Introduction
Disruption of the blood-brain barrier (BBB) and the
resulting edema are crucial initiating factors in cerebral
ischemia/reperfusion injury, as they increase intracranial
pressure, impair cerebral perfusion and oxygenation, and contribute
to additional ischemic injuries (1–3). A
potential therapeutic target in this process is vascular
endothelial growth factor (VEGF). As a pleiotropic growth factor,
VEGF is associated with angiogenesis, neurogenesis, axonal
plasticity, neuronal survival and vascular permeability (4). It has been demonstrated that VEGF
expression is upregulated in the peri-infarct regions within 3 h
following the onset of ischemia (5). In addition, the expression of VEGF is
closely associated with BBB leakage in the acute ischemic brain
(6). It has been reported that
early administration of VEGF to ischemic rat brains may enhance BBB
leakage, induce hemorrhagic transformation and increase infarct
size in the brain (6). Therefore,
applying an inhibitor of VEGF in the early acute phase of a stroke
may reduce the breakdown of the BBB, as well as the subsequent
edema.
Matrix metalloproteinases (MMPs) are zinc-containing
endopeptidases that digest the components of the extracellular
matrix that form the basal lamina surrounding the neurovascular
unit (7). Numerous proinflammatory
agents that are trigged by ischemia can activate MMP expression
immediately after a stroke (8). In
acute ischemia, MMP-2 and MMP-9 in particular, mediated degradation
of tight junction proteins (TJPs) in the BBB (9). It has been demonstrated that MMP
inhibition is an important therapeutic strategy to reduce brain
damage during acute stroke (8).
Several studies have suggested a strong relationship between the
induction of MMP-2 and MMP-9 and brain edema following cerebral
ischemic injury (10–12), indicating that MMP-2 and MMP-9 may
be important targets to prevent edema complications following
cerebral ischemia.
There is growing evidence to demonstrate that early
VEGF inhibition has a protective effect against cerebral ischemia
(13–16). However, it remains unclear whether
VEGF inhibition protects the BBB by regulating the expression of
MMPs following acute cerebral ischemia. Therefore, the aim of the
present study was to investigate the possible mechanisms underlying
early VEGF inhibition in the protection of the BBB following
ischemic brain injury, by evaluating ischemic lesions, brain edema,
neurological outcomes and MMP levels in a rat stroke model. The
results from the current study indicated that the possible
mechanisms underlying VEGF inhibition-mediated protection of the
BBB involved the MMP pathway.
Materials and methods
Animals
A total of 156 adult male Sprague-Dawley rats (age,
9 weeks; weight, 250–300 g) were purchased from the Animal
Experimental Center of Harbin Medical University (Harbin, China).
All of the animals were housed in individual cages under a 12-h
light/dark cycle at 22–24°C. The rats had free access to food and
water and were fasted for 12 h prior to surgery. The Harbin Medical
University Animal Supervision Committee approved all experimental
procedures. All efforts were made to minimize animal suffering and
the number of animals used.
Experimental transient middle cerebral
artery occlusion (MCAO) model
All 156 animals were anesthetized with an
intraperitoneal injection of 10% chloral hydrate (350 mg/kg;
Sinopharm Chemical Reagent Co., Ltd., Shanghai, China). Body
temperature was monitored continuously with a rectal probe and
maintained at 37±0.5°C by use of a heat pad. MCAO was induced using
the methods described previously (17). Following 90 min of MCAO,
reperfusion was initiated by withdrawing the monofilament. Sham
rats underwent anesthesia and surgery without the filament
insertion. At 1 h following reperfusion, neurological function was
evaluated according to Longa's neurological score criteria, with a
range from 0 (normal) to 4 (severe symptoms) (17). Rats with neurological scores ≤2,
breathing difficulties or serious bleeding (n=6) were excluded from
the study.
Experimental groups and in vivo drug
treatment
To investigate the role of VEGF in
ischemia-reperfusion-induced brain injury, the effects of a
neutralizing antibody against VEGF were investigated (RB-222;
NeoMarkers, Inc., Portsmouth, NH, USA). The rats were randomly
assigned into the following five groups (n=30 per group): Sham,
MCAO, IgG (MCAO + IgG), 5 µg RB-222 (MCAO + 5 µg RB-222) and 10 µg
RB-222 (MCAO + 10 µg RB-222). At 1 h following induction of
reperfusion, 5 or 10 µg/ml RB-222 [1 mg/ml, dissolved in 0.01 M
phosphate-buffered saline (PBS)] or an IgG control (anti-gp-120,
NeoMarkers, Inc.) was administered intracerebroventricularly.
RB-222 or an equal quantitity of IgG was injected into the right
lateral ventricle over a period of 5 min using a Hamilton syringe.
The location of the injection site was based on the following
coordinates: 1.0 mm rostral to bregma, 3.0 mm lateral to middle and
5.0 mm ventral to the skull surface.
Neurological tests
The tests for neurological function (sensory and
motor) and behavior (rotarod test and number of left turns) were
performed at 24 h after reperfusion as previously described
(18) to assess motor coordination
and balance changes after brain ischemia. The sums of the
neurological severity scores were graded on a scale from 0 (normal)
to 42 (severe symptoms). The tests were performed in a blinded
manner three times.
Measurement of infarct volume
Following neurological evaluation at 24 h following
reperfusion, the rats (n=6 per group) were sacrificed with an
overdose of 10% chloral hydrate (400 mg/kg), and the brains were
immediately removed. The brain tissue was cut into six 2-mm coronal
sections and immersed in 1% 2,3,5-triphenyltetrazolium chloride
(TTC; Sigma-Aldrich, Merck Millipore, Darmstadt, Germany) solution
at 37°C in the dark for 15 min. The infarction areas of all
sections were measured using an image analysis system (Image-Pro
Plus 6.0; Media Cybernetics, Inc., Rockville, MD, USA). To
compensate for the effect of brain edema, the actual infarct volume
was calculated using the following equation: Contralateral
hemisphere volume-(ipsilateral hemisphere volume-infarct
volume).
Measurement of brain edema
Brain edema was evaluated by calculating the brain
water content, which was defined as the difference in weight
between wet and dry samples. At 24 h following reperfusion, the
rats were sacrificed and their brains were removed, as mentioned
above. Subsequently, the brains were separated into the ipsilateral
ischemic hemisphere (ipsilateral) and the contralateral hemisphere
(contralateral). The brains were weighed to obtain the wet weight
(WW), and were weighed again after drying at 110°C for 24 h to
determine the dry weight (DW). The percentage brain water content
was calculated using the following formula: (WW-DW)/WWx100.
