Introduction
Chronic kidney disease (CKD) has become a worldwide
public health issue due to increased morbidity, premature mortality
and the associated health care costs (1). Globally, the mortality rate induced
by kidney disease has risen by 83% since 1990 (2), exerting a great pressure on the
health care system. Recently, clinical and experimental reports
confirmed that aldosterone (ALD) may initiate the development and
progression of chronic renal injury, in addition to the classical
effects on sodium and potassium transport in the renal tubules
(3–6). Furthermore, this mineralocorticoid
receptor has been localized to preglomerular vasculature, mesangial
cells (MCs) and fibroblasts, as well as distal tubular cells of the
nephron in the rat kidney (7),
suggesting a possible direct role of this mineralocorticoid hormone
in kidney damage. For example, infusion of ALD induces
glomerulosclerosis and tubulointerstitial fibrosis in rats
(8,9). Similarly, in hypertensive remnant
kidney rats, ALD infusion reverses the renoprotective effects of
angiotensin-converting enzyme inhibitors (4). ALD also shows a direct deleterious
influence on kidney cells, such as MCs, podocytes, proximal tubular
epithelial cells and fibroblasts (10–13).
Particularly, spironolactone, an ALD receptor antagonist, inhibits
ALD-induced MC apoptosis (14).
The underlying mechanism of ALD-induced cell
apoptosis has been increasingly investigated. Nagase and Fujita
(15) reported that ALD evoked
glomerular podocyte injury via oxidative stress and
serum/glucocorticoid regulated kinase 1 upregulation. Additionally,
evidence from animal studies indicated that renal injury in
ALD-infused rats is associated with increases in NADPH expression
and reactive oxygen species levels (8). There are also other signalling
pathways, such as the Wnt/wingless signalling pathway (16), and the p38 mitogen-activated
protein kinase and phosphatidylinositol 3-kinase (PI3K)/Akt
signalling pathways (17).
However, the precise mechanism for ALD-induced renal injury is not
well understood.
MCs are crucial in maintaining the structural
integrity of the glomerular microvascular bed, providing mesangial
matrix homeostasis and modulating glomerular filtration (18). They are considered to be one of the
main target cells of various pathogenic factors, including high
glucose levels, high pressure and high serum levels of transforming
growth factor-β and platelet-derived growth factor (19). Previous studies show that apoptosis
occurs during various types of chronic kidney disease, including
diabetic nephropathy, immunoglobulin (Ig)A nephropathy and lupus
nephritis (20–22). Previously, it was demonstrated that
MC apoptosis induces an increased severity of albuminuria in mice
(23) and MC apoptosis is directly
involved in the pathogenesis of progressive glomerulosclerosis
(24), which may eventually
progresses to end-stage renal disease. Furthermore, MC loss is also
observed in the late stage of diabetic nephropathy and apoptosis is
hypothesized to be involved (20).
These findings indicate that MC apoptosis is crucial for the
development of renal injury.
Cell apoptosis is regulated by multiple genes, among
which p53 has been the most widely investigated and has been shown
to have an important role in regulating apoptosis. As a tumour
suppressor gene, p53 acts as an apoptosis regulator by modulating
cell apoptosis via control of the G1 arrest checkpoint
of the cell cycle (25).
Accumulating evidence has indicated that p53 is critical for the
process of cell apoptosis, and p53 may be an upstream regulator of
the PI3K/Akt signalling pathway.
To the best of our knowledge, there are few studies
regarding the association between ALD and the upstream regulator of
the PI3K/Akt signalling pathway, and the underlying mechanism for
ALD-induced kidney injury has not been fully elucidated. Thus, the
aim of the present study was to evaluate the potential association
between ALD and p53. Furthermore, the current study may provide a
basis for clinical investigations into the underlying mechanism of
ALD-induced renal injury.
Materials and methods
Materials
Rat MCs were obtained from The Chinese Center for
Type Culture Collection (Wuhan, China) and ALD was obtained from
Sigma-Aldrich (St. Louis, MO, USA). Gibco fetal bovine serum (FBS),
Dulbecco's modified Eagle's medium (DMEM) and SYBR®
Green Mix were supplied by (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). Penicillin, streptomycin, TRIzol reagent and
Lipofectamine® 2000 were obtained from Invitrogen
(Thermo Fisher Scientific, Inc.). Rabbit anti-p53 polyclonal
antibody (cat. no. AF0879) was supplied by Affinity BIO (Burwood,
Victoria, AU) and rabbit anti-β-actin polyclonal antibody (cat. no.
AP0060) was purchased from Bioworld Technology, Inc. (St. Louis
Park, MN, USA). Horseradish peroxidase (HRP)-conjugated goat
anti-rabbit IgG polyclonal antibody (cat. no. DZP-03) was purchased
from Dizhao Biotech (Nanjing, China).
Cell culture
The cells were cultured in DMEM supplemented with
10% FBS, 100 U/ml penicillin and 100 mg/ml streptomycin at 37°C in
an atmosphere containing 5% CO2. Glomerular mesangial
cells (GMCs) between passages 3 and 6 were used in the present
study. To determine the effects of ALD on MCs, equal numbers of
growth-arrested MCs (preincubated in serum-free DMEM for 24 h at
37°C in 5% CO2) were incubated in medium (with 10% FBS)
containing either buffer (control) or ALD (10−6 M) for
24 h at 37°C. Cells were analysed at the end of the incubation
period.
RNA interference
MCs transfected with either an empty expression
vector (pGPU6-NC-shRNA) or a p53-silenced vector
(pGPU6/GFP/Neo-shRNA; GenePharma, Co., Ltd., Shanghai, China) were
established in our laboratory, the sequences of the two cDNA
fragments were as follows: Sense, 5′-GGAGGATTCACAGTCGGATAT-3′ for
p53 (siRNA); and sense, 5′-GTTCTCCGAACGTGTCACGT-3′ for the negative
control (NC). siRNA targeting p53 or its NC were transfected using
Lipofectamine® 2000. MCs in growth medium without
antibiotics were transfected in serum-free DMEM containing 10
µl Lipofectamine® 2000 with 4.0 ug siRNA per well
(6-well plate) for 4–6 h, and the medium was then replaced with
growth medium for 48 h. The MCs were serum-starved for 24 h and
stimulated with or without 10−6 M ALD for 24 h. Cells
were analysed at the end of the incubation period.
