyr185), total JNK (T-JNK), phospho-p38
mitogen-activated protein kinases (p-p38 MAPK; Thr180/Tyr182),
total p38 MAPK (T-p38 MAPK), phospho-p44/42 MAPK
[phospho-extracellular signal-related kinases 1 and 2 (p-ERK1/2);
Thr202/Tyr204] and total p44/42 MAPK (T-ERK1/2) were rabbit
monoclonal antibodies and were obtained from Cell Signaling
Technology (Beverly, MA, USA). Unless otherwise noted, all other
chemicals were purchased from Sigma (St. Louis, MO, USA).
Cell culture
Murine 3T3-L1 cells were plated in six-well plates
and maintained in high glucose (HG)-DMEM containing 10% FBS, 100
U/ml penicillin and 100 μg/ml streptomycin. Two days following 100%
confluence, preadipocytes were induced to differentiate by using
MDI media containing 520 μM isobutylmethylxanthine, 1 μM
dexamethasone and 1 μg/ml insulin in DMEM containing 10% fetal
bovine serum (FBS). After two days, the medium was replaced with
DMEM containing 10% FBS, antibiotics and insulin (insulin media).
Until >95% of the cells contained lipid droplets, HG-DMEM with
10% FBS was replaced every two days. Differentiated 3T3-L1
adipocytes were serum-starved in serum-free DMEM (free media) prior
to the TNF-α treatment. Fig. 1
illustrates the experimental design for TNF-α treatment and the
subsequent wash-off period with/without the inhibitor. The cells
were treated with 10 ng/ml TNF-α for the indicated incubation times
[16, 32 h or 48 h (Fig. 1A, C and
F, respectively)], rinsed and then incubated with DMEM
containing 10% FBS (complete media) for the indicated periods
according to TNF-α incubation time. In the adipocytes that received
16 h TNF-α treatment, the wash-off incubation was 16 h (Fig. 1B). TNF-α-treated 3T3-L1 adipocytes
for 32 h were cultured with complete media for either an additional
16 h (Fig. 1D) or 32 h (Fig. 1E). DMEM containing 10% FBS was used
for either 16 h (Fig. 1G) or 48 h
(Fig. 1H) following 48 h of TNF-α
incubation.
 | Figure 1Schematic of the experimental design.
3T3-L1 cells were seeded and differentiated. Differentiated
adipocytes were serum-starved for 6 h prior to TNF-α treatment (10
ng/ml). The cells were incubated with TNF-α for (A and B) 16 h, (C,
D, E) 32 h or (F, G, H) 48 h. After the indicated incubation times,
3T3-L1 cells were rinsed and re-fed with medium containing 10%
fetal bovine serum with/without inhibitors for (B, D, G) 16 h, (E)
32 h or (H) 48 h. Black arrow, TNF-α treatment; dotted arrow,
wash-off period with/without inhibitor following TNF-α treatment.
TNF-α, tumor necrosis factor-α. |
Total RNA isolation and quantitative
polymerase chain reaction (qPCR)
Total RNA was isolated using the RNA Mini kit
(Invitrogen Life Technologies) and cDNA was prepared using the
high-capacity cDNA kit (Applied Biosystems, Foster City, CA, USA)
following the manufacturer’s instructions. The reactions were
incubated initially at 37°C for 60 min and subsequently at 95°C for
5 min. The PCR primer design for qPCR was performed using the
Universal Probe Library (UPL) software (Roche Applied Science,
Mannheim, Germany). qPCR was conducted using the Roche real-time
PCR master mix in combination with UPL in at least triplicate using
a Roche Lightcycler 480 (Roche Applied Science) as follows: one
cycle of pre-denaturation at 95°C for 10 min, followed by 45 cycles
of denaturation at 95°C for 10 sec and annealing at 60°C for 20
sec, followed by one cycle of extension at 40°C for 30 sec. The
expression levels were determined using the ΔΔCt method. The
following sense and antisense primers were used for adiponectin:
5′-GGAGAGAAAGGAGATGCAGGT-3′ and 5′-CTTTCCTGCCAGGGGTTC-3′; and for
β-actin: 5′-ACTGCTCTGGCTCCTAGCAC-3′ and
5′-CCACCGATCCACACAGAGTA-3′.
