Introduction
Alzheimer's disease (AD), a progressive
neurodegenerative disorder, is the most common form of dementia
among the aging population (1).
More than 3.5 million individuals world-wide have been diagnosed
with AD and the proportion of people being diagnosed with AD after
85 years of age exceeds 1 in 3 (2). AD is clinically characterized by
cognitive impairment leading to dementia, immobility, and
eventually mortality (usually within a decade following the initial
diagnosis) (3). The
histopathological changes that occur in AD include wide-spread loss
of neurons, and formation of senile plaques and neurofibrillary
tangles (4). The predominant
theory for the molecular mechanism underlying AD is the amyloid
cascade hypothesis (5).
Extracellular aberrant generation and/or inad-equate clearance of
amyloid-β (Aβ), which originates from the amyloid precursor protein
(APP), are considered to be responsible for the death of neurons
and dementia in AD. An increased level of APP may increase the risk
of AD (6–8). However, the underlying mechanisms of
neurodegeneration, and the molecular and pathological components of
the disease remain to be elucidated (9).
A number of microRNAs (miRs) have been implicated in
AD (10–12). miRs are endogenous and
evolutionarily conserved non-coding small RNA molecules (length,
21–25 nt). They form partially complementary base pairs within the
3′-untranslated regions (UTR) of protein-encoding mRNAs, resulting
in the degradation of target transcripts or inhibition of
translation (13,14). miRs have been observed in various
biological processes, including embryogenesis, the immune response,
developmental timing, differentiation and organogenesis, cell-cycle
control, proliferation and apoptosis (15–20).
Multiple miRs have been observed to be aberrantly expressed in
patients with AD (21). The
dysregulation of miRs may participate in the progression of AD by
influencing the expression and functions of their targets (22). For example, miR-339-5p and miR-153
are significantly downregulated in AD specimens, and may inhibit
the expression of APP and BACE1. Thus, the identification of the
targets of miRs is critical to understand the function of miRs in
the development and progression of AD. It also suggested that miRs
may provide a target for therapeutic strategies for AD.
miR-26b was observed to be significantly upregulated
in the human temporal cortex in AD (23); however, the function of miR-26b has
not been verified. In the present study, miR-26b was upregulated in
a double transgenic mouse model of AD. It was also demonstrated
that upregulation of miR-26b in N2a/APP cells downregulated the
insulin-like growth factor 1 (IGF-1) protein level and promoted Aβ
production, whereas inhibition of miR-26b in N2a/APP cells
upregulated the IGF-1 protein level and suppressed Aβ production.
Furthermore, miR-26b target sites in IGF-1 were confirmed by a
luciferase assay in HEK293 cells. The results of the present study
may aid in the development of effective therapeutic strategies
against AD.
Materials and methods
Transgenic mice and sacrifice
A total of nine APP/PS1 double-transgenic mice, aged
3, 6 or 9-months-old, and nine age-matched controls, were purchased
from the Model Animal Research Center of Nanjing University
(Nanjing, China), originally obtained from The Jackson Laboratory
(Bar Harbor, ME, USA). They were maintained at 19–23°C under a 12-h
light/dark cycle with ad libitum access to sterile food and
water, and all animal handling was conducted in accordance with
institutional guidelines. The present study was approved by the
ethics committee of Yantai Yuhuangding Hospital (Yantai,
China).
The mice were sacrificed by intraperitoneal
injection with pentobarbital overdose (50 mg/kg; Beyotime Institute
of Biotechnology, Haimen, China), followed by the removal and
dissection of the brain tissues. The cortexes of the brains were
frozen in liquid nitrogen for further RNA extraction.
Cell culture and transfection
The HEK293 human embryonic kidney cell line was
purchased from the Shanghai Institute of Biochemistry and Cell
Biology (Shanghai, China). N2a/WT and N2a/APP cells were a gift
from the Tianjin Medical University (Tianjin, China). HEK293 cells
were cultured in Opti-MEM medium (Gibco; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) supplemented with 10% fetal bovine serum
(FBS; Gibco; Thermo Fisher Scientific, Inc.). N2a/WT and N2a/APP
cells were cultured in medium containing 50% Dulbecco's modified
Eagle's medium (Gibco; Thermo Fisher Scientific, Inc.), and 45%
Opti-MEM supplemented with 5% FBS. All cell lines were incubated in
a humidified air atmosphere of 5% CO2 at 37°C.