Reverse transcription-quantitative
polymerase chain reaction (qPCR)
Total RNA, obtained from rat MCs by TRIzol
extraction, was purified by processing with an RNeasy Mini kit
(Qiagen GmbH, Hilden, Germany), according to the manufacturer's
protocol. Quantification and quality checks were performed using
the NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific,
Inc., Pittsburgh, PA, USA) and Agilent 2100 Bioanalyzer (Agilent
Technologies, Inc., Santa Clara, CA, USA), respectively. RNA (1
µg) from each sample was reverse transcribed to cDNA using a
random hexamer primer and the Thermo Scientific™ RevertAid First
Strand cDNA Synthesis kit (Thermo Fisher Scientific, Inc.). Primers
for each long non-coding RNA were designed according to Primer 3
(http://sourceforge.net/projects/primer3/) and verified
against the National Center for Biotechnology Information Basic
Local Alignment Search Tool (http://blast.ncbi.nlm.nih.gov/Blast.cgi) to ensure a
unique amplification product. The following primers were used for
qPCR to measure p53 mRNA expression: Sense,
5′-TCTCCCCAGCAAAAGAAAAA-3′ and antisense,
5′-CTTCGGGTAGCTGGAGTGAG-3′ (expected product of ~168 bp) by. The
control rat glyceraldehyde-3-phosphate dehydrogenase (GAPDH)
primers were as follows: Sense, 5′-CAAGTTCAACGGCACAGTCAA-3′ and
antisense, 5′-TGGTGAAGACGCCAGTAGACTC-3′ (expected product of ~149
bp). PCR was performed on a ViiA™ 7 Dx Real-Time PCR Instrument
(Applied Biosystems; Thermo Fisher Scientific, Inc.) with 20
µl reaction mixtures consisting of 1 µl cDNA, 1
µl each of the forward and reverse primers, 10 µl
SYBR® Green Mix (containing Taq DNA polymerase
and dNTPs) and 7 µl dH2O. The cycling conditions
were as follows: 1 cycle at 95°C for 10 min, followed by 40 cycles
at 95°C for 15 sec, 56°C for 20 sec and 72°C for 1 min. The
relative expression levels were calculated using the
2−ΔΔCq method (26),
following normalization to GAPDH.
Western blotting
p53 protein expression was assessed by western
blotting. The cells were lysed in radioimmunoprecipitation assay
buffer containing 150 mM NaCl, 50 mM Tris-Cl (pH 7.6), 5 mM EDTA,
0.5% NP-40, 0.5% Triton X-100 containing 10 µg/ml each of
leupeptin, aprotinin, and antipain, 1 mM sodium orthovanadate and
0.5 mM phenylmethanesulfonyl fluoride (all Beyotime Institute of
Biotechnology, Shanghai, China). The protein concentration was
determined using the bicinchoninic acid assay (Thermo Fisher
Scientific, Inc.). Total protein (50 µg) was loaded onto
each lane, separated by 10% SDS-PAGE and transferred to a
polyvinylidene difluoride membrane (EMD Millipore, Billerica, MA,
USA). After being blocked with 5% skimmed milk in Tris-buffered
saline (pH 7.6; Beyotime Institute of Biotechnology) at room
temperature, the membrane was incubated at 4°C overnight with
rabbit anti-p53 (1:1,000) and anti-β-actin (1:1,000) primary
antibodies. Following incubation with HRP-conjugated goat
anti-rabbit secondary antibody (1:5,000), the immune complexes were
detected by enhanced chemiluminescence western blotting reagents
(Beyotime Institute of Biotechnology). Data were normalized to
β-actin protein expression levels and relative protein expression
levels were determined using AlphaView Stand Alone Analysis
Software 3.0 (ProteinSimple, San Jose, CA, USA).
Detection of apoptosis
Apoptosis was detected by annexin V/propidium iodide
(PI) staining using an Annexin V-FITC Apoptosis Detection kit
(Vazyme, Piscataway, NJ, USA). Briefly, cells (1×106 per
sample) were treated with 0.25% trypsin (Invitrogen; Thermo Fisher
Scientific, Inc.), washed twice with cold phosphate-buffered saline
(PBS) and resuspended in 100 µl 1X annexin V-binding buffer.
The cells were subsequently stained with 5 µl annexin
V-fluorescein isothiocyanate, and 5 µl PI at 4°C for 10 min
in the dark, then supplemented with 400 µl 1X binding buffer
and analysed by flow cytometry (BD Biosciences, Franklin Lakes, NJ,
USA). The data were processed using BD Accuri™ C6 software (BD
Biosciences).
Animal treatment
All experimental procedures were performed according
to the guidelines for the care and use of animals established by
Fudan University (Shanghai, China). The present study was approved
by the Medical Ethics Committee of Nanjing Medical University
(Nanjing, China). Male Sprague-Dawley rats (n=16; age, 5–6 weeks;
weight, ~190 g), obtained from the Animal Center of Fudan
University, were housed in individual cages in a
temperature-controlled room under a 12-h light/dark cycle and with
ad libitum access to food and water. After 2 weeks of
acclimatization, the rats underwent a right uninephrectomy or sham
surgical procedure following intraperitoneal injection with 1%
pentobarbital (40 mg/kg; Beyotime Institute of Biotechnology).
After 2 weeks of recovery from surgery or sham surgery, a
mini-osmotic pump (model 2004; Alzet, Cupertino, CA, USA) was
implanted subcutaneously (sc) to administer the vehicle (control)
or ALD (at 0 weeks), and the rats (weight, 260–290 g) were randomly
divided into two groups for 4 weeks: Group 1, vehicle [normal
saline (sc), n=8]; group 2, ALD [0.75 µg/h (sc), n=8]. The
rats in the two groups received 1% NaCl in drinking water
throughout the experimental period.