Western blot analysis
Differentiated 3T3-L1 adipocytes were serum-starved
for 6 h prior to treatment. Cells were incubated with TNF-α for 16,
32 or 48 h. After the indicated time periods, the cells were washed
with serum-free DMEM and re-fed with HG-DMEM containing the ERK1/2
inhibitor (PD98059; 50 μM) for the indicated times (Fig. 1). The cells were washed in ice-cold
PBS and lysed in radioimmunoprecipitation assay lysis buffer
(Amresco, Solon, OH, USA) containing freshly added protease
inhibitor cocktail and phosphatase inhibitor cocktail (Sigma). The
protein concentrations were measured using the bicinchoninic acid
assay (Pierce Biotechnology, Inc., Rockford, IL, USA). Bromophenol
blue and NuPage reducing agent (Invitrogen Life Technologies) were
added to the cell lysates and this mixture was heated at 95°C for 5
min. Equal protein levels were loaded into each lane of a 4–20% SDS
polyacrylamide gel. The proteins were separated by electrophoresis
and transferred to a polyvinylidene difluoride membrane (Millipore,
Billerica, MA, USA) with the Trans-Blot apparatus (Bio-Rad,
Hercules, CA, USA). For immunoblotting, the membranes were blocked
in 5% non-fat dry milk in TBST (0.05% Tween 20, 50 mM Tris-HCl, pH
7.5 and 150 mM NaCl), washed twice and incubated overnight with
primary antibodies. After washing, the blots were incubated with
horseradish peroxidase-conjugated secondary antibody for 1 h at
room temperature. The signal was detected using the Amersham
enhanced chemiluminescence plus system (Amersham-Pharmacia Biotech,
Arlington Heights, IL, USA). Densitometric analysis of the
individual bands was performed with Geliance Imaging software
(PerkinElmer Life and Analytical Sciences, Boston, MA, USA). The
data presented are representative of at least three independent
experiments with similar results.
Statistical analysis
Data are expressed as mean ± standard error (SE).
Statistical significance was assessed using PASW Statistics 18
(SPSS, Inc., Chicago, IL, USA). Student’s t-test or analysis of
variance was performed to compare between the groups. P<0.05 was
considered to indicate a statistically significant difference
between values.
Results
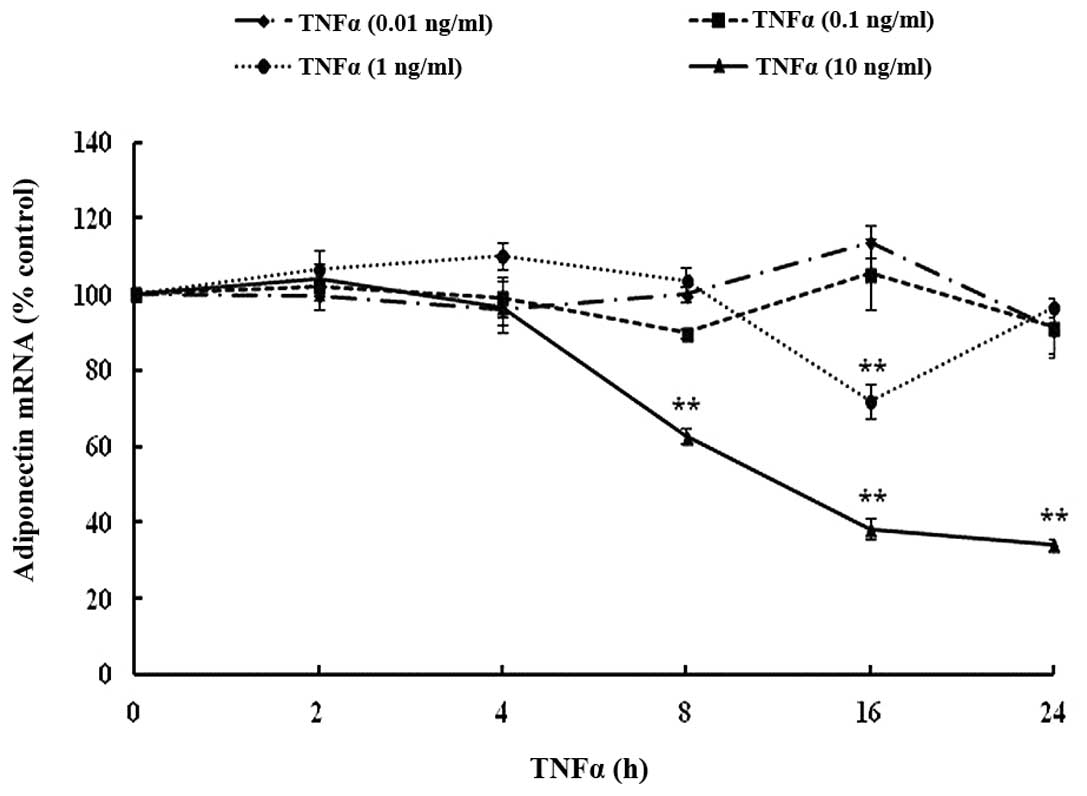
TNF-α inhibits mRNA expression of
adiponectin in a dose- and time-dependent manner
To investigate the impact of TNF-α on adiponectin,
the mRNA expression was first measured in differentiated 3T3-L1
adipocytes. TNF-α significantly suppressed adiponectin gene
expression in a dose- and time-dependent manner as compared with
the control group. There was a significant reduction in mRNA levels
by 37% following 8 h incubation at 10 ng/ml (Fig. 2). The maximal inhibition of
adiponectin mRNA expression was 62%, observed after 16 h incubation
at a TNF-α concentration of 10 ng/ml. There was no significant
difference between the expression from the 16 h and 24 h incubation
periods.