Cells were transfected with miR-26b mimic, negative
control (NC), miR-26b inhibitor or NC inhibitor (Shanghai
GenePharma, Co., Ltd., Shanghai, China), at a final concentration
of 50 nM, using Lipofectamine 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocols.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from tissues using mirVana
miRNA Isolation kit (Ambion; Thermo Fisher Scientific, Inc.) and
DNase I (Ambion; Thermo Fisher Scientific, Inc.), and treated
according to the manufacturer's protocol to obtain DNA-free RNA.
Equal quantities of RNA were subjected to cDNA synthesis using the
miScript Reverse Transcription kit (Qiagen, Inc., Valencia, CA,
USA). RT-qPCR for miR-26b was performed in a Lightcycler (Roche
Diagnostics GmbH, Mannheim, Germany), according to the SYBR Green
detection protocol, and all reactions were run in triplicate. Each
reaction was performed in a final volume of 20 µl. The PCR
cycling conditions were as follows: 95°C for 15 min, followed by 40
cycles of 94°C for 15 sec, 55°C for 30 sec and 70°C for 30 sec.
Primers for mature miR-26b and U6 snRNA were purchased from Qiagen,
Inc. Expression was determined in 18 mice (six APP/PS1 mice and six
control mice). Relative expression levels were calculated using the
2−ΔΔCq method (24).
Every sample was replicated three times.
Western blot analysis
Cells were washed with ice-cold phosphate-buffered
saline (PBS) and solubilized in cold radio-immunoprecipitation
lysis buffer (RIPA; Beyotime Institute of Biotechnology) 72 h after
transfection. Cells were incubated at 0°C for 15 min and
centrifuged at 2,000 × g for 10 min at 4°C. The supernatants were
collected, and the protein concentration was measured using a
bicinchoninic acid protein assay kit (Beyotime Institute of
Biotechnology). Samples were boiled for 5 min in loading buffer
(Beyotime Institute of Biotechnology) and then equal quantities of
the proteins (40 µg) were separated by 10% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (Beyotime Institute of
Biotechnology) and then transferred to a polyvinylidene difluoride
membrane (Beyotime Institute of Biotechnology). The membrane was
blocked with 5% non-fat dry milk for 2 h, followed by an overnight
incubation at 4°C with primary mouse anti-human IGF-1 (1:1,000;
sc-74116; Santa Cruz Biotechnology, Inc., Dallas, TX, USA) and
mouse anti-human β-actin (1:1,000; sc-130301; Santa Cruz
Biotechnology, Inc.) monoclonal antibodies. Following washing with
PBS three times (for 5 min each time), the membrane was incubated
for 1 h at room temperature with goat anti-mouse horseradish
peroxidase (HRP)-conjugated secondary antibody (1:500; sc-2005;
Santa Cruz Biotechnology, Inc.) in Tris-buffered saline with Tween
20 (Beyotime Institute of Biotechnology). The blot was detected
with an ECL kit (Pierce Biotechnology, Inc., Rockford, IL, USA) and
images were captured using a FluorChem imaging system
(ProteinSimple, San Jose, CA, USA). The protein intensities were
quantified using the AlphaEaseFC 4.1.0 software (Alpha Innotech,
San Leandro, CA, USA).
Enzyme-linked immunosorbent (ELISA)
assay
Aβ42 levels in cell lysates were quantified using a
mouse ELISA assay according to the manufacturer's protocols (Abcam,
Cambridge, UK), as described previously (25). Aβ42 in samples was captured with
G2-11, a monoclonal antibody specific for Aβ42 (Abeta GmbH,
Heidelberg, Germany). Aβ42 was then probed specifically with the
antibody Biotin-Wo2 (Abeta GmbH) overnight at 4°C, and finally
developed with NeutrAvidin-HRP (Pierce Biotechnology, Inc.). The
HRP activity was measured with the TMP Microwell Peroxidase system
(KPL, Inc., Gaithersburg, MD, USA).
Target Prediction of miR-26b
TargetScan 5.2 (http://www.targetscan.org/) was used to predict the
target genes of miR-26b.
Luciferase assay
The luciferase reporter plasmid and the wild-type
(WT)-pGL3-IGF-1-3′UTR Wt and mutant (Mut)-pGL3-IGF-1-3′UTR
expression vectors, were obtained from Shanghai GenePharma. The
HEK293 human embryonic kidney cells were plated at ~90% confluence
and transfected with the reporter plasmid, miR-26b mimics or NC in
a 12-well plate using Lipofectamine 2000, according to the
manufacturer's protocol. The Renilla and the firefly
luciferase activity were measured following 48-h incubation using
the Dual-Luciferase Reporter assay system (Promega Corporation,
Madison, WI, USA). The firefly and Renilla luciferase
activities were measured with a luminometer (Tecan Group, Ltd.,
Männedorf, Switzerland). The firefly luciferase activity was
normalized to Renilla luciferase activity for each
transfected well. Each assay was replicated three times.
