Introduction
Congenital fibrosis of the extraocular muscles
(CFEOM) is a hereditary ocular motility disorder characterized by
non-progressive restrictive ophthalmoplegia and ptosis (1). CFEOM can be classified into three
types, based on their phenotypes and inheritance patterns: CFEOM
type 1 (CFEOM1; OMIM 135700), type 2 (CFEOM2; OMIM602078) and type
3 (CFEOM3; OMIM 600638, 607034). Of these, CFEOM1 and CFEOM3 can be
inherited in an autosomal dominant manner, whereas CFEOM2 is
inheritable in an autosomal recessive manner. Three disease-causing
genes, Homo sapiens kinesin family member 21A (KIF21A),
paired-like homeobox 2a (ARIX or PHOX2A) and tubulin
β3 (TUBB3) have been identified to be associated with CFEOM
(2–4).
Known as 'classic CFEOM', CFEOM1 is the most common
form of CFEOM. It is a distinct, non-syndromic, congenital cranial
dysinnervation disorder, and is characterized by congenital
non-progressive bilateral external ophthalmoplegia, manifesting as
restricted vertical and horizontal ocular motility and ptosis,
leading to droopy eyelids and a chin-up position of the head
(5). Heterozygous missense
mutations in KIF21A have been identified to cause the
classic CFEOM1 phenotype, and sporadic cases result from de
novo mutations in KIF21A. In the 38-exon gene, only
three exons have been reported to harbor mutations: Exon 21
(2860C>T, 2861G>A, 2861G>T), exon 8 (1067T>C) and exon
20 (2830G>C, 2839A>G, 2840T>C, 2840T>G) (6,7).
CFEOM3 represents a form of CFEOM, which does not
meet the classic criteria for CFEOM1 or CFEOM2. It is less common
than CFEOM1 and, compared with CFEOM1, individuals with CFEOM3
demonstrate variable expressivity of CFEOM, and at least one
affected individual lacks or has only unilateral ophthalmoplegia or
ptosis, or has one or both eyes fixed above the midline, or has the
ability to raise one or both eyes above the midline (8,9).
CFEOM3 is often autosomal dominant with variable penetrance. The
majority of families with CFEOM3 have been mapped to 16qter
(10–12) and result from a heterozygous
mutation in TUBB3 (3).
However in 9% of families, CFEOM3 is reported to map to the CFEOM1
locus and harbor KIF21A mutations (9).
The present study identified two Chinese families,
one with CFEOM1 and one with CFEOM3. The genetic basis was
examined, and the clinical phenotypes of CFEOM1 and CFEOM3 were
described.
Materials and methods
Family recruitment and clinical
evaluation
Two families with CFEOM were identified. Following
the provision of informed consent from all participating
individuals or their parents, each participating individual was
subjected to comprehensive ophthalmic examinations. Venous blood
samples were collected from eight members of the two families and
from 200 unrelated control subjects from the same population.
Genomic DNA was extracted from peripheral blood leucocytes using
standard protocols. The present study was performed, according to
the principles of the Declaration of Helsinki and approved by the
Ethics Committee of Zhongshan Ophthalmic Center, Sun Yat-Sen
University (Guangzhou, China).