Measurement of BBB permeability
Disruption of the BBB was assessed at 24 h after
reperfusion by spectrophotometric measurement of Evans Blue (EB)
extravasation. EB dye (2%; 5 ml/kg; Sigma-Aldrich; Merck Millipore)
was injected into the right femoral vein over a period of 2 min and
allowed to circulate for 60 min. The animals were then anesthetized
with an intraperitoneal injection of 10% chloral hydrate (350
mg/kg) and perfused with PBS through the left ventricle until a
colorless perfusion fluid was observed from the right atrium. The
extravasated EB in the brain was measured by spectrophotometry at
an absorbance of 620 nm using a standard solution of EB dye (from 0
to 6.25 µg/ml). EB extravasation in the ischemic penumbra was also
observed using a FluoView-1000 Confocal Microscope (Olympus
Corporation, Tokyo, Japan).
Measurement of microvessel
density
Rats were sacrificed as above and perfused for
microvessel counting at 24 h post-reperfusion. The brains were
isolated and sliced into 5-mm-thick coronal slices in the
peri-bregma region. The brain slices were then fixed in a 4%
paraformaldehyde solution overnight at 4°C before they were
embedded in Tissue-Tek O.C.T. compound (Sakura Finetek Japan, Co.,
Ltd., Tokyo, Japan). After the sections were blocked with 5% bovine
serum albumin (Sigma-Aldrich; Merck Millipore) at 37°C for 30 min,
they were stained at 37°C for 30 min for the endothelial cell
marker CD31 (1:50; catalog no. sc-376764; Santa Cruz Biotechnology,
Inc., Dallas, TX, USA) and VEGF (1:25; catalog no. ab1316; Abcam,
Cambridge, MA, USA). Corresponding Alexa 488/594-conjugated
secondary antibodies (1:1,000; catalog nos. A-11029 and A-11032;
Invitrogen; Thermo Fisher Scientific, Inc.) were used to identify
the primary antibodies. Nuclei were counterstained with DAPI
(1:1,000; Abcam). The CD31-positive microvessels were observed in
six representative sections selected from each animal using a
FluoView-1000 Confocal Microscope (Olympus Corporation). Only
CD31-positive microvessels with a well-defined linear vessel shape
or a clear lumen were counted as one vessel. Single endothelial
cells were ignored. Microvessel number was calculated as the mean
microvessel count obtained from the six images. Microvessel numbers
were determined in a blind manner by two independent
investigators.
Western blot analysis
Brain samples were extracted from the ipsilateral
cerebral cortex with radioimmunoprecipitation buffer (Beyotime
Institute of Biotechnology, Haimen, China) and centrifuged to
remove the insoluble material (12,000 × g for 15 min at
4°C). Protein concentrations were determined using a BCA protein
assay kit (Beyotime Institute of Biotechnology) according to the
manufacturer's instructions. A total of 30 µg protein were
separated on 10% SDS-PAGE gels. Protein bands were then transferred
to polyvinylidene difluoride membranes and incubated for 2 h at
37°C in Tris-buffered saline plus 0.1% Tween 20 (TBST) containing
5% skim milk. Membranes were incubated overnight at 4°C with
primary antibodies against VEGF (1:1,000; catalog no. ab1316;
Abcam), MMP-2 (1:500; catalog no. sc-13594; Santa Cruz
Biotechnology, Inc.), MMP-9 (1:1,000; catalog no. ab76003; Abcam),
occludin (1:500; catalog no. sc-271842; Santa Cruz Biotechnology,
Inc.) and collagen-IV (1:500; catalog no. sc-11360; Santa Cruz
Biotechnology, Inc.). The membranes were then incubated with the
corresponding horseradish peroxidase-conjugated secondary
antibodies (1:500; catalog nos. ZDR-5306 and ZDR-5307; ZSGB-Bio,
Beijing, China) for 1 h at room temperature after washing the
membranes three times with TBST. β-actin (1:500; catalog no. TA-09;
ZSGB-Bio) expression was determined as a loading control. Labeled
proteins were visualized by chemiluminescence using an enhanced
chemiluminescence kit (Beyotime Institute of Biotechnology). The
intensity of the bands was measured using the ChemiDoc detection
system and Quantity One software version 4.6.8 (Bio-Rad
Laboratories, Inc., Hercules, CA, USA).
Statistical analysis
Data are presented as the mean ± standard deviation.