Statistical analysis
Data are presented as the mean ± standard deviation
and compared using Student's t-test in Stata, version 10.0
(StataCorp LP, College Station, TX, USA). P<0.05 was considered
to indicate a statistically significant difference.
Results
Expression of miR-26b in APP/PS1
double-transgenic mice
In previous miR profiling experiments, miR-26b was
observed to be upregulated in human brains from patients with AD
(23). In the present study,
APP/PS1 double-transgenic mice and age-matched controls were used
to investigate the expression levels of miR-26b by RT-qPCR. Results
of the present study demonstrated that the expression levels of
miR-26b were increased in 3, 6 and 9-month-old APP/PS1
double-transgenic mice compared with the age-matched controls
(Fig. 1). The results indicated
that miR-26b was upregulated in APP/PS1 double-transgenic mice.
IGF-1 is a direct target of miR-26b
In a previous study, the protein expression level of
IGF-1 was significantly lower in APP/PS1 double-transgenic mice, as
compared with control mice; however, no significant difference was
observed in the mRNA expression level of IGF-1 between the APP/PS1
double-transgenic and control mice (23). These findings suggested that the
regulation of IGF-1 expression occurred at the post-transcriptional
level in APP/PS1 mice. In order to further confirm the association
between IGF-1 and its regulators in APP/PS1 mice, as compared with
control mice, the present study used TargetScan 5.2 to assess the
complementarity of miR-26b to the IGF-1 3′-UTR. As presented in
Fig. 2A, one binding site of
miR-26b was observed within the 3′-UTR of IGF-1. In addition,
luciferase reporter assays were performed to investigate whether
IGF-1 is a direct target of miR-26b. As presented in Fig. 2B, the luciferase activity was
significantly inhibited in cells co-transfected with miR-26b and
WT-3′-UTR compared with the control vector group, whereas