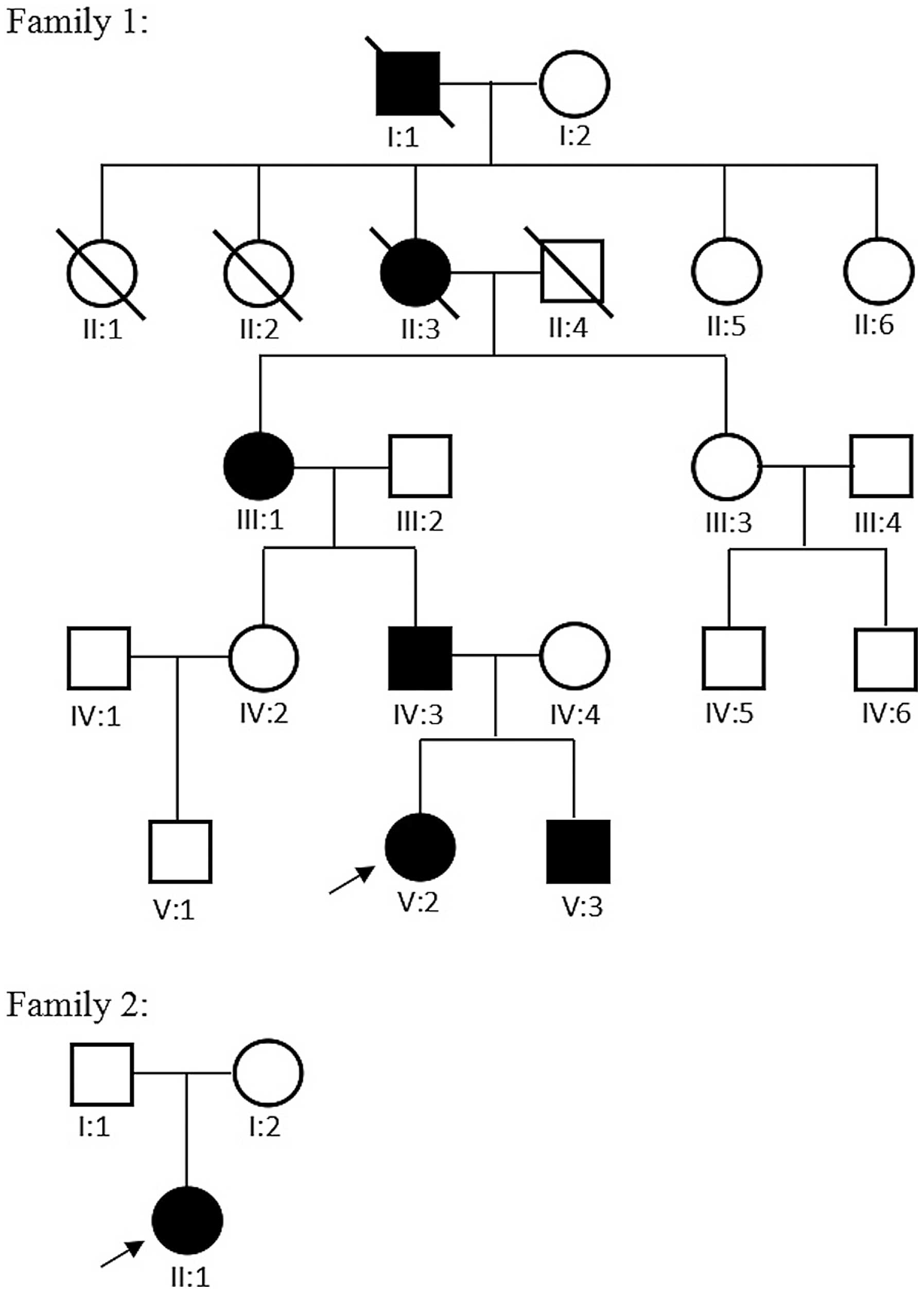
The two families were referred to Zhongshan
Ophthalmic Center, Sun Yat-Sen University, China in April 2015 for
severely restrictive strabismus and ptosis, present in the
probands. All eight family members, comprising IV:2, IV:3, IV:4,
V:2 and V:3 in family 1, and I:1, I:2 and II:1 in family 2
(Fig. 1), which included the four
affected individuals and their asymptomatic parents or siblings,
were subjected to comprehensive ophthalmic examinations (Table I), including routine ophthalmic
examination and strabismus assessments. The primary position of
gaze, ductions and versions with a cover test were analyzed and
quantified in six diagnostic positions. The affected individuals
underwent forced duction assessment under topical anesthesia. Globe
retraction or aberrant movements were observed. The width of the
palpebral fissure was measured. Levator function was measured from
the upper lid margin during attempted supraduction from the
infraducted position without recruitment of the frontalis muscle.