Comparisons between 2 groups were analyzed using an unpaired
Student's t-test, and comparisons among >2 groups were analyzed
by one-way analysis of variance with a post-hoc Tukey test. Data
analysis was performed using SPSS software, (version, 13.0; SPSS,
Inc., Chicago, IL, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
Neurobehavioral recovery
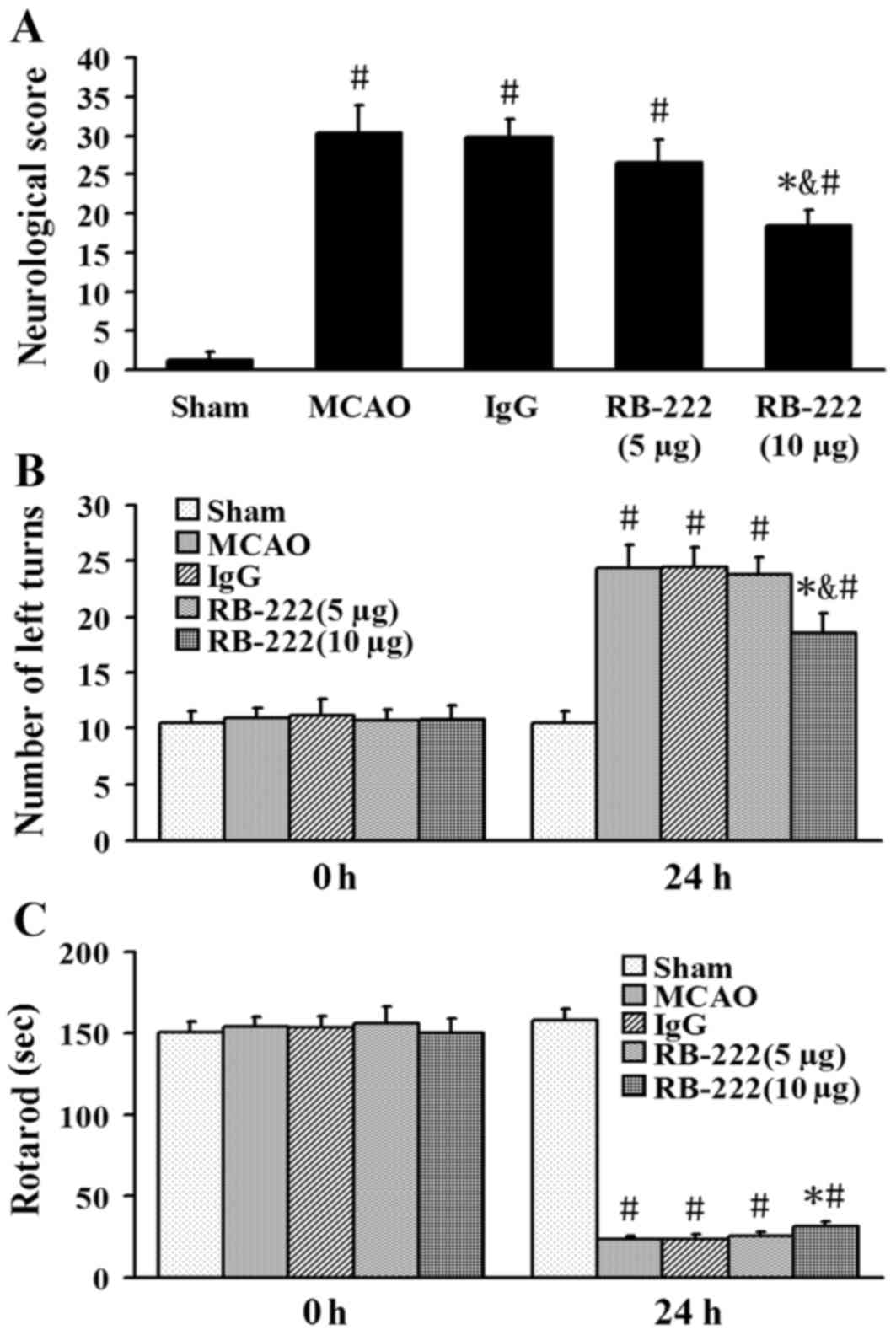
The neurological scores were 1.2±0, 30.3±3.5,
29.8±2.2, 26.5±3.1 and 18.5±1.9 in the Sham, MCAO, IgG, RB-222 (5
µg) and RB-222 (10 µg) groups, respectively (Fig. 1A). RB-222 treatment at a dose of 10
µg significantly reduced the neurological severity scores at 24 h
after reperfusion when compared with the MCAO (P<0.001) or IgG
groups (P<0.001); whereas no significant difference between the
MCAO group and the RB-222 (5 µg) group was observed (P=0.093). The
results of the elevated body swing test and the rotarod test
demonstrated no significant differences among the five groups prior
to surgery (0 h; Fig. 1B and C).
By contrast, neurobehavioral outcomes were significantly improved
in the RB-222 (10 µg) group compared with the MCAO (P<0.001 in
number of left turns and P=0.017 in rotarod test) and IgG groups
(P<0.001 in number of left turns and P=0.014 in rotarod test) at
24 h after surgery (Fig. 1).
RB-222 treatment at a dose of 5 µg did not have a significant
effect compared with the MCAO (P=0.985 in number of left turns and
P=0.961 in rotarod test) or IgG groups (P=0.958 in number of left
turns and P=0.944 in rotarod test; Fig. 1B and C).
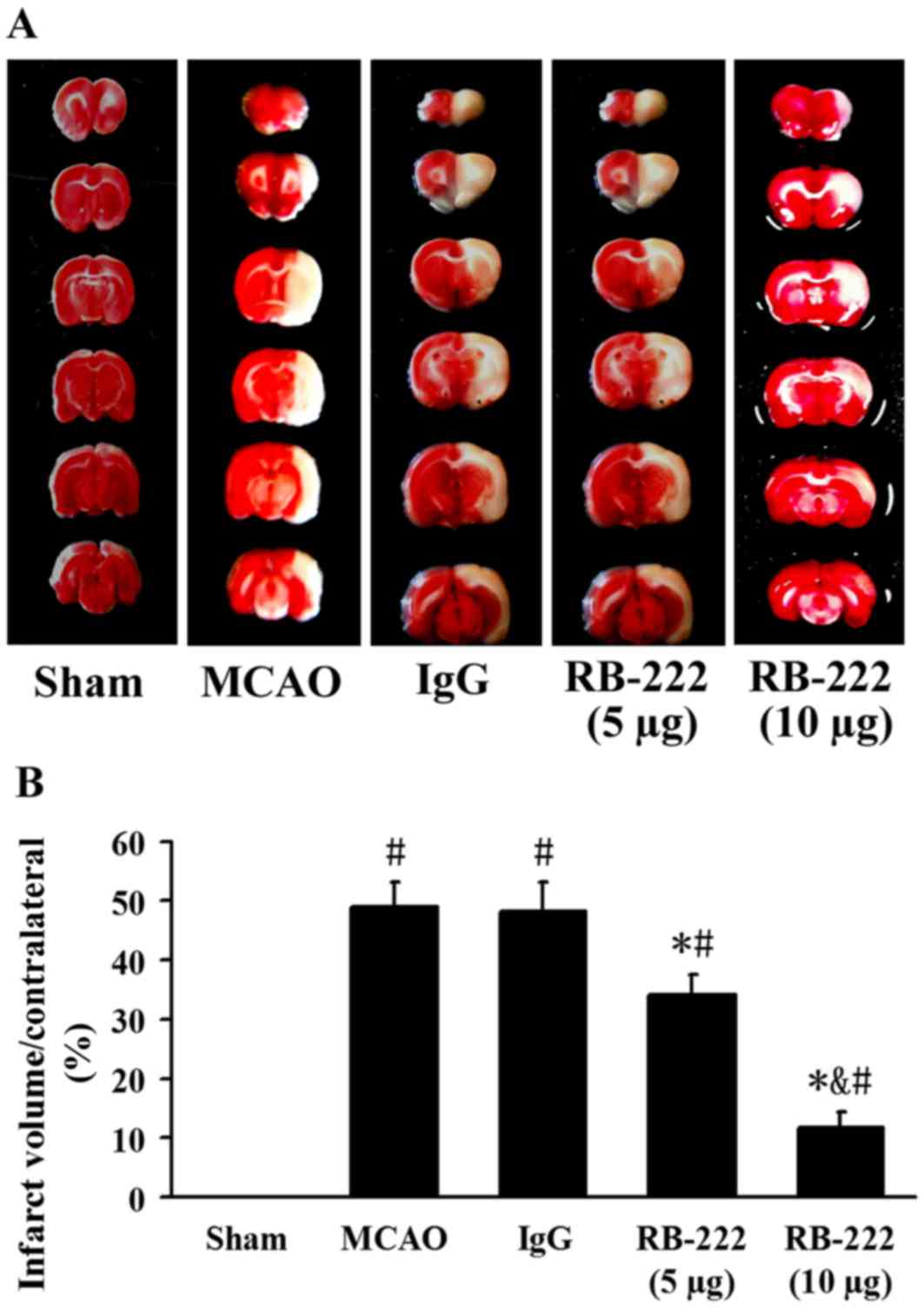
Infarct volume
Infarct volume was measured by TTC staining
(Fig. 2A). The infarct volume was
significantly increased in the MCAO (49.0±4.1%; P<0.001) and
IgG-treated (48.1±5.0%; P<0.001) groups compared with the Sham
group (Fig. 2). Treatment with
RB-222 at doses of 5 µg and 10 µg revealed a significant reduction
in infarct volume (34.2±3.3%; P<0.001 and 11.8±2.6%; P<0.001,
respectively) compared with the control IgG group. Furthermore,
RB-222 administration at a dose of 10 µg was associated with a
significant reduction in infarct volume when compared with
administration of RB-222 at a dose of 5 µg (P<0.001; Fig. 2).