Mut-3′-UTR luciferase activity changed only marginally, this
suggests that IGF-1 may be a direct target of miR-26b in
vitro.
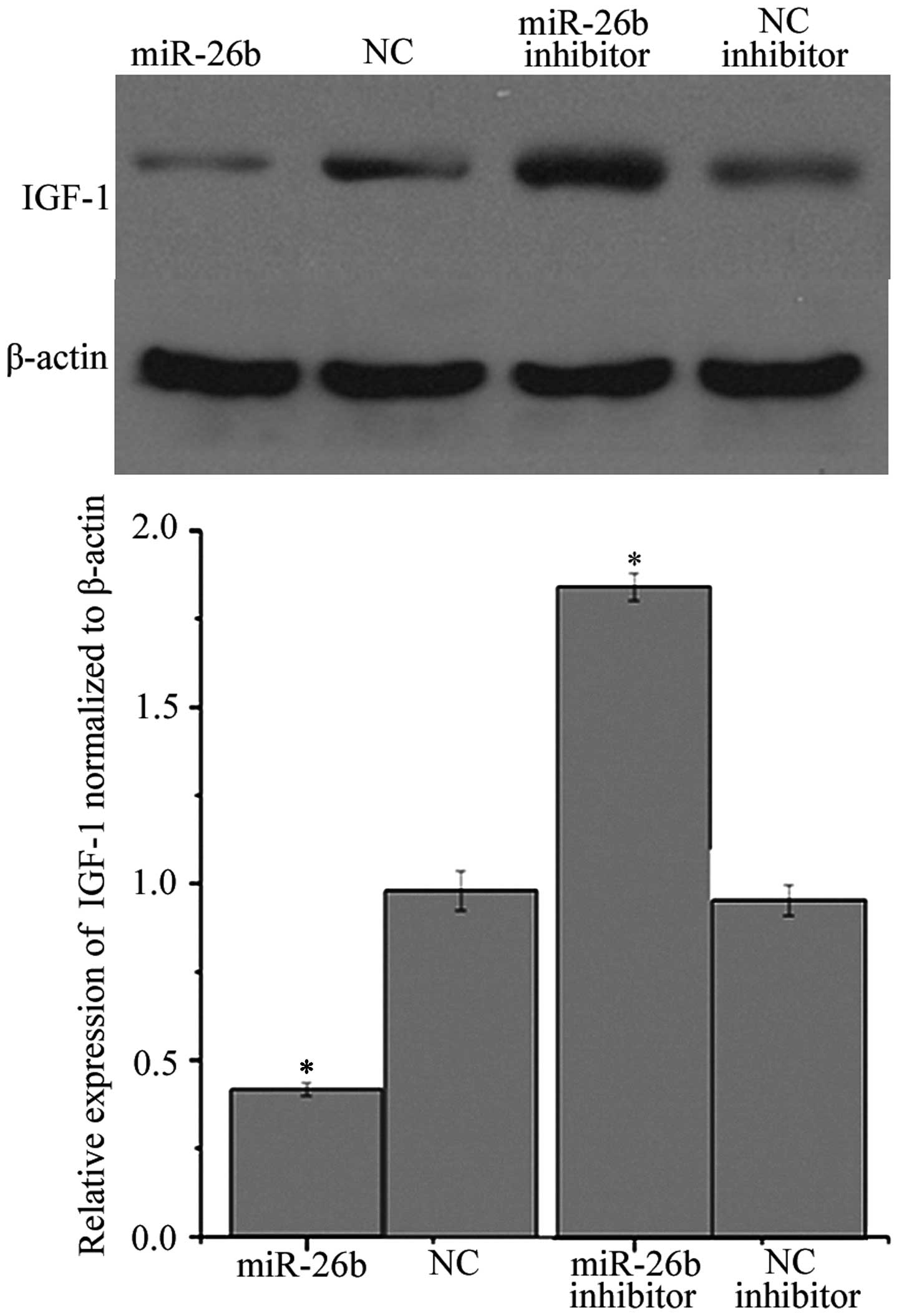
miR-26b reduces the level of IGF-1
To directly investigate whether miR-26b reduces the
expression of IGF-1, western blot analysis was performed. As
presented in Fig. 3, the IGF-1
protein level was significantly downregulated compared with that in
N2a/WT cells transfected with the NC. By contrast, treatment with
the miR-26b inhibitor led to a significant increase in the level of
IGF-1 protein compared with that in N2a/WT cells transfected with
NC inhibitor.
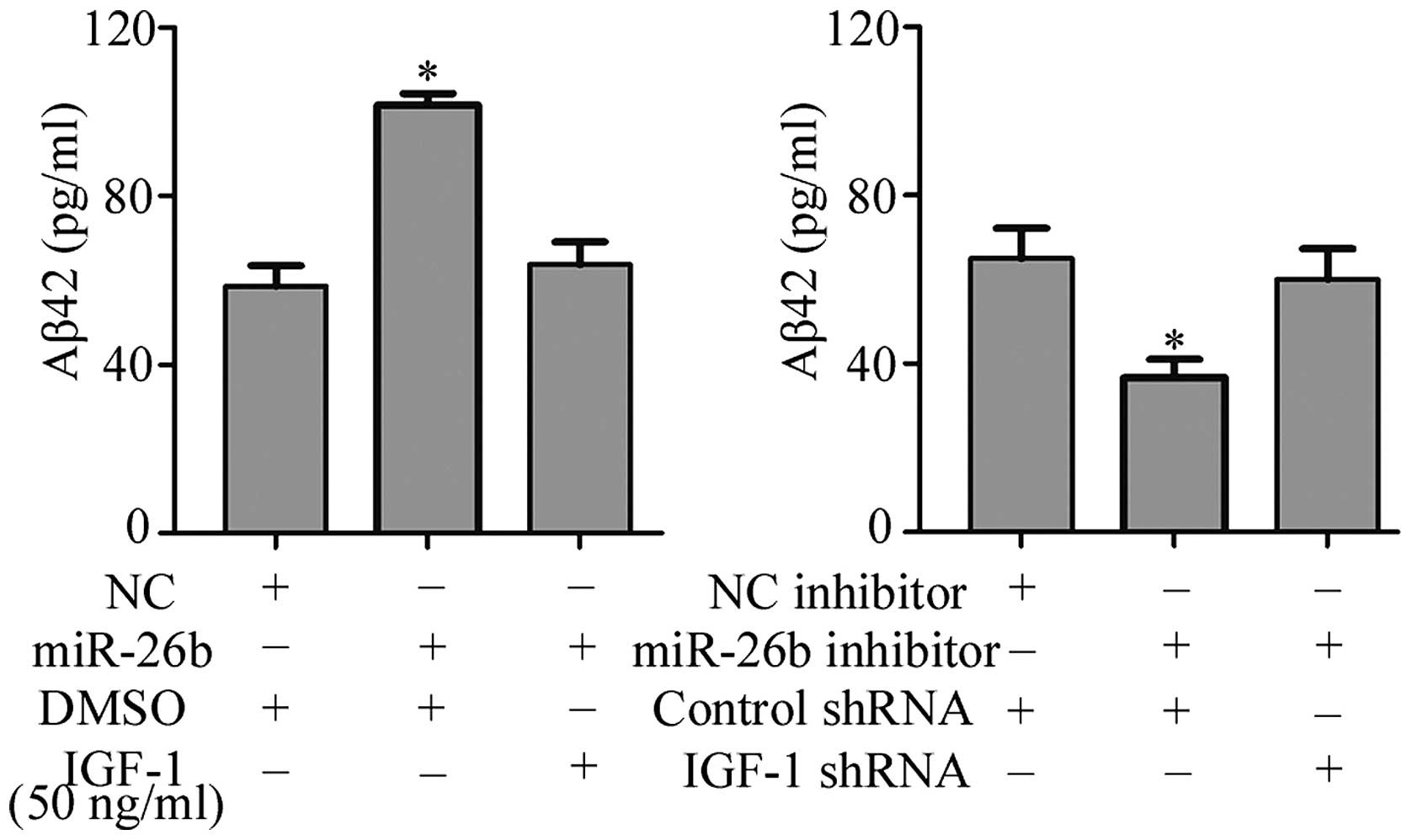
miR-26b increases Aβ production by
targeting IGF-1
To investigate the role of miR-26b in AD, ELISA
analysis was performed to observe the level of Aβ42 in lysates of
N2a/APP cells. As presented in Fig.
4, the level of Aβ42 in N2a/APP cell lysates was significantly
increased following transfection with miR-26b, whereas addition of
50 ng/ml IGF-1 supplementation reversed this upregulation. By
contrast, the level of Aβ42 in the lysates of N2a/APP cells was
downregulated following transfection with an miR-26b inhibitor.
These data indicate that miR-26b negatively regulates IGF-1
translation and induces Aβ production in vitro.
Discussion
Previous studies have demonstrated that miR-26b is
frequently downregulated in various tumors, including breast cancer
(26), nasopharyngeal carcinoma
(27), hepatocellular carcinoma
(28), squamous cell carcinoma of
the tongue (29), primary squamous
cell lung carcinoma (30) and
squamous cell carcinoma of glioma (31). It has been reported to be a
critical regulator in carcinogenesis and tumor progression by
acting as a tumor suppressor gene in various types of cancer
(27,31,32).
miR-26 has been observed to be upregulated in the human temporal
cortex in AD (23). In the present