Ptosis was defined by a ≥2 mm covering of the iris by the upper
lid, and was graded as mild if the upper lid covered the iris above
the upper pupillary margin, moderate if the lid occluded half the
pupil and severe if the lid occluded more than half the pupil
(13). Visual acuity and
cycloplegic refraction were also determined.
 | Table IClinical presentations of the four
affected members. |
Table I
Clinical presentations of the four
affected members.
| Parameter | Family 1: IV:3 | Family 1: V:2 | Family 1: V:3 | Family 2: II:1 |
|---|
| Age (years) | 34 | 6 | 3 | 4 |
| Gender | Male | Female | Male | Female |
| Birth history | Full-term | Full-term | Full-term | Full-term |
| BCVA (decimals) |
| OD | 0.4 | 0.2 | Unable | 0.1 |
| OS | 0.3 | 0.2 | Unable | 0.2 |
| Refractive error |
| OD | +0.50/+2.00 | +2.00/+5.00 | +2.00/+1.00 | −0.25/+1.00 |
| OS | +1.00/+2.50 | +1.50/+2.50 | +2.00/+1.50 | +1.25/+1.75 |
| Stereopsis
(butterfly) | Nila | Nil | Nil | Nil |
| Strabimus | Hypo with Eso | Hypo with Eso | Hypo | Hypo with Eso |
| Eye movement |
| OD | Restrictive UAHM | Fixed | Restrictive UAHM | Restrictive UAHM and
HM |
| OS | Restrictive UAHM and
HM | Fixed | Restrictive UAHM | Variable restrictive
upgaze and HM |
| Ptosis |
| OD | Moderate | Severe | Severe | No |
| OS | Severe | Severe | Severe | Severe |
| Bell's
phenomenon | Absent | Absent | Absent | Absent |
| Forced duction
test |
| OD | Positive | Unable | Unable | Unable |
| OS | Positive | Unable | Unable | Unable |
| Nystagmus | On upgaze | In all directions
of gaze | Absent | Absent |
| Synergistic
convergence | On upgaze | In all directions
of gaze | Absent | Absent |
| Pupil size | Normal | Normal | Normal | Normal |
| Pupillary light
reflex | Normal | Normal | Normal | Normal |
| Compensatory head
position | Chin up | Chin up and mouth
open | Chin up | Chin up |
| Proptosis
ocular | No | No | No | No |
| Upper eyelid
lag | No | No | No | No |
| Other ophthalmic
examinations | Unremarkable | Unremarkable | Unremarkable | Unremarkable |
Mutation screening and sequence
analysis
A 5 ml venous blood sample was collected from each
participant for genomic DNA extraction. DNA extraction from
peripheral blood leukocytes was performed using a DNA extraction
kit (Qiagen, Inc., Valencia, CA, USA) with standard protocols.
Ethylene diaminetetraacetic acid-treated tubes were used for blood
collection. Exons and flanking exon-intron boundaries of the
KIF21A gene (38 exons) were amplified using polymerase chain
reaction (PCR) analysis with the primers (14) (Beijing Genomics Institute,
Guangzhou, China). Briefly, PCR was performed in a 50 μl
reaction volume with 2 μl each primer, 2 μl DNA, 25
μl buffer mix and 19 μl H2O. All reagents
used for PCR were purchased from Takara Bio, Inc. (Tokyo, Japan).
The cycling profile was as follows: One cycle at 94°C for 5 min,
followed by 40 cycles at 94°C for 45 sec, 59°C for 45 sec and 72°C
for 45 sec, with a final cycle at 72°C for 10 min (14). The PCR products were sequenced from
both directions using an ABI3730 automated sequencer (PE
Biosystems, Foster City, CA, USA). The sequencing results were
analyzed using Seqman (version 2.3; Technelysium Pty, Ltd.,
Brisbane, QLD, Australia). Variations were identified by aligning
sequences to the reference sequences from the National Center for
Biotechnology Information database (NCBI; http://www.ncbi.nlm.nih.gov/). Detected variations
were further analyzed by cosegregation analysis in the eight
available family members and the normal control subjects.
Results
Clinical findings
Family and personal histories were carefully
reviewed. The two families were from the Guangdong province of
China. The diagnosis of CFEOM1 was based on inheritance patterns
and clinical phenotypes, and this family was designated as family
1. In family 1, there were six affected family members with CFEOM
in five generations (Fig. 1). The
three affected members available for investigation (IV:3, V:2, V:3)
shared the typical clinical features of CFEOM1, which have been
reported previously (5). These
features included bilateral congenital non-progressive ptosis,
severely impaired vertical motility with an inability to raise
either eye above the horizontal midline and variable impaired
horizontal motility, infraducted position in primary gaze, a
chin-up position of the head and an absence of Bell's phenomenon.