Systolic blood pressure (SBP) was measured in the
conscious state by tail-cuff plethysmography (BP-98A; Softron Co.,
Tokyo, Japan) at weeks 0 and 4 during the treatment period. 24-h
urine samples were collected following a 24-h acclimatization
period in metabolic cages. Urinary protein excretion was determined
using ELISA kits from Exocell, Inc. (Philadelphia, PA, USA; cat.
no. NR002). Urine and plasma creatinine levels were analysed using
an assay kit (Nanjing Jiancheng Bioengineering Institute, Nanjing,
China; cat. no. C011-1).
Immunohistochemistry
The rats were sacrificed by tail vein air embolism
following anesthetization with 1% pentobarbital (40 mg/kg), after
which the renal cortices of the rats were dissected, fixed with 4%
buffered formaldehyde, embedded in paraffin (both Beyotime
Institute of Biotechnology), deparaffinized and rehydrated.
Sections (thickness, 4 µm) were stained with periodic
acid-Schiff, and haematoxylin and eosin (both Beyotime Institute of
Biotechnology), according to standard protocol, and visualized
using a light microscope (CX32; Olympus Corporation, Tokyo, Japan).
Subsequently, the sections were pretreated with 0.3%
H2O2 in methanol (both Beyotime Institute of
Biotechnology) for 15 min to quench the endogenous peroxide
activity and boiled at 100°C for 10 min in a 10% citrate buffer
(Beyotime Institute of Biotechnology) for antigen retrieval.
Sections were then incubated overnight at 4°C with rabbit anti-p53
polyclonal antibody (1:200; cat. no. sc-1311-R; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA), followed by incubation with
HRP-conjugated goat anti-rabbit IgG antibody (1:100; cat. no.
BA1054; Boster Systems, Inc., Pleasanton, CA, USA) for 30 min at
37°C. Following washing three times with PBS, the sections were
incubated with 3,3′-diaminobenzidine substrate for visualization
under a microscope (CX32). NCs were prepared by replacing the
primary antibodies with PBS.
Terminal deoxynucleotidyl transferase
dUTP nick end labelling (TUNEL) studies
Apoptosis in renal tissue samples was analysed by
TUNEL assay using a kit from Roche Diagnostics (Indianapolis, IN,
USA; cat. no. 11684795910). Briefly, 4-µm deparaffinised
renal sections were exposed to terminal transferase in the presence
of a nucleotide mixture containing fluorescein-2′-deoxyuridine
5′-triphosphate and were examined by fluorescence microscopy (Axio
Observer. A1; Carl Zeiss AG, Oberkochen, Germany).
Statistical analysis
Data are presented as the mean ± standard deviation
of at least three independent experiments. Statistical analysis was