study, miR-26b was observed to be upregulated in APP/PS1
double-transgenic mice, suggesting that miR-26b may function in the
development of AD.
The potential benefit of the analysis of miRs in the
diagnosis and treatment of numerous diseases, including cancer,
infection and neurodegenerative disease, has been previously
evaluated in numerous studies (9,11,33).
The expression of miRs is known to be altered in multiple regions
of the brain in AD, however, the cause and consequence in the
pathology of the disease remains to be elucidated (8). Downregulation of IGF-1 associated
with accelerated accumulation of Aβ in the brain is a feature of AD
(34). In the present study, it
was demonstrated that upregulation of miR-26b in N2a/APP cells
downregulated the IGF-1 protein level and promoted Aβ production,
whereas inhibition of miR-26b in N2a/APP cells upregulated the
IGF-1 protein level and suppressed Aβ production. Furthermore,
miR-26b target sites in IGF-1 were confirmed by a luciferase assay
in HEK293 cells. These results suggested that miR-26b may be
considered a novel therapy for patients with AD.
AD is the most common form of dementia in the
elderly. It is pathologically characterized by synaptic impairment,
accumulation of neurofibrillary tangles, and Aβ deposition
(35). IGF-1 is a member of the
insulin family of hormones (36),
and-1 is part of an evolutionarily conserved signaling pathway. It
is involved in neuronal growth, survival and differentiation, and
it promotes neurite outgrowth, migration, protein synthesis,
neuronal cytoskeletal protein expression, and nascent synapse
formation (37–41). Previous studies have demonstrated
the role of IGF-1 signaling in AD-pathogenesis using various animal
and human models of AD (42–44);
the serum IGF-1 levels were significantly downregulated in patients
with AD, as compared with patients with vascular dementia or
age-matched non-demented elderly subjects (43,44).