Synergistic convergence on attempted upgaze was also observed in
IV:3 and V:2. Of the three patients, one exhibited nystagmoid
movements in all directions of gaze, one exhibited nystagmoid
movements only in upgaze and the third exhibited no nystagmoid
movements. The forced duction test demonstrated marked restrictions
in passive elevation of the globes in IV:3. Family members V:2 and
V:3 were unable to participate in the forced duction test. IV:3 and
V:2 had poor visual acuity, and V:3 was unable to be assessed for
visual acuity. No notable findings were detected in the
examinations of the anterior segments or fundus.
In family 2, there were no other affected family
members in the four generations. The proband presented with the
typical clinical features of CFEOM3. Specifically, the proband had
unilateral, congenital, non-progressive ptosis, variable restricted
upgaze and horizontal eye movements, and was able to elevate left
eye above the midline. The chin-elevated position of the head and
the absence of Bell's phenomenon were similar to the observations
with CFEOM1. The clinical presentations of the four affected
members are listed in Table I.
Examples of the compensatory head position and primary images of
the unaltered eyelid and nine-gaze positions of the probands are
shown in Figs. 2 and 3. The range of extraocular movements and
the position of the eyes in the primary position of the four
affected members are shown in Fig.
4.
 | Figure 2Images of the proband from family 1.
Images were captured of the (A) compensatory head position, in (B)
primary gaze with unaltered eyelids and in (C–K) nine-gaze
positions with lifted eyelids (in sequence C–K, looking up and
right, straight up, up and left, straight right, straight, straight
left, down and left, straight down, down and left). This individual
exhibited typical CFEOM1 features, including bilateral ptosis,
infraducted globe position in primary gaze, severely restricted
upgaze with an inability to raise the eyes above horizontal
midline, variable impaired horizontal motility and chin-up position
of the head. |
 | Figure 3Images of the proband from family 2.
Images were captured of the (A) compensatory head position, in (B)
primary gaze with unaltered eyelids and in (C–K) nine-gaze
positions with lifted eyelids (in sequence C–K, looking up and
right, straight up, up and left, straight right, straight, straight
left, down and left, straight down, down and left). This individual
exhibited typical CFEOM3 features, including unilateral ptosis,
variable restricted upgaze and horizontal eye movements, with an
ability to elevate the left eye above the midline, and chin-up
position of the head. |
Mutation screening
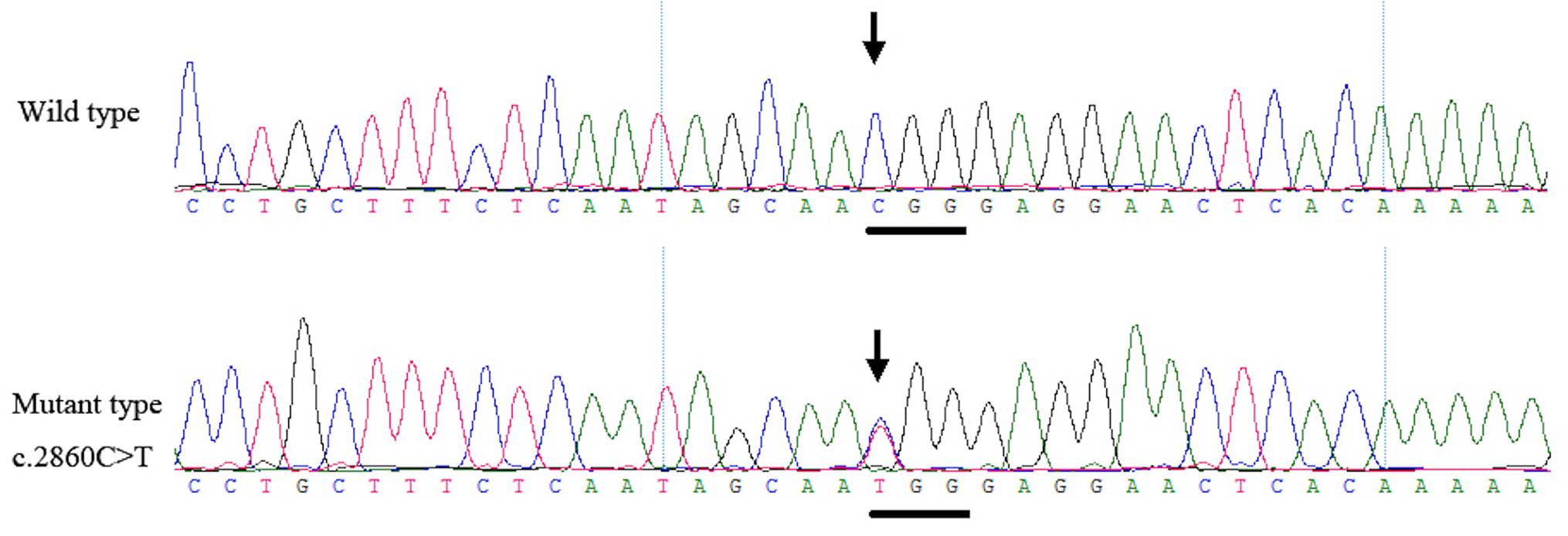
Genetic analysis documented the presence of a
heterozygous mutation, c.2860C>T, in the KIF21A gene
within all four patients of the two families. This mutation was not
observed in the unaffected family members or in the 200 unrelated
control subjects from the same population (Fig. 5).