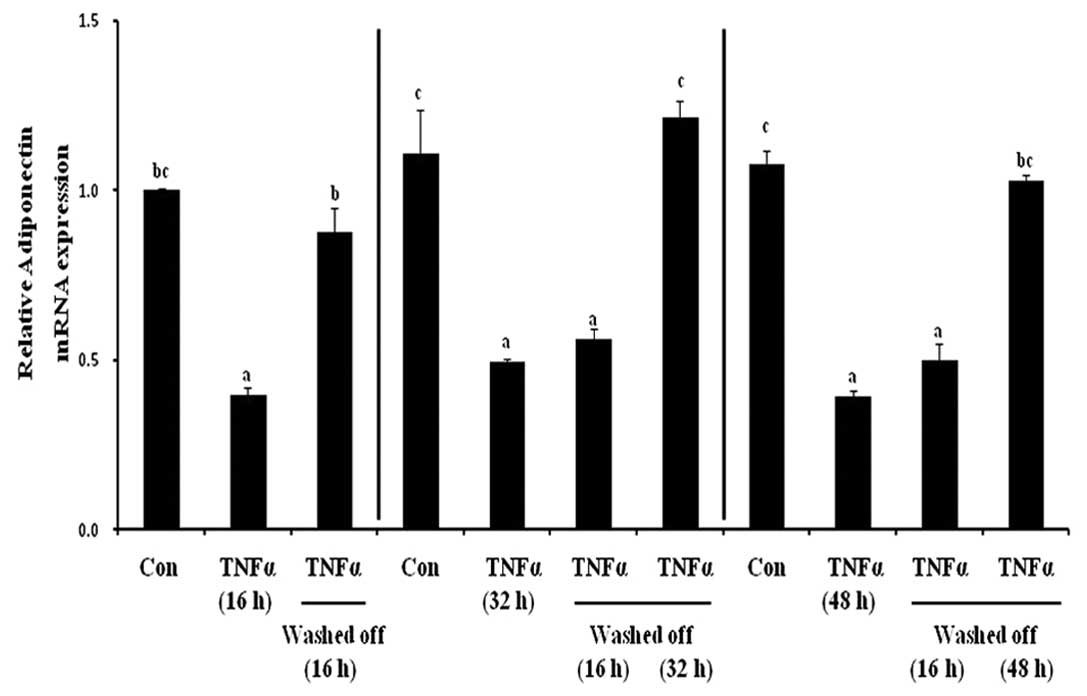
Inhibitory effect of TNF-α on gene
expression of adiponectin is reversible in 3T3-L1 cells
To investigate the reversibility in the inhibitory
effects of the proinflammatory adipokine, TNF-α, on adiponectin
mRNA expression, the effect of TNF-α at different exposure and
removal time periods in differentiated 3T3-L1 adipocytes was
examined. After three different TNF-α incubation periods (16, 32
and 48 h), the cells were rinsed and re-fed with HG-DMEM containing
10% FBS for an additional time as follows: i) 16 h wash-off
incubation following 16 h TNF-α treatment; ii) 16 or 32 h recovery
period following 32 h TNF-α treatment and iii) 16 or 48 h TNF-α
removal following 48 h TNF-α exposure (Fig. 1). Chronic TNF-α incubation
significantly reduced adiponectin gene expression by 60% compared
with the untreated control cells (Fig.
3). Following removal of TNF-α from the medium, there was a
time-dependent reversal of the TNF-α-mediated reduction in
adiponectin levels. In Fig. 3,
column 3, the 16 h wash-off phase completely reversed the decrease
in adiponectin levels following 16 h of TNF-α treatment, returning
to the levels observed in the control. The 16 h TNF-α withdrawal
did not induce a significant effect (column 6), but the 32 h
wash-off period fully reversed the decrease in adiponectin
expression following the 32 h TNF-α treatment (column 7). Similar
results were observed for 48 h TNF-α incubation, a 16 h restoration
did not have a significant impact (column 10); however, a 48 h
wash-off period fully reversed the negative effect of TNF-α on
adiponectin mRNA expression (column 11). These findings suggested
that the inhibitory effect of TNF-α on adiponectin is reversible
and the adverse effects are dependent on the length of TNF-α
exposure and the subsequent recovery period.