assessed using one-way analysis of variance followed by
Student-Newman-Keuls q test or Dunnett's multiple comparison tests,
with the SPSS 16.0 statistical software package (SPSS Inc.,
Chicago, IL, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
Clinical characteristics and renal
function
The kidney/body weight ratio, BP, urine volume,
final urinary ALD, and urinary albumin/creatinine were all
significantly increased in the ALD group when compared with control
rats. However, no significant differences in body weight and
creatinine clearance were observed between the two groups (Table I).
 | Table IBiological parameters in control and
ALD-infused rats at 4 weeks. |
Table I
Biological parameters in control and
ALD-infused rats at 4 weeks.
| Parameter | Group
|
|---|
| Control | ALD |
|---|
| Body weight
(g) | 460.00±10.00 | 452.00±11.00 |
| Kidney weight/body
weight ratio (mg/g) | 5.70±0.30 | 9.10±0.50a |
| Blood pressure
(mmHg) | 142.00±3.00 | 18.01±5.00a |
| Creatinine
clearance (ml/min) | 3.20±0.30 | 3.00±0.30 |
| Urine volume
(ml) | 13.00±3.00 | 4.01±9.00a |
| Final urinary ALD
(mg/24 h) | 0.04±0.02 | 0.16±0.04a |
| Albumin/creatinine
(mg/mg) | 1.50±0.70 | 9.00±1.20a |
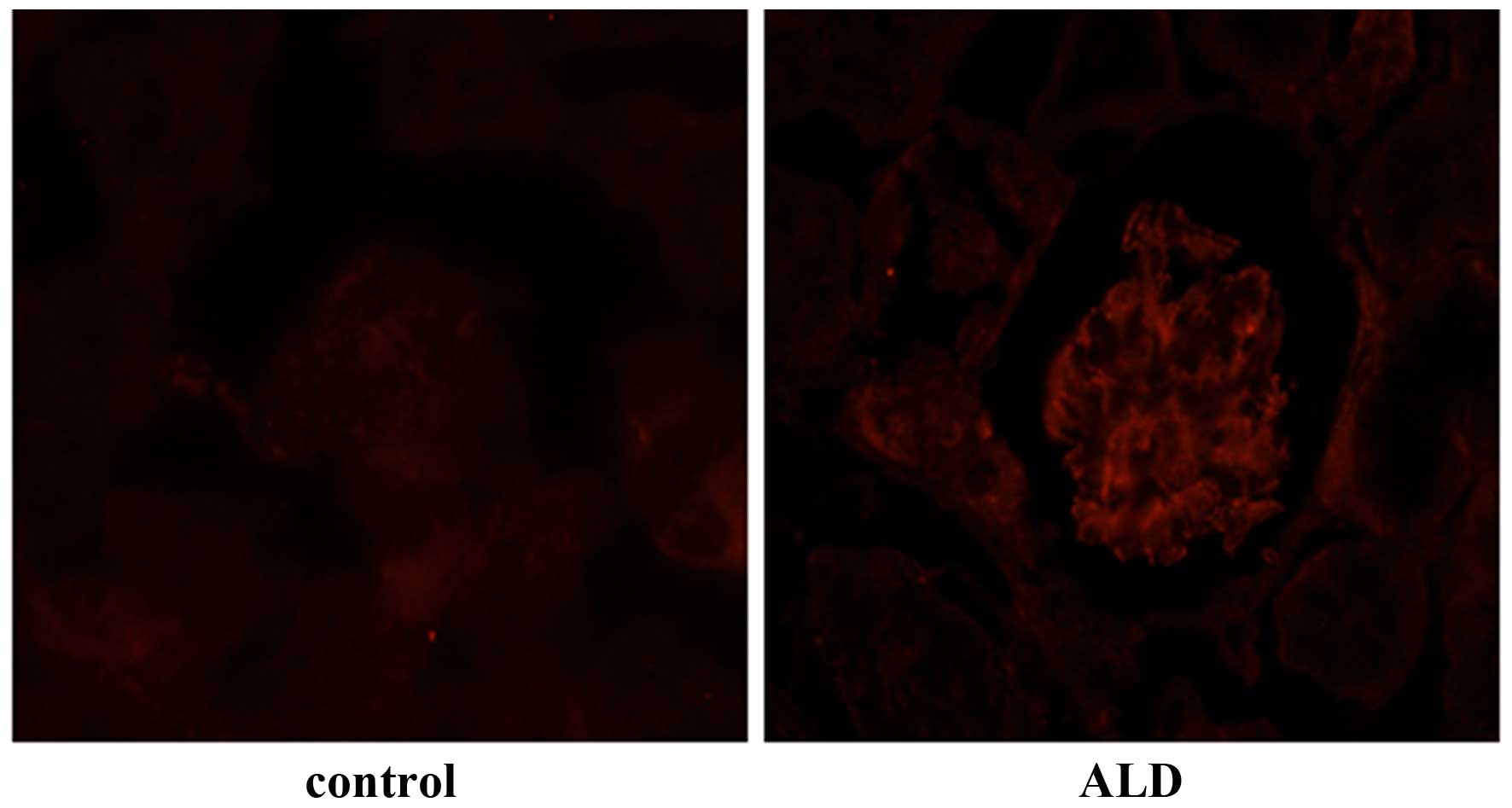
Effects of ALD on MC apoptosis in
vivo
To investigate the effects of ALD on MC apoptosis,
rats were randomly assigned to receive normal saline or ALD for 4
weeks and the ratio of MC apoptosis was analysed using a TUNEL
assay. As shown in Fig. 1, renal
cortical sections of ALD-treated rats demonstrated a greater number
of TUNEL-positive MCs (red arrows) than that in the control rat
sections. The data indicated that treatment with ALD induced MC
apoptosis in vivo. The present study was unable to
differentiate the MCs from other types of cell that may be
simultaneously undergoing apoptosis. However, as the apoptotic
cells were also located in the mesangium it was hypothesized that
ALD induced MC apoptosis.
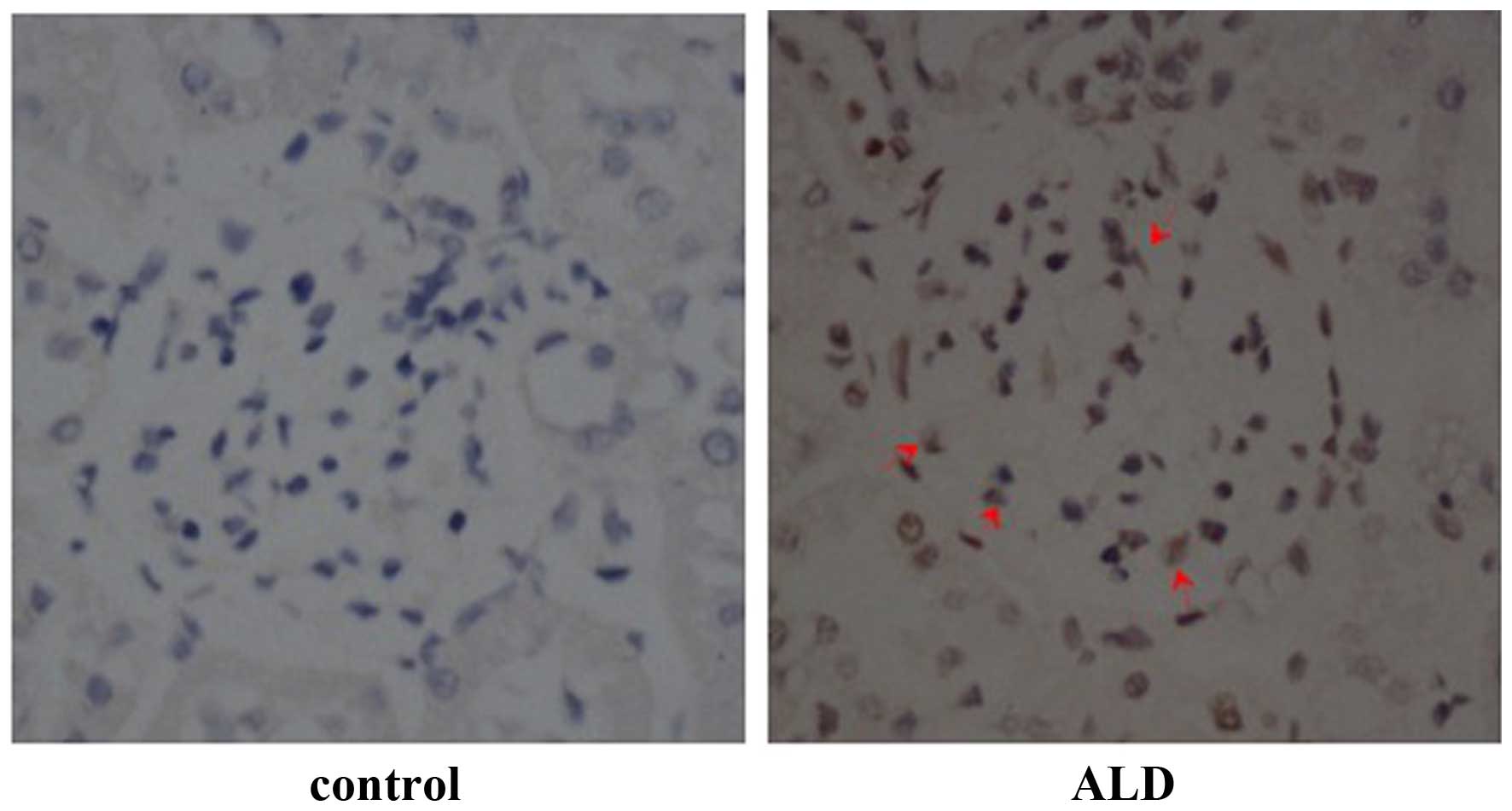
Effects of ALD on p53 expression
levels
To determine the effects of ALD on the expression
level and localization of p53, rats were randomly assigned to
receive normal saline or ALD for 4 weeks and the expression levels
of p53 were detected by immunohistochemistry. The level of p53
expression was markedly higher in the renal cortical sections from
ALD-infused rats, as compared with those from the control group,
and p53 protein was predominantly expressed in the renal cortex
(Fig. 2). Conversely, there was
little p53 protein expressed in the normal saline tissue samples.