Discussion
In the present study, a known mutation
(c.2860C>T) in KIF21A was identified in two Chinese
families with CFEOM1 and CFEOM3. The clinical findings revealed the
phenotypic heterogeneity present within the three members of family
1 affected with CFEOM1.
The diagnosis of CFEOM1 in family 1 was based upon
an autosomal dominant inheritance pattern, and was characterized by
bilateral congenital ptosis, bilateral infraducted globe position
in the primary gaze and a severely restricted upgaze with variable
horizontal eye movements. In CEEOM1, all affected members of a
family exhibit classic presentations of CFEOM and show dominant
inheritance. By contrast, in families with CFEOM3, one or more
affected individuals fail to exhibit the classic characteristics of
this disorder. For example, the individual's eyes may not be
infraducted or may elevate above the midline, the individual may be
unilaterally affected, or ptosis may be absent. In the proband of
family 2, unilateral ptosis and variable restrictive upgaze
indicated a diagnosis of CFEOM3, rather than CFEOM1. Among the
three affected members in family 1, IV:3 and V:2 exhibited
nystagmus and synergistic convergence, whereas no such aberrant eye
movements were recorded in V:3. It appears likely that aberrant
innervation may be responsible for the severity of restricted eye
movements. Among the three affected members in family 1, both IV:3
and V:2 exhibited nystagmus as well as synergistic convergence,
whereas no such aberrant eye movements were present in V:3.
CFEOM1 is inherited as a fully penetrant, autosomal
dominant trait and maps to the CFEOM1 locus on chromosome 12cen
(15–18). The disease-causing gene,
KIF21A, belongs to a family of kinesin motor proteins, which
are responsible for the transport of membranous organelles, protein
complexes and mRNAs to specific destinations within the cell, in a
microtubule and ATP-dependent manner. These functions are essential
for the normal morphogenesis and functioning of the cell (19). Results from previous studies have
indicated that the KIF21A gene product may have a
generalized effect in the development of all the ocular motor
nuclei of cranial nerves III, IV and VI (6,7).
To date, 12 missense mutations and one deletion have
been identified in the KIF21A gene in patients presenting
with CFEOM1 (4,5,7,20,21).
Among these, the c.2860C>T mutation has been identified as the
most common mutation, with a prevalence of between 72 and 75%
(4,22). The c.2860C>T mutation is located
in a methylated CpG (mCpG) dinucleotide of the gene (23) and these mCpG sequences represent
mutational hotspots in human genetic diseases. Random deamination
of 5-methylcytosines produces these mutations (24). Considerable evidence exists
indicating that the CFEOM1 phenotype results from mutations in
KIF21A and that sporadic cases are due to de novo
mutations in the same gene (7,19).
The occurrence of this mutation in the families in the present
study suggests a mutation hot spot.
The proband in family 2 also harbored the most
commonly observed KIF21A mutation, c.2860C>T.