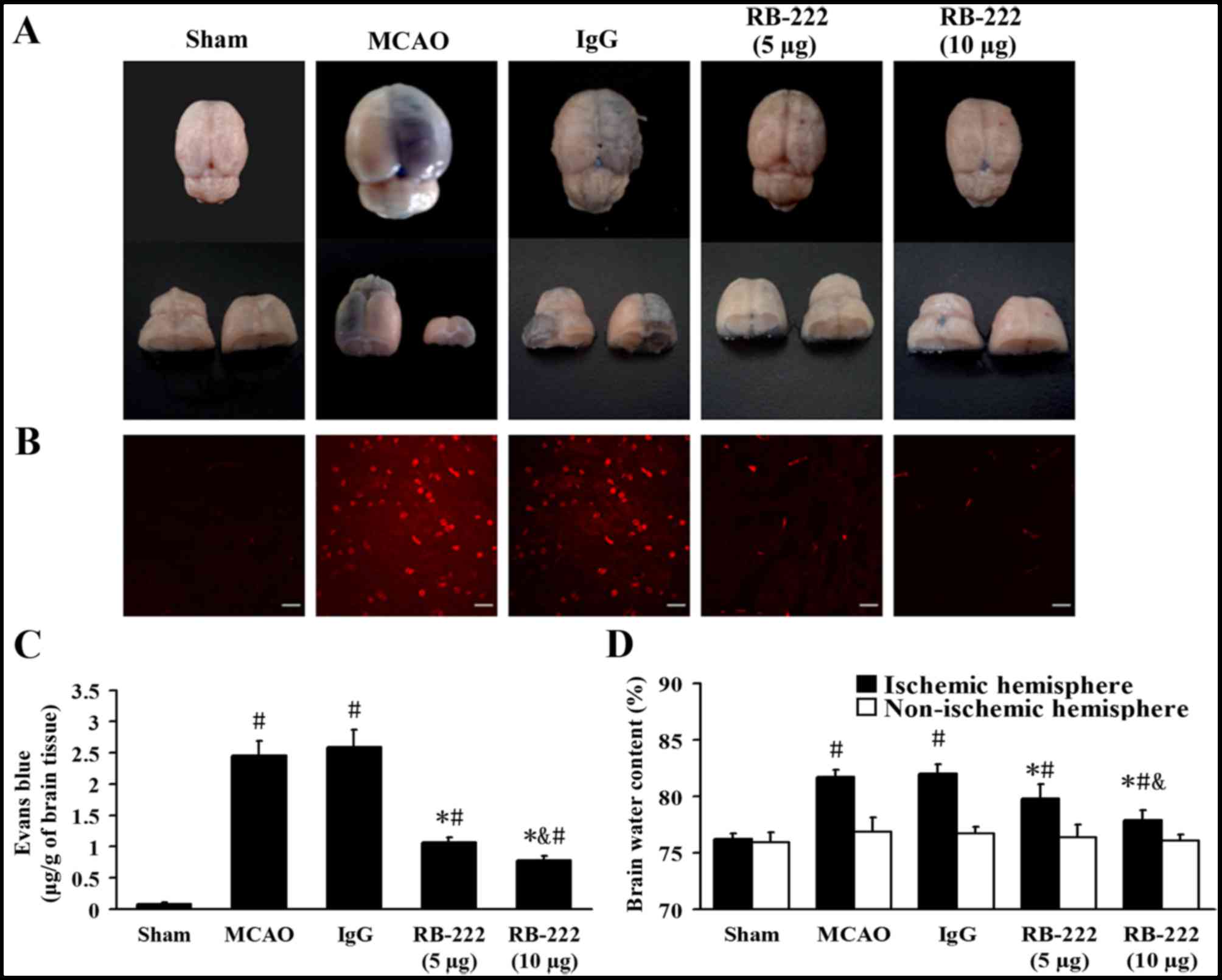
BBB leakage and brain water
content
Representative samples of brain sections with EB
extravasation are shown in Fig. 3A and
B. At 24 h following reperfusion, the EB content of brain
tissues from the Sham, MCAO, IgG, RB-222 (5 µg) and RB-222 (10 µg)
groups were 0.08±0.02, 2.46±0.23, 2.59±0.28, 1.07±0.08 and
0.78±0.72 µg/g, respectively (Fig.
3C). MCAO induced significant extravasation of EB in the MCAO
(P<0.001) and IgG (P<0.001) groups compared with the Sham
group. Treatment with 5 and 10 µg RB-222 significantly reduced the
EB extravasation (P<0.001 and P<0.001, respectively) compared
with the IgG group, with 10 µg RB-222 associated with a greater
reduction in EB extravasation (P=0.049 vs. 5 µg RB-222; Fig. 3C).