These results suggested that ALD induced the expression of p53 that
was observed in the renal cortical sections.
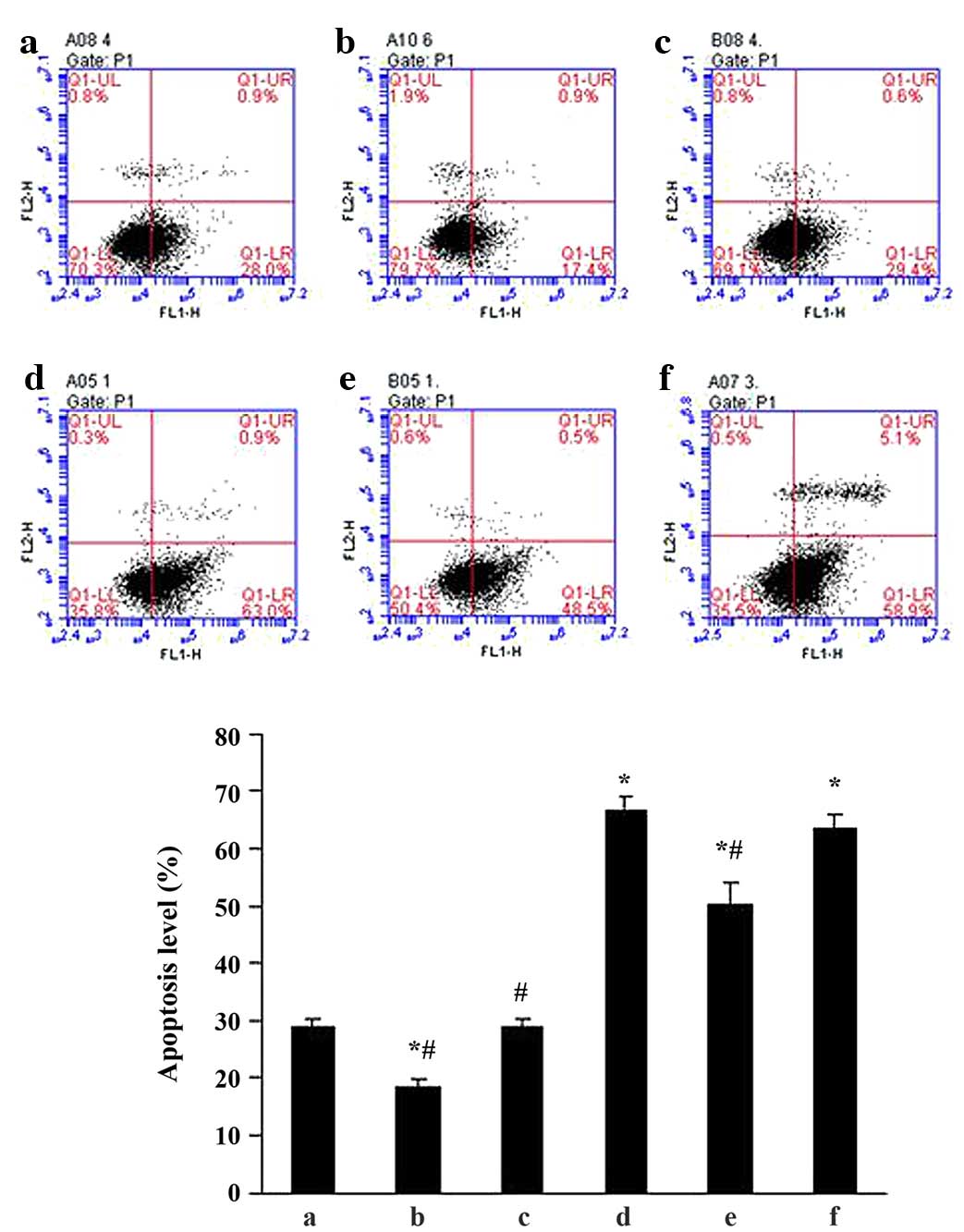
Effects of ALD on MC apoptosis in
vitro
To determine the effects of ALD on MC apoptosis,
standard flow cytometry was performed. MCs were transfected with
either an empty expression vector (pGPU6-NC-shRNA) or a
p53-silenced vector (pGPU6/GFP/Neo-shRNA) for 48 h separately, and
co-cultured with or without 10−6 M ALD for 24 h. MC
apoptosis was measured by flow cytometry (Fig. 3) and the ALD group demonstrated a
significantly higher level of apoptotic cells than the control
group (29.11±1.33% vs. 66.55±2.55%; P<0.05). However, this
effect of ALD was significantly inhibited by knockdown of p53 with
siRNA (50.30±3.60%; P<0.05). The results indicated that ALD
increased the MC apoptosis ratio by upregulating p53 expression
(Fig. 3).