KIF21A mutations are reported to be a rare cause of CFEOM3
(9). However, the KIF21A
mutation has been reported in a Taiwanese pedigree (25) and in families from other regions of
China with CFEOM3 (5). To date,
five Chinese families with CFEOM3 have been reported with
identified mutations (5,25–27),
with four sharing the same KIF21A mutation and the fifth
harboring the TUBB3 gene mutation. This suggests that the
KIF21A mutation may be a hot spot mutation of CFEOM3 in
Chinese individuals.
In the present study, a heterozygous KIF21A
c.2860C>T mutation was found to be present in a family with
CFEOM1 and a family with CFEOM3. Identical gene mutations causing
distinct phenotypes were observed in CFEOM. Taken together, it is
reasonable to conclude that the KIF21A mutation may be a
major disease-causing gene for CFEOM3 in Chinese individuals.
Abbreviations:
|
CFEOM
|
congenital fibrosis of the extraocular
muscles
|
|
PCR
|
polymerase chain reaction
|
References
|
1
|
Brown HW: Congenital structural muscle
anomalies. Strabismus Ophthalmic Symposium. Allen JH: CV Mosby; St
Louis: pp. 205–236. 1950
|
|
2
|
Nakano M, Yamada K, Fain J, Sener EC,
Selleck CJ, Awad AH, Zwaan J, Mullaney PB, Bosley TM and Engle EC:
Homozygous mutations in ARIX (PHOX2A) result in congenital fibrosis
of the extraocular muscles type 2. Nat Genet. 29:315–320. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tischfield MA, Baris HN, Wu C, Rudolph G,
Van Maldergem L, He W, Chan WM, Andrews C, Demer JL, Robertson RL,
et al: Human TUBB3 mutations perturb microtubule dynamics, kinesin
interactions and axon guidance. Cell. 140:74–87. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yamada K, Andrews C, Chan WM, McKeown CA,
Magli A, de Berardinis T, Loewenstein A, Lazar M, O'Keefe M, Letson
R, et al: Heterozygous mutations of the kinesin KIF21A in
congenital fibrosis of the extraocular muscles type 1 (CFEOM1). Nat
Genet. 35:318–321. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lu S, Zhao C, Zhao K, Li N and Larsson C:
Novel and recurrent KIF21A mutations in congenital fibrosis of the
extraocular muscles type 1 and 3. Arch Ophthalmol. 126:388–394.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Traboulsi EI and Engle EC: Mutations in
KIF21A are responsible for CFEOM1 worldwide. Ophthalmic Genet.
25:237–239. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chan WM, Andrews C, Dragan L, Fredrick D,
Armstrong L, Lyons C, Geraghty MT, Hunter DG, Yazdani A, Traboulsi
EI, et al: Three novel mutations in KIF21A highlight the importance
of the third coiled-coil stalk domain in the etiology of CFEOM1.
BMC Gent. 8:262007. View Article : Google Scholar
|
|
8
|
Engle EC: The molecular basis of the
congenital fibrosis syndromes. Strabismus. 10:125–128. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yamada K, Chan WM, Andrews C, Bosley TM,
Sener EC, Zwaan JT, Mullaney PB, Ozturk BT, Akarsu AN, Sabol LJ, et
al: Identification of KIF21A mutations as a rare cause of
congenital fibrosis of the extraocular muscles type 3 (CFEOM3).
Invest Ophthalmol Vis Sci. 45:2218–2223. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Doherty EJ, Macy ME, Wang SM, Dykeman CP,
Melanson MT and Engle EC: CFEOM3: A new extraocular congenital
fibrosis syndrome that maps to 16q24.2–q24.3. Invest Ophthalmol Vis
Sci. 40:1687–1694. 1999.PubMed/NCBI
|
|
11
|
Gillies WE, Harris AJ, Brooks AM, Rivers
MR and Wolfe RJ: Congenital fibrosis of the vertically acting
extraocular muscles. A new group of dominantly inherited ocular
fibrosis with radiologic findings. Ophthalmology. 102:607–612.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mackey DA, Chan WM, Chan C, Gillies WE,
Brooks AM, O'Day J and Engle EC: Congenital fibrosis of the
vertically acting extraocular muscles maps to the FEOM3 locus.
Human Genet. 110:510–512. 2002. View Article : Google Scholar
|
|
13
|
Sener EC, Lee BA, Turgut B, Akarsu AN and
Engle EC: A clinically variant fibrosis syndrome in a Turkish
family maps to the CFEOM1 locus on chromosome 12. Arch Ophthalmol.