Length of recovery phase affects
TNF-α-induced MAPK activation in differentiated 3T3-L1
adipocytes
There is a positive correlation between TNF-α and
activation of three different MAPKs, JNK, ERK1/2 and p38 MAPK, in
the development of obesity-associated insulin resistance.
Therefore, in the present study, the effect of a wash-off period
following exposure to the proinflammatory cytokine TNF-α and the
subsequent activation of MAPKs was examined. Similar to the pattern
of adiponectin mRNA expression (Fig.
4A), the decrease in adiponectin protein expression after 32 h
treatment with TNF-α was completely reversed following an
equivalent (32 h) wash-out period (Fig. 4B and C). In addition, the reduction
in adiponectin levels following 32 h TNF-α treatment was
concomitantly accompanied by activation of JNK, ERK1/2 and p38
MAPK, expressed as phospho-protein/total protein. A 16 h wash-off
phase following 32 h TNF-α-treatment reversed the TNF-α-activated
JNK and p38 MAPK, but not ERK1/2 (Fig.
4B). ERK1/2 activation was attenuated following a 32 h wash-off
period (Fig. 4B and D). This
restoration pattern of ERK1/2 in response to chronic TNF-α exposure
and wash-off was similar to that of adiponectin protein and mRNA
expression. These results suggested that the effect of TNF-α on
adiponectin expression was reversible through ERK activation.
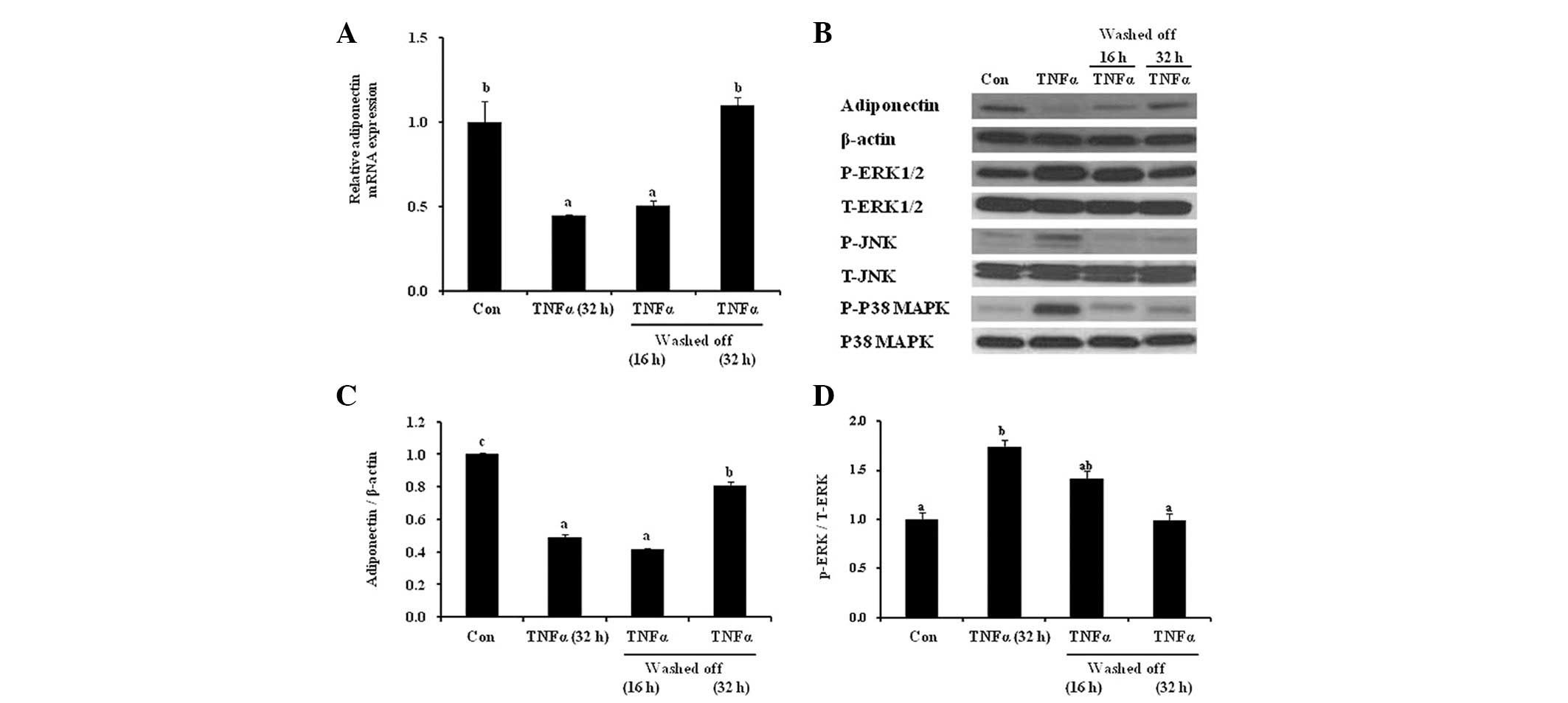 | Figure 4Recovery phase following TNF-α
treatment restores adiponectin levels and ERK activation.