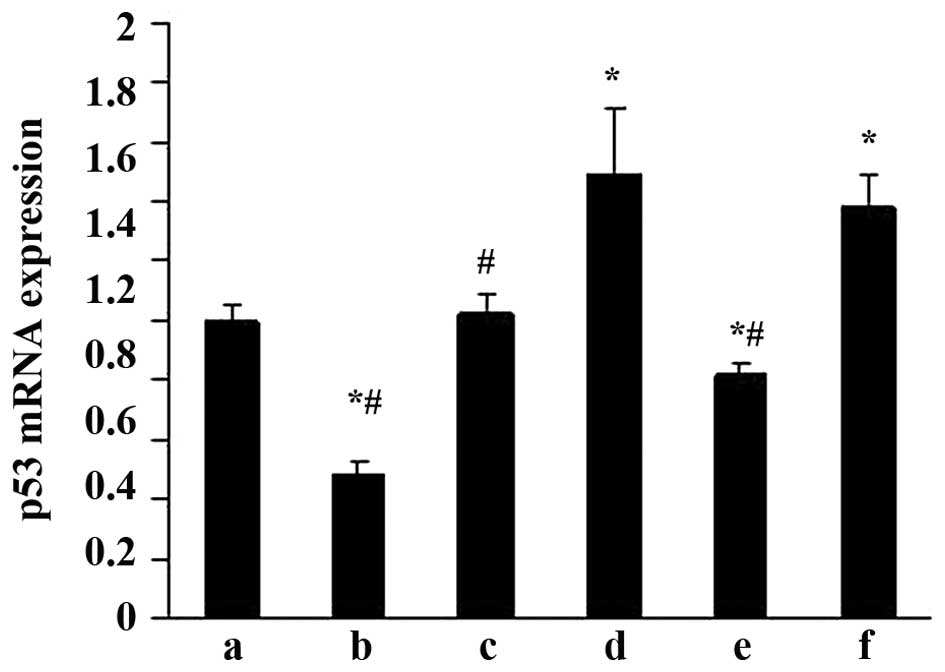
Effects of ALD on p53 mRNA expression in
rat MCs
RT-qPCR was performed to evaluate the effects of ALD
on p53 mRNA expression. MCs were transfected with either an empty
expression vector (pGPU6-NC-shRNA) or a p53-silenced vector
(pGPU6/GFP/Neo-shRNA) for 48 h separately. The MCs were then
co-cultured with or without 10−6 M ALD for 24 h and the
p53 mRNA expression level was measured by RT-qPCR (Fig. 4). Taking the mRNA level in the
control as 1, the p53 mRNA level was significantly increased by ALD
(1.49±0.23; P<0.05); however, these changes were significantly
suppressed by knockdown of p53 with siRNA (0.82±0.03;
P<0.05).
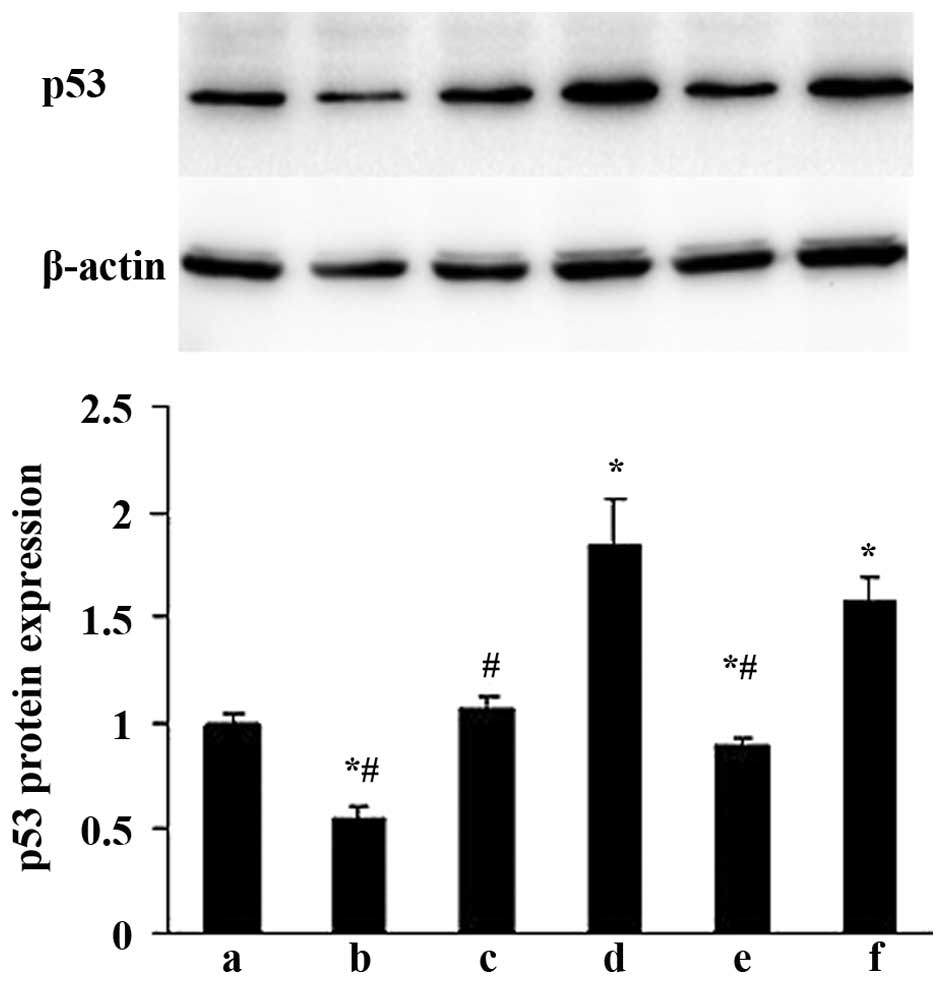
Effects of ALD on p53 protein expression
in rat MCs
The effects of ALD on p53 protein expression were
examined by western blotting. MCs were transfected with either an
empty expression vector (pGPU6-NC-shRNA) or a p53-silenced vector
(pGPU6/GFP/Neo-shRNA) for 48 h separately, and co-cultured with or
without 10−6 M ALD for 24 h. The p53 protein expression
was measured by western blotting (Fig.
5). Compared with the control group, the p53 protein level was
significantly upregulated in the ALD group at the end of the
incubation period (1.00±0.05 vs. 1.83±0.24; P<0.05); whereas the
expression level of the p53 protein was significantly decreased
following knockdown of p53 with siRNA (0.89±0.03; P<0.05).
Discussion
In the present study, the role of p53 in ALD-induced
MC injury was investigated. In vivo studies demonstrated
that ALD infusion significantly increased SBP, the kidney/body
weight ratio, and the urinary protein/creatinine ratio.
Additionally, renal cortical sections from ALD-infused rats showed
a greater numbers of TUNEL-positive MCs and higher levels of p53
expression. Furthermore, ALD promoted cell apoptosis in cultured
MCs, and increased the expression levels of p53 mRNA and protein.
The pro-apoptotic effect of ALD was attenuated by knockdown of p53
with siRNA. The results of the present study indicate that the
pro-apoptotic effect of ALD on MCs is mediated through p53.