The mean brain water content in the ischemic and
non-ischemic hemispheres was examined and is shown in Fig. 3D. MCAO produced a significant
increase in brain water content in the ischemic hemisphere at 24 h
following reperfusion in the MCAO (P<0.001) and IgG groups
(P<0.001) compared with the Sham group. Treatment with RB-222 at
doses of 5 and 10 µg significantly decreased brain water content in
the ischemic hemisphere (P=0.001 and P<0.001, respectively)
compared with the IgG group, although 10 µg RB-222 was more
effective than 5 µg RB-222 in reducing brain water content
(P=0.006; Fig. 3D). No significant
difference in brain water content was observed among the groups in
the non-ischemic hemisphere (Fig.
3D).
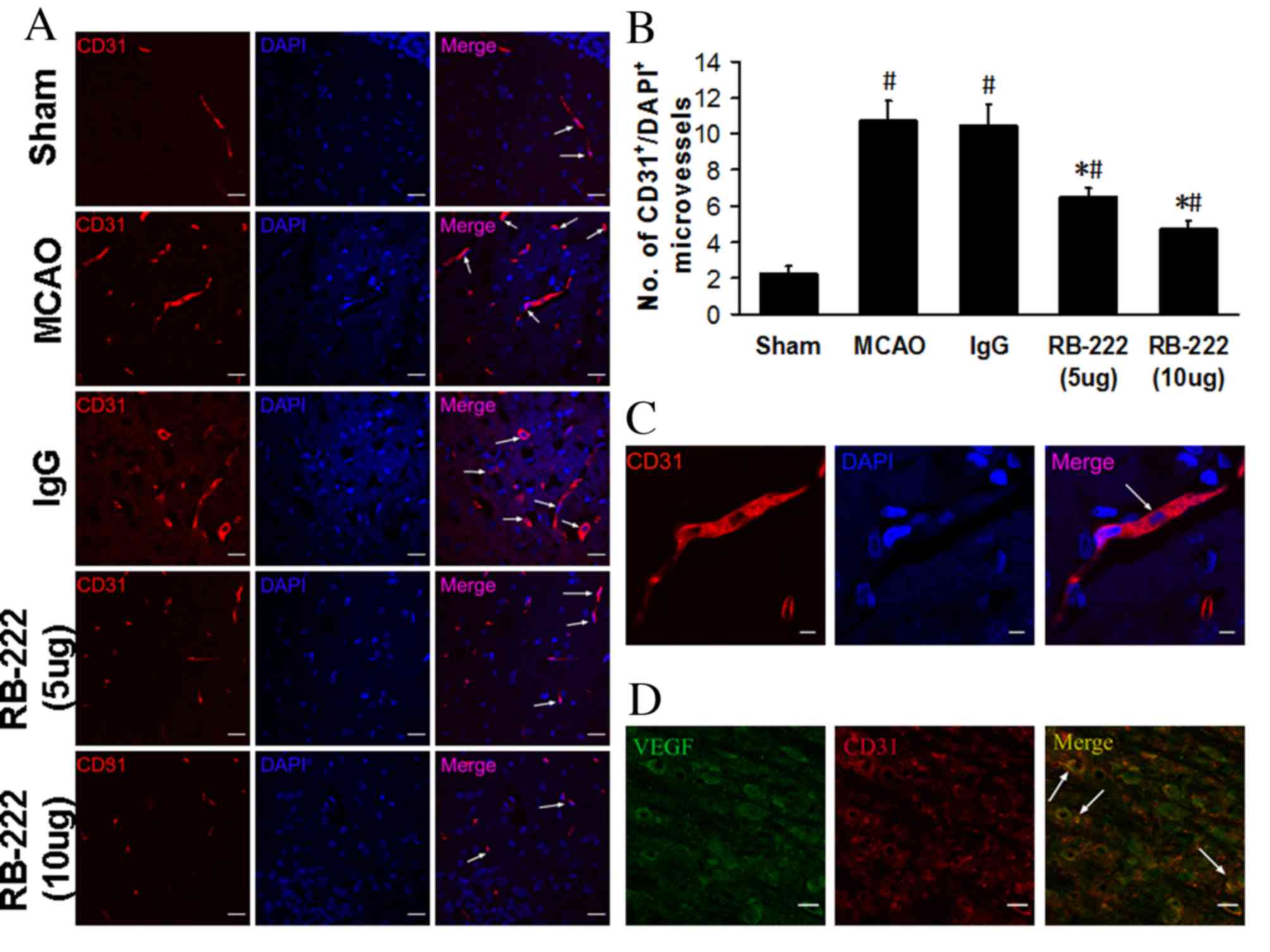
Microvessel counts
The number of microvessels was analyzed in the
peri-infarct area at 24 h after reperfusion by CD31 immunostaining
(Fig. 4A). Microvessel number was
significantly higher in the MCAO (10.75±1.09; P<0.001) and IgG
(10.50±1.12; P<0.001) groups compared with the Sham group
(2.25±0.43; Fig. 4B). RB-222
treatment at doses of 5 and 10 µg significantly reversed the
MCAO-induced increase in microvessel number (6.50±0.50 and
4.75±0.43, respectively; Fig. 4B)
when compared with MCAO and IgG groups (P<0.001 for all
comparisons). Furthermore, confocal images demonstrated that, in
CD31-stained microvessels (Fig.
4C), VEGF colocalized with CD31 in the microvessels (Fig. 4D).