Differentiated adipocytes were treated with TNF-α (10 ng/ml) for 32
h and then the cells were subjected to a wash-off period (16 or 32
h). (A) mRNA expression of adiponectin was assessed using
quantitative polymerase chain reaction. (B) The cell lysates were
analyzed by western blot analysis and probed for adiponectin,
β-actin, p-JNK (Thr183/Tyr185), T-JNK, p-p38 MAPK (Thr180/Tyr182),
T-p38 MAPK, p-ERK1/2 (Thr202/Tyr204) and T-ERK1/2. The signal
density was quantified and expressed as (C) adiponectin/β-actin and
(D) phospho-/total ERK. The results are expressed as the mean ±
standard error. Different letters indicate statistical difference
(P<0.05). TNF-α, tumor necrosis factor-α; MAPK,
mitogen-activated protein kinases; JNK, c-Jun N-terminal kinase;
ERK1/2, extracellular signal-related kinase 1 and 2; p, phospho; T,
total. |
Recovery phase facilitates TNF-α-induced
suppression of adiponectin expression via ERK1/2 activation
To delineate the effects of the ERK pathway on the
recovery of adiponectin expression following TNF-α exposure, 3T3-L1
adipocytes were treated with TNF-α (32 h, 10 ng/ml) and co-treated
with the ERK inhibitor PD98059 (50 μM) in HG-DMEM containing 10%
FBS for an additional restoration time period (16 or 32 h)
(Fig. 5). The 16 h recovery period
did not completely reverse the inhibitory effects of 32 h TNF-α
treatment, whereas the use of PD98059 during this recovery period
fully restored adiponectin levels to the control levels. The
recovery effect of the ERK inhibitor on adiponectin levels was
equivalent to the effect of the 32 h wash-off time without the
inhibitor. However, there were no additional effects on restoration
in the 32 h restoration phase with PD98059. Furthermore, the
addition of the ERK1/2 inhibitor during the 16 h restoration period
significantly increased the protein expression of adiponectin
compared with the control and concomitantly reduced ERK
activation.
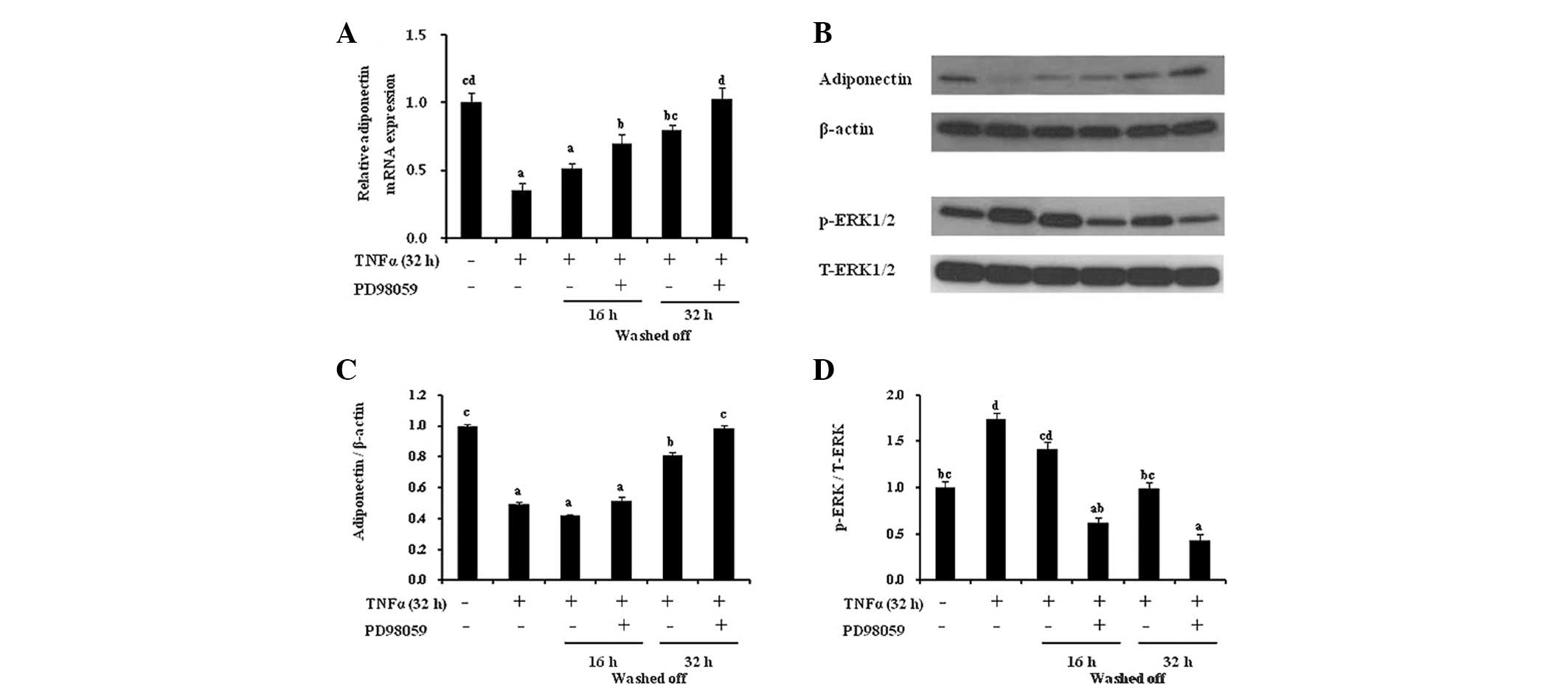 | Figure 5ERK inhibition is involved in the
TNF-α-induced decline and recovery of adiponectin expression in
adipocytes. The 6 h serum-starved 3T3-L1 adipocytes were treated
with TNF-α for 32 h, rinsed with DMEM and co-treated with PD98059