IGF-1 may contribute to the regulation of τ phosphorylation,
amyloid precursor protein (APP) cleavage, Aβ transport and
degradation, memory formation, aging and longevity in AD (45).
The major neuropathological finding in AD is
considered to be the presence of high levels of Aβ in the brain
samples. These peptides are neurotoxic and form amyloid plaques
(46). The association between
serum IGF-1 and brain amyloidosis was established by a previous
study indicating a potential role of IGF-1 in the clearance of Aβ
from the brain (34). Systemic
IGF-1 administration proved effective in lowering brain Aβ levels,
while blockade of systemic IGF-1 action was sufficient to induce
brain amyloidosis (34,47). Furthermore, reduced IGF-1 input
lowers neuronal resistance to Aβ peptide toxicity, increases
cellular susceptibility to cell death signals and leads,
ultimately, to the accumulation of Aβ in the brain (48). The findings of these previous
studies suggest that IGF-1 is a potential therapeutic target in AD.
Upregulation of IGF-1 may, therefore, provide neuroprotection,
facilitate Aβ clearance, antagonize the deleterious effects of
tumor necrosis factor-α and inhibit certain features of the
inflammatory reaction (49).
miR-based therapy is expected to be more efficient than the
traditional single target therapy, since miRs regulate multiple
target genes simultaneously (50).
The results of the present study may aid the development effective
therapeutic strategies against AD.
In conclusion, this is the first study to
demonstrate that miR-26b was downregulated in APP/PS1
double-transgenic mice and negatively regulated the expression of
IGF-1 in vitro. As IGF-1 is critical in Aβ formation,
increasing IGF-1 expression levels is suggested as a potential
therapeutic strategy for AD. Results of the present study suggest
that miR-26b is a potential therapeutic target for the
downregulation of Aβ formation. However, further studies in
vivo are required to address the delivery of the miR-26b
inhibitor into the mouse hippocampus using adenoviruses to
determine whether this upregulates IGF-1 protein levels and
reduces Aβ formation.
References
|
1
|
Wang LL, Huang Y, Wang G and Chen SD: The
potential role of microRNA-146 in Alzheimer's disease: Biomarker or
therapeutic target? Med Hypotheses. 78:398–401. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Alzheimer's Association: 2013 Alzheimer's
disease facts and figures. Alzheimers Dement. 9:208–245. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Banzhaf-Strathmann J, Benito E, May S,
Arzberger T, Tahirovic S, Kretzschmar H, Fischer A and Edbauer D:
MicroRNA-125b induces tau hyperphosphorylation and cognitive
deficits in Alzheimer's disease. EMBO J. 33:1667–1680. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Giannakopoulos P, Hof PR and Bouras C:
Selective vulnerability of neocortical association areas in
Alzheimer's disease. Microsc Res Tech. 43:16–23. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Karran E, Mercken M and De Strooper B: The
amyloid cascade hypothesis for Alzheimer's disease: An appraisal
for the development of therapeutics. Nat Rev Drug Discov.
10:698–712. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lewczuk P, Kamrowski-Kruck H, Peters O,
Heuser I, Jessen F, Popp J, Bürger K, Hampel H, Frölich L, Wolf S,
et al: Soluble amyloid precursor proteins in the cerebrospinal
fluid as novel potential biomarkers of Alzheimer's disease: A
multicenter study. Mol Psychiatry. 15:138–145. 2010. View Article : Google Scholar
|
|
7
|
Weiner MW: Dementia in 2012: Further
insights into Alzheimer disease pathogenesis. Nat Rev Neurol.
9:65–66. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu CG, Wang JL, Li L and Wang PC:
MicroRNA-384 regulates both amyloid precursor protein and
β-secretase expression and is a potential biomarker for Alzheimer's
disease. Int J Mol Med. 34:160–166. 2014.PubMed/NCBI
|
|
9
|
Liu CG, Song J, Zhang YQ and Wang PC:
MicroRNA-193b is a regulator of amyloid precursor protein in the
blood and cerebrospinal fluid derived exosomal microRNA-193b is a
biomarker of Alzheimer's disease. Mol Med Rep. 10:2395–2400.