ALD, a key component of the circulating
renin-angiotensin-ALD system, has been increasingly recognized as
an important factor in the development and progression of CKD,
independent of its haemodynamic effects (4,5,27,28).
Previous studies show that ALD is associated with hypertension
(29), albuminuria (30) and glomerulosclerosis (31) as an independent agent in renal
failure and diabetic nephropathy models. In the present study, the
effects of ALD on MC apoptosis were investigated. It was found that
ALD-treated rats showed a greater number of TUNEL-positive MCs,
which is indicative of the pro-apoptotic effect of ALD on MCs in
vivo. Furthermore, the ALD group showed higher levels of
apoptotic cells than the control group in cultured MCs, which
demonstrates that ALD promotes MC apoptosis in vivo and
in vitro, indicating the possible involvement of ALD in the
progression of renal injury. Although there have been numerous
studies on the role of ALD-induced renal injury, the exact
mechanisms responsible for the pro-apoptotic effects of ALD on
renal cells remain unclear.
Apoptosis is essential in the developmental
processes and tissue homeostasis of the majority of multicellular
organisms; deregulation of apoptosis has been implicated in the
pathogenesis and progression of many disease states (32). Cell apoptosis is an active response
to an altered microenvironment and is modulated by families of
proteins with a multitude of positive and negative regulators
acting at serial steps along a programmed pathway (33). The tumour suppressor, p53 is
crucial in the regulation of the cell cycle and apoptosis. In
response to a variety of stresses, such as DNA damage, the
wild-type p53 protein induces growth arrest at the G1/S
phase of the cell cycle until damage is repaired or until cell
suicide is triggered via apoptosis depending on the severity of the
damage. While muted p53 lost this function and cells with damaged
DNA were able to proliferate (34). p53 triggers apoptosis via
transcription-dependent and -independent signalling pathways. As a
transcriptional regulator, the activation of p53 may downregulate
the anti-apoptotic gene product, B-cell lymphoma 2 and upregulate
the pro-apoptotic gene product, BCL2-associated X protein to
trigger myocyte apoptosis (35–37).
The cyclin dependent kinase inhibitor,
p21Waf1/Cip1 is also a direct p53-dependent
target gene that induces the cell cycle arrest response to p53
(38). p53 is capable of
initiating apoptosis via transcription-independent pathways. Recent
studies demonstrated that WW domain-containing oxidoreductase is
involved in binding and stabilizing p53 in mitochondria and
deletion of this gene significantly abolishes p53 apoptotic
function (39).
Since p53 is an important molecule that regulates
cell apoptosis, the expression of p53 was evaluated in renal
cortical tissue samples and in cultured MCs. The results of the
current study indicated that ALD-treated rats and ALD-cultured MCs
exhibited higher levels of p53 expression when compared with normal
controls, which indicated that ALD promotes p53 expression in
vivo and in vitro. Apoptosis is induced in MCs exposed
to ALD, while knockdown of p53 with siRNA significantly inhibited
ALD-induced apoptosis, which suggests the involvement of p53 in the
pro-apoptotic effect of ALD. Han et al (32) reported that cyclosporine A, also
induced MC apoptosis by upregulating the pro-apoptotic factor, p53.
In addition to MCs, ALD exerts a direct pro-apoptotic influence on
other kidney cells, including podocytes and proximal tubular
epithelial cells (10,11).
In conclusion, the present study indicates that ALD
induced MC apoptosis via p53 in vitro and in vivo.
These findings partly elucidate the potential mechanism by which
ALD induces kidney injury; however, additional studies involving a
mineralocorticoid receptor antagonist are required. Furthermore,
inhibiting the ALD system may represent a novel therapeutic
strategy to prevent MC injury in the progression of CKD.
Acknowledgments
The present study was supported by the Scientific
Research Program from Nanjing Medical University (grant nos.
2011NJMU251 and 2014NJMUZD068) and the Health department of
Nanjing, China (grant no. QYK11226).
References
|
1
|
White SL, Cass A, Atkins RC and Chadban
SJ: Chronic kidney disease in the general population. Adv Chronic
Kidney Dis. 12:5–13. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lozano R, Naghavi M, Foreman K, Lim S,
Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY, et
al: Global and regional mortality from 235 causes of death for 20
age groups in 1990 and 2010: A systematic analysis for the global
burden of disease study 2010. Lancet. 380:2095–2128. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bolignano D, Palmer SC, Navaneethan SD and
Strippoli GF: Aldosterone antagonists for preventing the
progression of chronic kidney disease. Cochrane Database Syst Rev.
4:CD0070042014.PubMed/NCBI
|
|
4
|
Greene EL, Kren S and Hostetter TH: Role
of aldosterone in the remnant kidney model in the rat. J Clin
Invest. 98:1063–1068. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lai L, Chen J, Hao CM, Lin S and Gu Y:
Aldosterone promotes fibronectin production through a
Smad2-dependent TGF-beta1 pathway in mesangial cells. Biochem
Biophys Res Commun. 348:70–75. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liang W, Chen C, Shi J, Ren Z, Hu F, van
Goor H, Singhal PC and Ding G: Disparate effects of eplerenone,
amlodipine and telmisartan on podocyte injury in
aldosterone-infused rats. Nephrol Dial Transplant. 26:789–799.
2011. View Article : Google Scholar :
|
|
7
|
Del Vecchio L, Procaccio M, Vigano S and
Cusi D: Mechanisms of disease: The role of aldosterone in kidney
damage and clinical benefits of its blockade. Nat Clin Pract
Nephrol. 3:42–49. 2007. View Article : Google Scholar
|
|
8
|
Nishiyama A, Yao L, Nagai Y, Miyata K,
Yoshizumi M, Kagami S, Kondo S, Kiyomoto H, Shokoji T, Kimura S, et
al: Possible contributions of reactive oxygen species and
mitogen-activated protein kinase to renal injury in
aldosterone/salt-induced hypertensive rats. Hypertension.
43:841–848. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fan YY, Baba R, Nagai Y, Miyatake A,
Hosomi N, Kimura S, Sun GP, Kohno M, Fujita M, Abe Y and Nishiyama
A: Augmentation of intrarenal angiotensin II levels in
uninephrectomized aldosterone/salt-treated hypertensive rats;
renoprotective effects of an ultrahigh dose of olmesartan.