(50 μM) for the additional incubation period. (A) The gene
expression of adiponectin was determined by quantitative polymerase
chain reaction and normalized to β-actin expression for all of the
samples. (B) Western blot analysis for adiponectin, β-actin,
p-ERK1/2 and T-ERK1/2 was performed. Densitometric analysis of (C)
adiponectin or (D) p-ERK1/2 was normalized to the expression of
β-actin or T-ERK1/2, respectively. The results are expressed as the
mean ± standard error. Different letters indicate statistical
difference (P<0.05). TNF-α, tumor necrosis factor-α; MAPK,
mitogen-activated protein kinases; JNK, c-Jun N-terminal kinases;
ERK, extracellular signal-related kinases; DMEM, Dulbecco’s
modified Eagle’s medium; p, phospho; T, total. |
Discussion
The results of the present study demonstrated the
regulation and restoration of adiponectin expression by an
obesity-induced proinflammatory adipokine in adipocytes. It was
identified that TNF-α significantly decreased the expression of
both adiponectin protein and mRNA, and this inhibitory effect was
completely reversed in a time-dependent manner. In order to reverse
the inhibitory effects of TNF-α on adiponectin, the reversal
periods were required to be at least as long as the TNF-α treatment
period. Concurrently, it was identified that the reversal pattern
of ERK activation was consistent with the TNF-α-induced changes in
adiponectin expression. Further investigation revealed that the
addition of an ERK inhibitor during restoration following chronic
TNF-α treatment resulted in a complete reversal of the inhibitory
effects of the proinflammatory cytokine on adiponectin, and the
recovery was notably quicker than that in the absence of the ERK
inhibitor. These results suggested that the ERK pathway may
modulate recovery of adiponectin levels following TNF-α
exposure.
TNF-α is the strongest candidate for the link
between obesity and insulin resistance. There is a close
association between proinflammatory adipokines and the deleterious
effects of obesity-associated comorbidities, and numerous studies
have described the inhibitory effects of TNF-α on the regulation of
another cytokine, adiponectin (16,17,23).