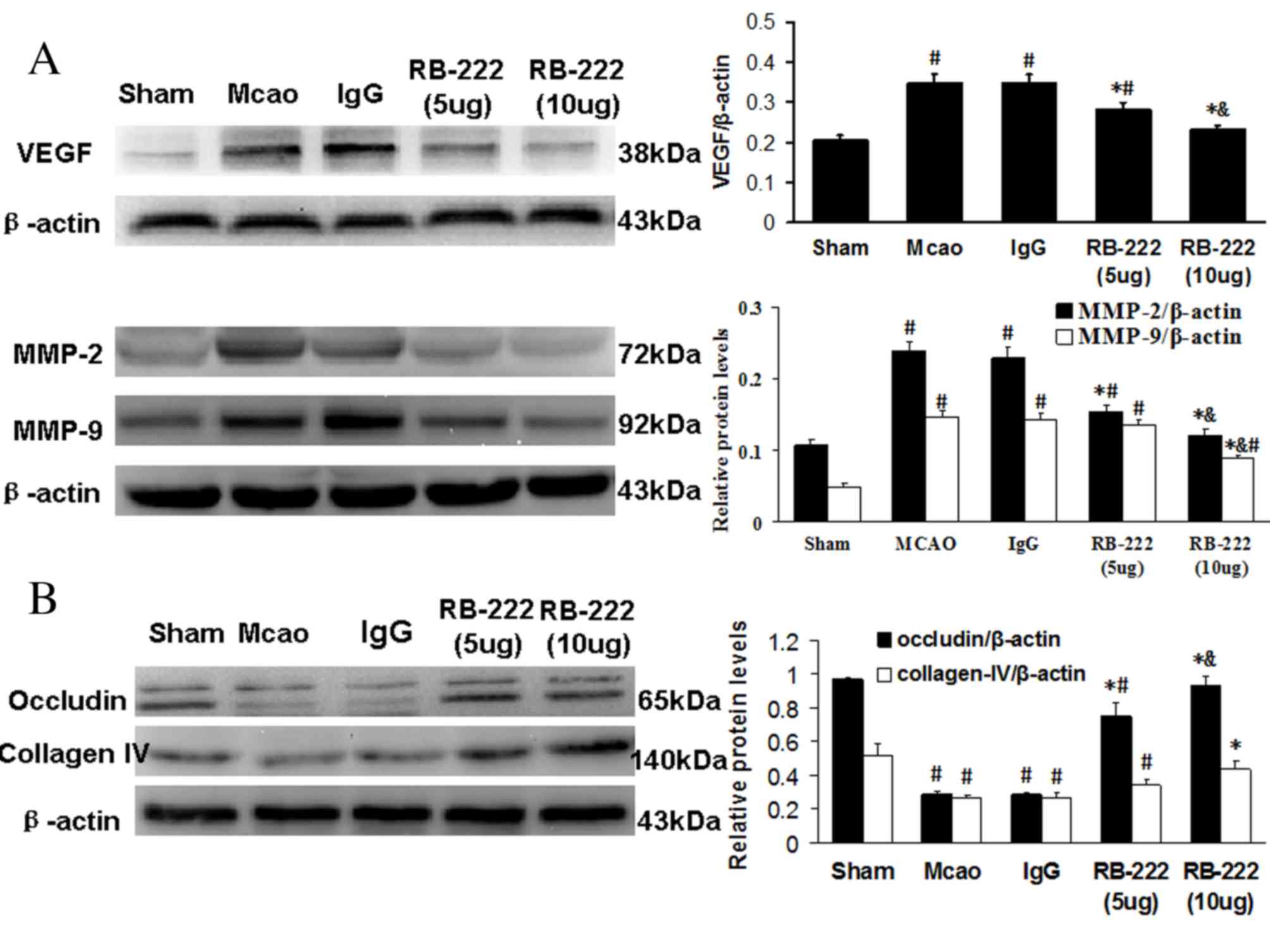
Western blot analysis
The western blotting results demonstrated that
cerebral ischemia significantly increased VEGF expression in the
MCAO (P<0.001) and IgG groups (P<0.001) at 24 h following
reperfusion compared with the Sham group (Fig. 5A). However, RB-222 treatment at
doses of 5 and 10 µg significantly attenuated VEGF expression when
compared with the MCAO (P=0.001 vs. 5 µg RB-222; P<0.001 vs. 10
µg RB-222) and IgG groups (P=0.001 vs. 5 µg RB-222; P<0.001 vs.
10 µg RB-222). The expression of MMP-2 and MMP-9 was significantly
increased in the MCAO and IgG groups when compared with the Sham
group, and was significantly decreased following treatment with 5
and 10 µg RB-222 compared with the MCAO and IgG groups (P<0.001
for all comparisons; Fig. 5A). As
expected, the expression levels of occludin and collagen-IV were
significantly decreased in the MCAO and IgG groups when compared
with the Sham group (P<0.001 for all comparisons; Fig. 5B). Treatment with RB-222 at 5 and
10 µg significantly increased the levels of occludin when compared
with the MCAO (P<0.001) and IgG (P<0.001) groups. However, a
significant increase in collagen-IV was only observed following
treatment with 10 µg RB-222 compared with the MCAO (P=0.001) and
IgG (P=0.001) groups.
Discussion
The results of the current study demonstrated the
following: i) Ischemia-induced vascular leakage may be partially
induced by early VEGF secretion; ii) early VEGF inhibition may
alleviate vascular permeability following ischemic stroke; iii)
early VEGF inhibition may reduce infarct volume and improve
neurobehavioral recovery in the focal cerebral ischemic rats; and
iv) this protective effect may have been mediated by a reduction in
MMP-2 and MMP-9 expression, together with the upregulation of TJP
expression and improved BBB integrity. To the best of our
knowledge, this is the first study investigating early VEGF
inhibition through intracerebroventricular treatment, and these
findings demonstrated a neuroprotective effect, potentially
involving the MMP pathway.
Brain infarction and cerebral edema are major
life-threatening pathophysiological alterations that may cause
neurological deterioration or mortality in the acute phase of
ischemic stroke (2,19). The rapid induction of VEGF is a
major inducer of the vascular permeability that is involved in BBB
disruption and brain edema in the acute phase of ischemic stroke
(6). Consistent with previous
studies, the current study demonstrated that the upregulation of
VEGF expression induces BBB damage and brain edema following acute
ischemic stroke. However, the aggravated brain injury was reversed
by early administration of RB-222, as demonstrated by decreased EB
extravasation and reduced ischemic brain water content. Early
inhibition of VEGF has been demonstrated to decrease infarction
following stroke in rats (14). In
addition, the present study indicated that RB-222, at doses of 5
and 10 µg, reduced infarct size by 30 and 75%, respectively,
compared with the MCAO group. Infarct size is widely used to
determine the effectiveness of a drug for the treatment of ischemic
stroke in most animal studies (20,21).
However, the neuroprotective efficacy of RB-222 was determined by
measuring neurological function in clinical trials (21). In the present study, treatment with
RB-222 at a dose of 10 µg significantly improved neurological
outcome, as evidenced by the decrease in severity of neurological
response. These results confirmed the neuroprotective effects of
VEGF inhibition in a rat model of ischemic stroke.
Angiogenesis provides a natural defense mechanism
after stroke by enhancing oxygen and nutrient supply to the
ischemic brain tissue (22).