Hypertens Res. 29:169–178. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kiyomoto H, Rafiq K, Mostofa M and
Nishiyama A: Possible underlying mechanisms responsible for
aldosterone and mineralocorticoid receptor-dependent renal injury.
J Pharmacol Sci. 108:399–405. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shibata S, Nagase M, Yoshida S, Kawachi H
and Fujita T: Podocyte as the target for aldosterone: Roles of
oxidative stress and Sgk1. Hypertension. 49:355–364. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nagase M and Fujita T: Endocrinological
aspects of proteinuria and podocytopathy in diabetes: Role of the
aldosterone/mineralocorticoid receptor system. Curr Diabetes Rev.
7:8–16. 2011. View Article : Google Scholar
|
|
13
|
Epstein M: Aldosterone blockade: An
emerging strategy for abrogating progressive renal disease. Am J
Med. 119:912–919. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhu D, Yu H, He H, Ding J, Tang J, Cao D
and Hao L: Spironolactone inhibits apoptosis in rat mesangial cells
under hyperglycaemic conditions via the Wnt signalling pathway. Mol
Cell Biochem. 380:185–193. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nagase M and Fujita T: Aldosterone and
glomerular podocyte injury. Clin Exp Nephrol. 12:233–242. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Merkel CE, Karner CM and Carroll TJ:
Molecular regulation of kidney development: Is the answer blowing
in the Wnt? Pediatr Nephrol. 22:1825–1838. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen C, Liang W, Jia J, van Goor H,
Singhal PC and Ding G: Aldosterone induces apoptosis in rat
podocytes: Role of PI3-K/Akt and p38MAPK signaling pathways.
Nephron Exp Nephrol. 113:e26–e34. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Abboud HE: Mesangial cell biology. Exp
Cell Res. 318:979–985. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Brunskill EW and Potter SS: Changes in the
gene expression programs of renal mesangial cells during diabetic
nephropathy. BMC Nephrol. 13:702012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dalla Vestra M, Saller A, Mauer M and
Fioretto P: Role of mesangial expansion in the pathogenesis of
diabetic nephropathy. J Nephrol. 14(Suppl 4): S51–S57. 2001.
|
|
21
|
Kitamura H, Shimizu A, Masuda Y, Ishizaki
M, Sugisaki Y and Yamanaka N: Apoptosis in glomerular endothelial
cells during the development of glomerulosclerosis in the
remnant-kidney model. Exp Nephrol. 6:328–336. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shimizu A, Masuda Y, Kitamura H, Ishizaki
M, Sugisaki Y and Yamanaka N: Apoptosis in progressive crescentic
glomerulonephritis. Lab Invest. 74:941–951. 1996.PubMed/NCBI
|
|
23
|
Mishra R, Emancipator SN, Kern T and
Simonson MS: High glucose evokes an intrinsic proapoptotic
signaling pathway in mesangial cells. Kidney Int. 67:82–93. 2005.
View Article : Google Scholar
|
|
24
|
Sugiyama H, Kashihara N, Makino H,
Yamasaki Y and Ota A: Apoptosis in glomerular sclerosis. Kidney
Int. 49:103–111. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ford HL, Sclafani RA and DeGregori J: Cell
cycle and growth control: Biomolecular regulation and cancer. Stein
GSaP AB: Cell cycle regulatory cascades Hoboken, New Jersey:
Wiley-Liss; pp. 95–128. 2004
|
|
26
|
Livak and Schmittgen: Analysis of relative
gene expression data using real-time quantitative PCR and the
2−ΔΔCt method. Methods. 25:402–408. 2001. View Article : Google Scholar
|
|
27
|
Chrysostomou A and Becker G:
Spironolactone in addition to ACE inhibition to reduce proteinuria
in patients with chronic renal disease. N Engl J Med. 345:925–926.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rocha R, Chander PN, Zuckerman A and Stier
CT Jr: Role of aldosterone in renal vascular injury in stroke-prone
hypertensive rats. Hypertension. 33:232–237. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Blasi ER, Rocha R, Rudolph AE, Blomme EA,
Polly ML and McMahon EG: Aldosterone/salt induces renal
inflammation and fibrosis in hypertensive rats. Kidney Int.
63:1791–1800. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Miric G, Dallemagne C, Endre Z, Margolin
S, Taylor SM and Brown L: Reversal of cardiac and renal fibrosis by
pirfenidone and spironolactone in streptozotocin-diabetic rats. Br
J Pharmacol. 133:687–694. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Aldigier JC, Kanjanbuch T, Ma LJ, Brown NJ
and Fogo AB: Regression of existing glomerulosclerosis by
inhibition of aldosterone. J Am Soc Nephrol. 16:3306–3314. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Han SY, Chang EJ, Choi HJ, Kwak CS, Park
SB, Kim HC and Mun KC: Apoptosis by cyclosporine in mesangial
cells. Transplant Proc. 38:2244–2246. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Oltvani ZN and Korsmeyer SJ: Checkpoints
of dueling dimers foli death wishes. Cell. 79:189–192. 1994.
View Article : Google Scholar
|
|
34
|
Vogelstein B and Kinzler KW: p53 function
and dysfunction. Cell. 70:523–526. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Miyashita T and Reed JC: Tumor suppressor
p53 is a direct transcriptional activator of the human bax gene.
Cell. 80:293–299. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pierzchalski P, Reiss K, Cheng W, Cirielli
C, Kajstura J, Nitahara JA, Rizk M, Capogrossi MC and Anversa P:
p53 induces myocyte apoptosis via the activation of the
renin-angiotensin system. Exp Cell Res. 234:57–65. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Leri A, Fiordaliso F, Setoguchi M, Limana
F, Bishopric NH, Kajstura J, Webster K and Anversa P: Inhibition of
p53 function prevents renin-angiotensin system activation and
stretch-mediated myocyte apoptosis. Am J Pathol. 157:843–857. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
el-Deiry WS: Regulation of p53 downstream
genes. Semin Cancer Biol. 8:345–357. 1998. View Article : Google Scholar
|
|
39
|
Chang NS: A potential role of p53 and WOX1
in mitochondrial apoptosis (review). Int J Mol Med. 9:19–24.
2002.
|