Adiponectin deletion results in increased TNF-α expression and
diet-induced insulin resistance. Adiponectin administration in
adiponectin knockout transgenic mice ameliorates high TNF-α
concentrations in the plasma and adipose TNF-α mRNA expression, and
also increases insulin sensitivity (24). Furthermore, an in vitro
study demonstrated that the inhibitory effect of TNF-α on
adiponectin gene expression is fully reversible following the
removal of TNF-α (17). These
studies demonstrated that TNF-α and adiponectin antagonistically
regulate each other and in turn affect obesity-associated insulin
sensitivity. As expected, in the present study, TNF-α induced a
significant reduction in adiponectin mRNA expression in a dose- and
time-dependent manner. As reported in a previous investigation, it
was also identified that the inhibitory effects of TNF-α on
adiponectin expression are reversible. Furthermore, this
restoration is dependent on the TNF-α incubation time in 3T3-L1
adipocytes, as the decrease in adiponectin levels following 16 h
TNF-α treatment was completely reversed after a 16 h restoration
phase. In 3T3-L1 cells treated with TNF-α for 32 or 48 h, a 16 h
wash-off period resulted in partial restoration of 55 and 45%,
respectively, compared with the untreated control adipocytes. It
appears that, in order to fully reverse the inhibitory effect of
TNF-α on adiponectin expression, the reversal period is required to
be at least as long as the TNF-α treatment period. These data
indicated that normalization of the proinflammatory cytokine,
TNF-α, by weight loss, decreased adiposity or improved energy
homeostasis may increase adiponectin levels and thereby reduce
obesity-associated comorbidities.
In the development of obesity-associated insulin
resistance, the direct actions of TNF-α on insulin signaling and
insulin action occur via the activation of ERK1/2, p38 MAPK and JNK
(25), all of which suppress
adiponectin levels (26,27). As previously reported, in the
present study, chronic TNF-α exposure in 3T3-L1 adipocytes
activated JNK, ERK1/2 and p38 MAPK. However, the reversal of ERK
activation only matched the TNF-α-associated decrease and
restoration of adiponectin expression. The use of PD98059 had
favorable effects on adiponectin recovery, as the restoration of
the TNF-α-induced decrease in adiponectin levels was accelerated in
the presence of PD98059. This evidence indicated that ERK
activation may regulate the recovery duration following chronic
cytokine exposure.
The present study was limited by the fact that it
did not address how exposure to TNF-α and withdrawal may affect
body weight, adiposity and whole body insulin sensitivity by
modulating adiponectin expression and the ERK pathway. Therefore,
further study is required to demonstrate this using an in
vivo model. However, the present study provided evidence that
the inhibitory effect of TNF-α on adiponectin mRNA expression was
reversible depending on the length of the wash-off phase and ERK
activation in adipocytes. The restoration of adiponectin following
chronic exposure to TNF-α was accelerated by the presence of an ERK
inhibitor. In conclusion, the present study suggested that
reduction of TNF-α by energy balance, weight loss, decreased
adiposity or blockage of TNF-α or its receptors, and the inhibition
of ERK activation may increase adiponectin levels and thereby
result in increased insulin sensitivity.
Acknowledgements
The present study was partially supported by the
Korean Diabetes Association (Won Jun Kim, 2010).
Abbreviations:
|
ERK1/2
|
extracellular signal-regulated kinases
1 and 2
|
|
JNK
|
c-Jun N-terminal kinases
|
|
p38 MAPK
|
p38 mitogen-activated protein
kinases
|
|
TNF-α
|
tumor necrosis factor-α
|
References
|
1
|
Trayhurn P and Beattie JH: Physiological
role of adipose tissue: white adipose tissue as an endocrine and
secretory organ. Proc Nutr Soc. 60:329–339. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Frühbeck G, Gómez-Ambrosi J, Muruzábal FJ,
et al: The adipocyte: a model for integration of endocrine and
metabolic signaling in energy metabolism regulation. Am J Physiol
Endocrinol Metab. 280:E827–E847. 2001.PubMed/NCBI
|
|
3
|
Berg AH and Scherer PE: Adipose tissue,
inflammation, and cardiovascular disease. Circ Res. 96:939–949.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hotamisligil GS, Arner P, Caro JF, et al:
Increased adipose tissue expression of tumor necrosis factor-alpha
in human obesity and insulin resistance. J Clin Invest.
95:2409–2415. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hotamisligil GS, Shargill NS and
Spiegelman BM: Adipose expression of tumor necrosis factor-alpha:
direct role in obesity-linked insulin resistance. Science.
259:87–91. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kern PA, Ranganathan S, Li C, et al:
Adipose tissue tumor necrosis factor and interleukin-6 expression
in human obesity and insulin resistance. Am J Physiol Endocrinol
Metab. 280:E745–E751. 2001.PubMed/NCBI
|
|
7
|
Souza SC, Palmer HJ, Kang YH, et al:
TNF-alpha induction of lipolysis is mediated through activation of
the extracellular signal related kinase pathway in 3T3-L1
adipocytes. J Cell Biochem. 89:1077–1086. 2003. View Article : Google Scholar
|
|
8
|
Zhang B, Berger J, Hu E, et al: Negative
regulation of peroxisome proliferator-activated receptor-gamma gene
expression contributes to the antiadipogenic effects of tumor
necrosis factor-alpha. Mol Endocrinol. 10:1457–1466. 1996.
|
|
9
|
Hotamisligil GS, Budavari A, Murray D, et
al: Reduced tyrosine kinase activity of the insulin receptor in
obesity-diabetes. Central role of tumor necrosis factor-alpha. J
Clin Invest. 94:1543–1549. 1994. View Article : Google Scholar
|
|
10
|
Hotamisligil GS, Peraldi P, Budavari A, et
al: IRS-1-mediated inhibition of insulin receptor tyrosine kinase
activity in TNF-alpha- and obesity-induced insulin resistance.