Furthermore, angiogenesis is activated in the peri-infarct areas as
early as 12–24 h following ischemic stroke, which correlates with
longer survival following cerebral ischemia (22,23).
VEGF, as a vascular growth factor, serves a critical role in this
process, and is primarily secreted in astrocytes, endothelial cells
and neural stem cells in ischemic rats (24). However, the neurorestorative
process is likely to be effective after the acute phase of stroke
(25). Previous studies have
indicated that there may be an immediate increase in VEGF
expression after stroke; which is a potent vascular permeability
factor associated with the formation of brain edema following acute
stroke (13,16). Moreover, microvessels formed in
response to VEGF at the onset of stroke are more permeable, with an
immature endothelial permeability barrier and basal lamina, both of
which contribute to edema formation (1,6).
Results of the current study demonstrated that treatment with
RB-222 significantly reduced microvessel number in the peri-infarct
region at 24 h after ischemic/reperfusion injury. In addition,
colocalization of VEGF and CD31 in endothelial cells was observed,
indicating the critical role of VEGF in angiogenesis. Therefore,
early VEGF inhibition decreased focal immature angiogenesis and
reduced the BBB permeability and brain edema in ischemic rats.
To investigate the possible molecular mechanisms
involved in the protective effects of early VEGF inhibition on
acute ischemia-induced brain injury, the expression of MMP-2 and
MMP-9 in the cerebral cortex was examined in the present study. A
reduction in MMP-2 and MMP-9 expression was observed following
RB-222 treatment. It has been reported that the instant elevation
of MMPs in acute ischemia-induced brain injury is involved in the
digestion of the endothelial basal lamina and the triggering of
secondary injury, including brain edema in acute stroke (8). The inhibition of MMPs promotes BBB
restoration and neurovascular remodeling following stroke (9). Furthermore, TJPs, which are
considered to be a major part of the BBB, were significantly
decreased following ischemic stroke (26). This is consistent with prior
studies demonstrating that the loss of TJPs following brain
ischemia is responsible for BBB disruption by an MMP-dependent step
(7,9). Among the TJPs, occludin is a
regulatory protein that assembles with claudin-5 between
endothelial cells and is associated with endothelial permeability
(27–29). In addition, collagen-IV contributes
to 90% of the components of the basal lamina and is critically
involved in the integrity of the vessel wall (7,10,30).
The results of the present study demonstrated that MMP-2 and MMP-9
expression were decreased and occludin and collagen-IV levels were
increased following VEGF inhibition. This suggests that,
RB-222-mediated inhibition of VEGF expression in the
microvasculature at 24 h after cerebral reperfusion, resulted in
decreased MMP-2 and MMP-9 levels. This may have led to an increase
in occludin levels, thereby maintaining closure of tight junctions,
as well as an increase in collagen-IV levels, thus maintaining the
structure of the basal lamina.
The present study has limitations. Although MMP-2
and MMP-9 expression levels were significantly decreased following
VEGF inhibition, the underlying mechanisms involved remains largely
unknown. A previous study indicated that tumor necrosis factor-α
(TNF-α) participates in the BBB deterioration that occurs in
ischemia/reperfusion injury models (31). Therefore, the role of TNF-α in this
process of ischemic injury requires further investigation in the
current study. In particular, following translocation to the
nucleus, the NF-kB promoter domains present in numerous
pro-inflammatory genes, including TNF-α, have been shown to induce
the expression of MMP-9 (32).
Based on the results presented in the current study, the authors
hypothesized that the TNF-α/NF-kB/MMP signaling pathway may be a
potential pathway by which RB-222 preserves the BBB following acute
ischemic stroke. Further investigation is required in order to
clarify this hypothesis. It was observed in the present study, that
inhibition of VEGF during the acute phase of stroke attenuates BBB
disruption by regulating the expression of MMPs and reducing
permeable microvessel formation. This inhibition further decreased
infarct size and improved the outcome after ischemia. However, the
critical roles of VEGF in the processes of angiogenesis,
neurogenesis and functional recovery during the later phase of
ischemic injury have been well studied (33). In different phases of ischemic
stroke, VEGF serves different roles, therefore it is important to
determine whether early administration of RB-222 may affect
outcomes in the chronic phase. Further studies are necessary to
evaluate the long-term effects of VEGF inhibition and the optimal
duration of treatment during the acute phase of stroke.
In conclusion, the results of the present study
confirmed that the VEGF signaling pathway serves an important role
in BBB damage following acute cerebral ischemic stroke, partially
by regulating the expression of occludin and collagen-IV
potentially via the MMP signaling pathway. Early inhibition of VEGF
may have vast potential for the treatment of ischemic stroke.
Acknowledgements
This study was supported by the Natural Science
Foundation of Heilongjiang Province (grant no. D201279), the
National High Technology Research and Development Program 863
(grant no. 2012AA02A508), the National Natural Science Foundation
of China (grant no. 81372700) and the International Science &
Technology Cooperation Program of China (grant no.
2011DFA31470).
References
|
1
|
Krueger M, Bechmann I, Immig K,
Reichenbach A, Härtig W and Michalski D: Blood-brain barrier
breakdown involves four distinct stages of vascular damage in
various models of experimental focal cerebral ischemia. J Cereb
Blood Flow Metab. 35:292–303. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ayata C and Ropper AH: Ischaemic brain
oedema. J Clin Neurosci. 9:113–124. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Battey TW, Karki M, Singhal AB, Wu O,
Sadaghiani S, Campbell BC, Davis SM, Donnan GA, Sheth KN and
Kimberly WT: Brain edema predicts outcome after nonlacunar ischemic
stroke. Stroke. 45:3643–3648. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Greenberg DA and Jin K: Vascular
endothelial growth factors (VEGFs) and stroke. Cell Mol Life Sci.
70:753–1761. 2013.
|
|
5
|
Hayashi T, Abe K, Suzuki H and Itoyama Y:
Rapid induction of vascular endothelial growth factor gene
expression after transient middle cerebral artery occlusion in
rats. Stroke. 28:2039–2044. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang ZG, Zhang L, Jiang Q, Zhang R,
Davies K, Powers C, Bruggen Nv and Chopp M: VEGF enhances
angiogenesis and promotes blood-brain barrier leakage in the
ischemic brain. J Clin Invest. 106:829–838. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yang Y, Estrada EY, Thompson JF, Liu W and
Rosenberg GA: Matrix metalloproteinase-mediated disruption of tight
junction proteins in cerebral vessels is reversed by synthetic
matrix metalloproteinase inhibitor in focal ischemia in rat. J
Cereb Blood Flow Metab. 27:697–709. 2007.PubMed/NCBI
|
|
8
|
Kurzepa J, Kurzepa J, Golab P, Czerska S
and Bielewicz J: The significance of matrix metalloproteinase
(MMP)-2 and MMP-9 in the ischemic stroke. Int J Neurosci.