118:1090–1097. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Khan AO, Shinwari J, Omar A, Al-Sharif L,
Khalil DS, Alanazi M, Al-Amri A and Al Tassan N: Lack of KIF21A
mutations in congenital fibrosis of the extraocular muscles type I
patients from consanguineous Saudi Arabian families. Mol Vis.
17:218–224. 2011.PubMed/NCBI
|
|
15
|
Engle EC, Kunkel LM, Specht LA and Beggs
AH: Mapping a gene for congenital fibrosis of the extraocular
muscles to the centromeric region of chromosome 12. Nat Genet.
7:69–773. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Engle EC, Marondel I, Houtman WA, de Vries
B, Loewenstein A, Lazar M, Ward DC, Kucherlapati R and Beggs AH:
Congenital fibrosis of the extraocular muscles (autosomal dominant
congenital external ophthalmoplegia): Genetic homogeneity, linkage
refinement, and physical mapping on chromosome 12. Am J Hum Genet.
57:1086–1094. 1995.PubMed/NCBI
|
|
17
|
Engle EC, McIntosh N, Yamada K, Lee BA,
Johnson R, O'Keefe M, Letson R, Lodon A, Ballard E, Ruttum M, et
al: CFEOM1, the classic familial form of congenital fibrosis of the
extraocular muscles, is genetically heterogeneous but does not
result from mutations in ARIX. BMC Genet. 3:32002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Uyama E, Yamada K, Kawano H, Chan WM,
Andrews C, Yoshioka M, Uchino M and Engle EC: A Japanese family
with FEOM1-linked congenital fibrosis of the extraocular muscles
type 1 associated with spinal canal stenosis and refinement of the
FEOM1 critical region. Neuromuscul Disord. 13:472–478. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Miki H, Setou M, Kaneshiro K and Hirokawa
N: All kinesin superfamily protein, KIF, genes in mouse and human.
Proc Natl Acad Sci USA. 98:7004–7011. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yamada K, Hunter DG, Andrews C and Engle
EC: A novel KIF21A mutation in a patient with congenital fibrosis
of the extraocular muscles and Marcus Gunn jaw-winking phenomenon.
Arch Ophthalmol. 123:1254–1259. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang P, Li S, Xiao X, Guo X and Zhang Q:
KIF21A novel deletion and recurrent mutation in patients with
congenital fibrosis of the extraocular muscles-1. Int J Mol Med.
28:973–975. 2011.PubMed/NCBI
|
|
22
|
Li ND, Zhao J, Wang LM, Chen X, Ma HZ, Zhu
LN, Guo X and Zhao KX: R954 mutations in KIF21A gene in Chinese
patients with congenital fibrosis of extraocular muscles. Zhonghua
Yan Ke Za Zhi. 48:1077–1082. 2012.In Chinese.
|
|
23
|
Ali M, Venkatesh C, Ragunath A and Kumar
A: Mutation analysis of the KIF21A gene in an Indian family with
CFEOM1: Implication of CpG methylation for most frequent mutations.
Ophthalmic Genet. 25:247–255. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pfeifer GP: Mutagenesis at methylated CpG
sequences. Curr Top Microbiol Immunol. 301:259–281. 2006.PubMed/NCBI
|
|
25
|
Lin LK, Chien YH, Wu JY, Wang AH, Chiang
SC and Hwu WL: KIF21A gene c.2860C>T mutation in congenital
fibrosis of extraocular muscles type 1 and 3. Mol Vis. 11:245–248.
2005.PubMed/NCBI
|
|
26
|
Yang X, Yamada K, Katz B, Guan H, Wang L,
Andrews C, Zhao G, Engle EC, Chen H, Tong Z, et al: KIF21A
mutations in two Chinese families with congenital fibrosis of the
extra-ocular muscles (CFEOM). Mol Vis. 16:2062–2070.
2010.PubMed/NCBI
|
|
27
|
Zhang J, Zhou L, Zha Y, Liu T, Tian M,
Yuan J and Xing Y: The gene mutation screening of a family with
congenital fibrosis of the extraocular muscles associated with
corpus callosum agenesis. Zhonghua Yan Ke Za Zhi. 49:621–626.
2013.In Chinese. PubMed/NCBI
|