Science. 271:665–668. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yamauchi T, Hara K, Kubota N, et al: Dual
roles of adiponectin/Acrp30 in vivo as an anti-diabetic and
anti-atherogenic adipokine. Curr Drug Targets Immune Endocr Metabol
Disord. 3:243–253. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wu X, Motoshima H, Mahadev K, et al:
Involvement of AMP-activated protein kinase in glucose uptake
stimulated by the globular domain of adiponectin in primary rat
adipocytes. Diabetes. 52:1355–1363. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hotta K, Funahashi T, Bodkin NL, et al:
Circulating concentrations of the adipocyte protein adiponectin are
decreased in parallel with reduced insulin sensitivity during the
progression to type 2 diabetes in rhesus monkeys. Diabetes.
50:1126–1133. 2001. View Article : Google Scholar
|
|
14
|
Hotta K, Funahashi T, Arita Y, et al:
Plasma concentrations of a novel, adipose-specific protein,
adiponectin, in type 2 diabetic patients. Arterioscler Thromb Vasc
Biol. 20:1595–1599. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lihn AS, Richelsen B, Pedersen SB, et al:
Increased expression of TNF-alpha, IL-6, and IL-8 in HALS:
implications for reduced adiponectin expression and plasma levels.
Am J Physiol Endocrinol Metab. 285:E1072–E1080. 2003.PubMed/NCBI
|
|
16
|
Ajuwon KM and Spurlock ME: Adiponectin
inhibits LPS-induced NF-kappaB activation and IL-6 production and
increases PPARgamma2 expression in adipocytes. Am J Physiol Regul
Integr Comp Physiol. 288:R1220–R1225. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fasshauer M, Klein J, Neumann S, et al:
Hormonal regulation of adiponectin gene expression in 3T3-L1
adipocytes. Biochem Biophys Res Commun. 290:1084–1089. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lee YH and Pratley RE: The evolving role
of inflammation in obesity and the metabolic syndrome. Current Diab
Rep. 5:70–75. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Heilbronn LK and Campbell LV: Adipose
tissue macrophages, low grade inflammation and insulin resistance
in human obesity. Curr Pharm Des. 14:1225–1230. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Monzillo LU, Hamdy O, Horton ES, et al:
Effect of lifestyle modification on adipokine levels in obese
subjects with insulin resistance. Obes Res. 11:1048–1054. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fontana L, Eagon JC, Trujillo ME, et al:
Visceral fat adipokine secretion is associated with systemic
inflammation in obese humans. Diabetes. 56:1010–1013. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fain JN, Madan AK, Hiler ML, et al:
Comparison of the release of adipokines by adipose tissue, adipose
tissue matrix, and adipocytes from visceral and subcutaneous
abdominal adipose tissues of obese humans. Endocrinology.
145:2273–2282. 2004. View Article : Google Scholar
|
|
23
|
Kern PA, Di Gregorio GB, Lu T, et al:
Adiponectin expression from human adipose tissue relation to
obesity, insulin resistance, and tumor necrosis factor-alpha
expression. Diabetes. 52:1779–1785. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Maeda N, Shimomura I, Kishida K, et al:
Diet-induced insulin resistance in mice lacking adiponectin/ACRP30.
Nat Med. 8:731–737. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fujishiro M, Gotoh Y, Katagiri H, et al:
Three mitogen-activated protein kinases inhibit insulin signaling
by different mechanisms in 3T3-L1 adipocytes. Mol Endocrinol.
17:487–497. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kim KY, Kim JK, Jeon JH, et al: c-Jun
N-terminal kinase is involved in the suppression of adiponectin
expression by TNF-alpha in 3T3-L1 adipocytes. Biochem Biophys Res
Commun. 327:460–467. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhao T, Hou M, Xia M, et al: Globular
adiponectin decreases leptin-induced tumor necrosis factor-alpha
expression by murine macrophages: involvement of cAMP-PKA and MAPK
pathways. Cell Immunol. 238:19–30. 2005. View Article : Google Scholar
|