124:707–716. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yang Y, Thompson JF, Taheri S, Salayandia
VM, McAvoy TA, Hill JW, Yang Y, Estrada EY and Rosenberg GA: Early
inhibition of MMP activity in ischemic rat brain promotes
expression of tight junction proteins and angiogenesis during
recovery. J Cereb Blood Flow Metab. 33:1104–1114. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rosenberg GA, Estrada EY and Dencoff JE:
Matrix metalloproteinases and TIMPs are associated with blood-brain
barrier opening after reperfusion in rat brain. Stroke.
29:2189–2195. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Seo JH, Guo S, Lok J, Navaratna D, Whalen
MJ, Kim KW and Lo EH: Neurovascular matrix metalloproteinases and
the blood-brain barrier. Curr Pharm Des. 18:3645–3648. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lenglet S, Montecucco F and Mach F: Role
of matrix metalloproteinases in animal models of ischemic stroke.
Curr Vasc Pharmacol. 13:161–166. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
van Bruggen N, Thibodeaux H, Palmer JT,
Lee WP, Fu L, Cairns B, Tumas D, Gerlai R, Williams SP, van
Lookeren Campagne M and Ferrara N: VEGF antagonism reduces edema
formation and tissue damage after ischemia/reperfusion injury in
the mouse brain. J Clin Invest. 104:1613–1620. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kimura R, Nakase H, Tamaki R and Sakaki T:
Vascular endothelial growth factor antagonist reduces brain edema
formation and venous infarction. Stroke. 36:1259–1263. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chi OZ, Hunter C, Liu X and Weiss HR:
Effects of anti-VEGF antibody on blood-brain barrier disruption in
focal cerebral ischemia. Exp Neurol. 204:283–287. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kanazawa M, Igarashi H, Kawamura K,
Takahashi T, Kakita A, Takahashi H, Nakada T, Nishizawa M and
Shimohata T: Inhibition of VEGF signaling pathway attenuates
hemorrhage after tPA treatment. J Cereb Blood Flow Metab.
31:1461–1474. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Longa EZ, Weinstein PR, Carlson S and
Cummins R: Reversible middle cerebral artery occlusion without
craniectomy in rats. Stroke. 20:84–91. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Reglodi D, Tamás A and Lengvári I:
Examination of sensorimotor performance following middle cerebral
artery occlusion in rats. Brain Res Bull. 59:459–466. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wijdicks EF, Sheth KN, Carter BS, Greer
DM, Kasner SE, Kimberly WT, Schwab S, Smith EE, Tamargo RJ and
Wintermark M: American Heart Association Stroke Council:
Recommendations for the management of cerebral and cerebellar
infarction with swelling: A statement for healthcare professionals
from the American Heart Association/American Stroke Association.
Stroke. 45:1222–1238. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bae ON, Serfozo K, Baek SH, Lee KY,
Dorrance A, Rumbeiha W, Fitzgerald SD, Farooq MU, Naravelta B,
Bhatt A and Majid A: Safety and efficacy evaluation of carnosine,
an endogenous neuroprotective agent for ischemic stroke. Stroke.
44:205–212. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Schaar KL, Brenneman MM and Savitz SI:
Functional assessments in the rodent stroke model. Exp Transl
Stroke Med. 2:132010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Beck H and Plate KH: Angiogenesis after
cerebral ischemia. Acta Neuropathol. 117:481–496. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Krupinski J, Kaluza J, Kumar P, Kumar S
and Wang JM: Role of angiogenesis in patients with cerebral
ischemic stroke. Stroke. 25:1794–1798. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tang Y, Wang J, Lin X, Wang L, Shao B, Jin
K, Wang Y and Yang GY: Neural stem cell protects aged rat brain
from ischemia-reperfusion injury through neurogenesis and
angiogenesis. J Cereb Blood Flow Metab. 34:1138–1147. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sun Y, Jin K, Xie L, Childs J, Mao XO,
Logvinova A and Greenberg DA: VEGF-induced neuroprotection,
neurogenesis, and angiogenesis after focal cerebral ischemia. J
Clin Invest. 111:1843–1851. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu J, Jin X, Liu KJ and Liu W: Matrix
metalloproteinase-2-mediated occludin degradation and
caveolin-1-mediated claudin-5 redistribution contribute to
blood-brain barrier damage in early ischemic stroke stage. J
Neurosci. 32:3044–3057. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tsukita S and Furuse M: Pores in the wall:
Claudins constitute tight junction strands containing aqueous
pores. J Cell Biol. 149:13–16. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hirase T, Kawashima S, Wong EY, Ueyama T,
Rikitake Y, Tsukita S, Yokoyama M and Staddon JM: Regulation of
tight junction permeability and occludin phosphorylation by
Rhoa-p160ROCK-dependent and -independent mechanisms. J Biol Chem.
276:10423–10431. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hawkins BT and Davis TP: The blood-brain
barrier/neurovascular unit in health and disease. Pharmacol Rev.
57:173–185. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sellner J and Leib SL: In bacterial
meningitis cortical brain damage is associated with changes in
parenchymal MMP-9/TIMP-1 ratio and increased collagen type IV
degradation. Neurobiol Dis. 21:647–656. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wiggins-Dohlvik K, Merriman M, Shaji CA,
Alluri H, Grimsley M, Davis ML, Smith RW and Tharakan B: Tumor
necrosis factor-α disruption of brain endothelial cell barrier is
mediated through matrix metalloproteinase-9. Am J Surg.
208:954–960. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kim JY, Kawabori M and Yenari MA: Innate
inflammatory responses in stroke: Mechanisms and potential
therapeutic targets. Curr Med Chem. 21:2076–2097. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Crafts TD, Jensen AR, Blocher-Smith EC and
Markel TA: Vascular endothelial growth factor: Therapeutic
possibilities and challenges for the treatment of ischemia.
Cytokine. 71:385–393. 2015. View Article : Google Scholar : PubMed/NCBI
|