2014.PubMed/NCBI
|
|
10
|
Chan AW and Kocerha J: The path to
microRNA therapeutics in psychiatric and neurodegenerative
disorders. Front Genet. 3:822012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Delay C, Mandemakers W and Hébert SS:
MicroRNAs in Alzheimer's disease. Neurobiol Dis. 46:285–290. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Delay C and Hébert SS: MicroRNAs and
Alzheimer's disease mouse models: Current insights and future
research avenues. Int J Alzheimers Dis. 2011:8949382011.PubMed/NCBI
|
|
13
|
Lee RC, Feinbaum RL and Ambros V: The C.
elegans heterochronic gene lin-4 encodes small RNAs with antisense
complementarity to lin-14. Cell. 75:843–854. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang SE and Lin RJ: MicroRNA and
HER2-overexpressing cancer. Microrna. 2:137–147. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Alvarez-Garcia I and Miska EA: MicroRNA
functions in animal development and human disease. Development.
132:4653–4662. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Decembrini S, Bressan D, Vignali R, Pitto
L, Mariotti S, Rainaldi G, Wang X, Evangelista M, Barsacchi G and
Cremisi F: MicroRNAs couple cell fate and developmental timing in
retina. Proc Natl Acad Sci USA. 106:21179–21184. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rosa A and Brivanlou AH: MicroRNAs in
early vertebrate development. Cell Cycle. 8:3513–3520. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lu LF and Liston A: MicroRNA in the immune
system, microRNA as an immune system. Immunology. 127:291–298.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Trang P, Weidhaas JB and Slack FJ:
MicroRNAs as potential cancer therapeutics. Oncogene. 27(Suppl 2):
S52–S57. 2008. View Article : Google Scholar
|
|
20
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang WX, Huang Q, Hu Y, Stromberg AJ and
Nelson PT: Patterns of microRNA expression in normal and early
Alzheimer's disease human temporal cortex: White matter versus gray
matter. Acta Neuropathol. 121:193–205. 2011. View Article : Google Scholar :
|
|
22
|
Eacker SM, Dawson TM and Dawson VL:
Understanding microRNAs in neurodegeneration. Nat Rev Neurosci.
10:837–841. 2009.PubMed/NCBI
|
|
23
|
Absalon S, Kochanek DM, Raghavan V and
Krichevsky AM: MiR-26b, upregulated in Alzheimer's disease,
activates cell cycle entry, tau-phosphorylation and apoptosis in
postmitotic neurons. J Neurosci. 33:14645–14659. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔCt method. Methods. 25:402–408. 2001. View Article : Google Scholar
|
|
25
|
Zhang YC, Wang ZF, Wang Q, Wang YP and
Wang JZ: Melatonin attenuates beta-amyloid-induced inhibition of
neuro-filament expression. Acta Pharmacol Sin. 25:447–451.
2004.PubMed/NCBI
|
|
26
|
Li J, Kong X, Zhang J, Luo Q, Li X and
Fang L: MiRNA-26b inhibits proliferation by targeting PTGS2 in
breast cancer. Cancer Cell Int. 13:72013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ji Y, He Y, Liu L and Chong X: MiRNA-26b
regulates the expression of cyclooxygenase-2 in
desferrioxamine-treated CNE cells. FEBS Lett. 584:961–967. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ji J, Shi J, Budhu A, Yu Z, Forgues M,
Roessler S, Ambs S, Chen Y, Meltzer PS, Croce CM, et al: MicroRNA
expression, survival and response to interferon in liver cancer. N
Engl J Med. 361:1437–1447. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cao J, Guo T, Dong Q, Zhang J and Li Y:
miR-26b is downregulated in human tongue squamous cell carcinoma
and regulates cell proliferation and metastasis through a
COX-2-dependent mechanism. Oncol Rep. 33:974–980. 2015.
|
|
30
|
Gao W, Shen H, Liu L, Xu J, Xu J and Shu
Y: MiR-21 over-expression in human primary squamous cell lung
carcinoma is associated with poor patient prognosis. J Cancer Res
Clin Oncol. 137:557–566. 2011. View Article : Google Scholar
|
|
31
|
Wu N, Zhao X, Liu M, Liu H, Yao W, Zhang
Y, Cao S and Lin X: Role of microRNA-26b in glioma development and
its mediated regulation on EphA2. PLoS One. 6:e162642011.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li J, Li X, Kong X, Luo Q, Zhang J and
Fang L: MiRNA-26b inhibits cellular proliferation by targeting CDK8
in breast cancer. Int J Clin Exp Med. 7:558–565. 2014.PubMed/NCBI
|
|
33
|
Dassow H and Aigner A: MicroRNAs (miRNAs)
in colorectal cancer: From aberrant expression towards therapy.
Curr Pharm Des. 19:1242–1252. 2013.PubMed/NCBI
|
|
34
|
Carro E, Trejo JL, Gomez-Isla T, LeRoith D
and Torres-Aleman I: Serum insulin-like growth factor I regulates
brain amyloid-beta levels. Nat Med. 8:1390–1397. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Xiao AW, He J, Wang Q, Luo Y, Sun Y, Zhou
YP, Guan Y, Lucassen PJ and Dai JP: The origin and development of
plaques and phosphorylated tau are associated with axonopathy in
Alzheimer's disease. Neurosci Bull. 27:287–299. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jones JI and Clemmons DR: Insulin-like
growth factors and their binding proteins: Biological actions.
Endocr Rev. 16:3–34. 1995.PubMed/NCBI
|
|
37
|
van Exel E, Eikelenboom P, Comijs H, Deeg
DJ, Stek ML and Westendorp RG: Insulin-like growth factor-1 and
risk of late-onset Alzheimer's disease: Findings from a family
study. Neurobiol Aging. 35:725e7–e10. 2014. View Article : Google Scholar
|
|
38
|
Ye P, Xing Y, Dai Z and D'Ercole AJ: In
vivo actions of insulin-like growth factor-I (IGF-I) on cerebellum
development in transgenic mice: Evidence that IGF-I increases
proliferation of granule cell progenitors. Brain Res Dev Brain Res.
95:44–54. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
D'Ercole AJ, Ye P and O'Kusky JR: Mutant
mouse models of insulin-like growth factor actions in the central
nervous system. Neuropeptides. 36:209–220. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Popken GJ, Hodge RD, Ye P, Zhang J, Ng W,
O'Kusky JR and D'Ercole AJ: In vivo effects of insulin-like growth
factor-I (IGF-I) on prenatal and early postnatal development of the
central nervous system. Eur J Neurosci. 19:2056–2068. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
O'Kusky JR, Ye P and D'Ercole AJ:
Insulin-like growth factor-I promotes neurogenesis and
synaptogenesis in the hippocampal dentate gyrus during postnatal
development. J Neurosci. 20:8435–8442. 2000.PubMed/NCBI
|
|
42
|
Wang W, Yu JT, Tan L, Liu QY, Wang HF and
Ma XY: Insulin-like growth factor 1 (IGF1) polymorphism is
associated with Alzheimer's disease in Han Chinese. Neurosci Lett.
531:20–23. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Rollero A, Murialdo G, Fonzi S, Garrone S,
Gianelli MV, Gazzerro E, Barreca A and Polleri A: Relationship
between cognitive function, growth hormone and insulin-like growth
factor I plasma levels in aged subjects. Neuropsychobiology.
38:73–79. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Watanabe T, Miyazaki A, Katagiri T,
Yamamoto H, Idei T and Iguchi T: Relationship between serum
insulin-like growth factor-1 levels and Alzheimer's disease and
vascular dementia. J Geriatr Soc. 53:1748–1753. 2005. View Article : Google Scholar
|
|
45
|
Freude S, Schilbach K and Schubert M: The
role of IGF-1 receptor and insulin receptor signaling for the
pathogenesis of Alzheimer's disease: From model organisms to human
disease. Curr Alzheimer Res. 6:213–223. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yankner BA, Dawes LR, Fisher S,
Villa-Komaroff L, Oster-Granite ML and Neve RL: Neurotoxicity of a
fragment of the amyloid precursor associated with Alzheimer's
disease. Science. 245:417–420. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Carro E, Trejo JL, Gerber A, Loetscher H,
Torrado J, Metzger F and Torres-Aleman I: Therapeutic actions of
insulin-like growth factor I on APP/PS2 mice with severe brain
amyloidosis. Neurobiol Aging. 27:1250–1257. 2006. View Article : Google Scholar
|
|
48
|
Carro E and Torres-Aleman I: Insulin-like
growth factor I and Alzheimer's disease: Therapeutic prospects?
Expert Rev Neurother. 4:79–86. 2004. View Article : Google Scholar
|
|
49
|
Gasparini L and Xu H: Potential roles of
insulin and IGF-1 in Alzheimer's disease. Trends Neurosci.
26:404–406. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Daválos A and Suárez Y: MiRNA-based
therapy: From bench to bedside. Pharmacol Res. 75:1–2. 2013.
View Article : Google Scholar : PubMed/NCBI
|