Introduction
It is well established that tumorigenesis is
attributed to chromosomal instability or accumulated genetic
changes, including structure variations, genetic copy number
variants, single nucleotide variants (SNVs) and small insertions
and deletions (indels) (1–3). Somatic mutations are defined by
mutations that are absent in corresponding adjacent tissues;
however, they are present in all tumors (4). Somatic mutation calling is a critical
step for cancer genome characterization and clinical genotyping.
Next-generation sequencing (NGS) has become a popular strategy for
genotyping, enabling more precise mutation detection compared with
traditional methods due to its high resolution and high throughput.
Whole-genome sequencing reveals overall genetic information about
the variants, whereas whole-exome sequencing (WES) with effective
strategy only points economically at coding regions and is
currently offered by more laboratories (5). WES of tumor samples and matched
normal controls has the potential to rapidly identify
protein-altering mutations across hundreds of patients, potentially
enabling the discovery of recurrent events that drive tumor
development and growth. Identification of somatic mutations from
WES data is an increasingly common technique in the study of cancer
genomics, and a large number of somatic alterations have been
identified by WES in extensive tumor types (6–9). The
most prevalent mutations observed are in the p53 tumor suppressor
gene (TP53), Wnt/β-catenin signaling pathway regulatory genes
(catenin β1 and AXIN 1), chromatin remodeling complex components
[AT-rich interactive domain (ARID) 2 and ARID1A], Janus kinase
(JAK)/signal transducer and activator of transcription
pathway-regulated JAK1, as well as hepatitis B (HBV) integrations
into myeloid/lymphoid or mixed-lineage leukemia 4, telomerase
reverse transcriptase and cyclin E1 (10,11).
The calling of accurate somatic mutations using WES
data remains one of the major challenges in cancer genomics due to
various sources of errors, including artifacts occurring during
polymerase chain reaction (PCR) amplification or targeted capture,
machine sequencing errors and incorrect local alignments of reads
(12). Tumor heterogeneity and
normal tissue contamination generate additional difficulties for
the identification of tumor-specific somatic mutations (12,13).
In recent years, several methods have been developed to improve the
accuracy of somatic mutation calling. Despite the variations in the
methodology of somatic mutation algorithms, the aim of each program
is to identify tumor-specific variants by comparing the tumor
variant data with the dbSNP of paired adjacent tissue and germline
variant data in the same patient. Currently the most popular
computational algorithms are MuTect (14), VarScan2 (15) and Genome Analysis Toolkit (GATK)
(16). GATK calculates the
variants in tumors and adjacent tissues separately, and then
subtracts the variants identified in the adjacent tissues from
those in the tumors. MuTect and VarScan2 directly compare the tumor
tissues with the adjacent tissues at each mutation point, which in
some cases improves the accuracy of variant calling. MuTect detects
somatic mutation sensitively with a Bayesian model at low
allele-fractions, whereas VarScan2 applies a powerful
heuristic/statistic approach to identify high-quality variants
(12). However, it is unclear
which is the best strategy for identifying and accurately calling
genome variations as well as how well these different tools improve
the true positive mutations when they are combined.
The present study integrated the resources of
different somatic mutation algorithms and optimized their own
parameters in order to identify novel and recurrent mutations more
effectively and faster. The present study used one case of
hepatocellular carcinoma (HCC) to explain the whole-exome analysis
pipeline and identify the key somatic mutations of HCC.
Materials and methods
Patient
A punctured HCC tumor and paired adjacent tissue was
obtained from a patient (57 years, male) at the Youan Hospital,
Capital Medical University of China (Beijing, China) and complied
with the principles of The Declaration of Helsinki. The patient was
infected with HBV and received no radiation and chemotherapy prior
to radiofrequency ablation.
NGS platforms
The DNA was extracted using an E.Z.N.A.®
Tissue DNA Kit (Omega Bio-Tek, Inc., Norcross, GA, USA) and the
extracted DNA was captured using Agilent Human All Exon 50 M kit
(Agilent Technologies, Inc., Santa Clara, CA, USA) following the
protocols recommended by the manufacturer. Sequencing machines
generated a large volume of data at a rapid speed by sequencing
paired-end DNA fragments in parallel using Illumina His-seq2,000
(Illumina, Inc., San Diego, CA, USA) (17,18).
Following a series of library construction and actual sequencing, a
large quantity of raw data was produced.
Quality evaluation of the raw
reads
Raw reads generated by a sequenator are usually
affected by adverse factors, including adaptor contamination, poor
base sequence quality and guanine-cytosine (GC) bias (19). Once the raw data was obtained, the
quality of raw reads was assessed and the adaptor was clipped using
fastq-mcf (version 1.04.636; www.github.com/ExpressionAnalysis/ea-utils/blob/wiki/FastqMcf.md).
The sequencing data was then processed using the FastQC tool
(http://www.bioinformatics.babraham.ac.uk/projects/fastqc/)
to analyze the distribution of base GC content and sequence quality
scores.
Alignment and duplicated PCR
removal
Following the quality control analyses, the
processed reads were aligned to an established reference genome
(version hg19), which was provided by the University of California
Santa Cruz (Santa Cruz, CA, USA) (20). Millions of short reads were aligned
efficiently to the reference genome using Burrows-Wheeler Aligner
(BWA) software with default parameters, which were based on the
Burrows-Wheeler transform (21).
The aligned reads were then stored in BAM file (.bam) using
samtools software (22), which was
able to sort and index the BAM file to save space and help
subsequent process. For the assembled genome data, the picard tool
(http://picard.sourceforge.net/index.shtml) was
combined with bamtools to filter out the mismatching and
inappropriate reads. In addition, picard removed the read
duplicates derived from library PCR. The data distribution and
reads coverage were then evaluated using the CalculateHsMetrics
package. Recalibration and realignment were performed using GATK
(version 2.8; Broad Institute, Cambridge, MA, USA; www.broadinstitute.org/gatk/). Finally, the
resulting data were used for further variation identification.
Variant identification
A key step in the analysis of cancer exome
sequencing data is the identification of variants. The depth of
sequence coverage determines the choice of somatic mutation
algorithms used for identification of variants mutation. The
different identification abilities in different allele frequencies
of GATK (version 2.8.1), MuTect (version 1.1.4; Broad Institute;
http://www.broadinstitute.org/cancer/cga/mutect), and
VarScan (version 2.3.6; http://varscan.sourceforge.net/), and the joint
analysis strategy by combining the three softwares (the
three-caller pipeline approach), were taken into consideration when
identifying somatic mutations.
Variant annotation
Oncotator (http://portals.broadinstitute.org/oncotator/) was used
to annotate the screened variations (23). All of the candidate mutations were
validated visually using the Integrated Genomics Viewer (IGV)
(24) and were confirmed using
Sanger sequencing in paired samples. The tools, Polyphen-2
(www.genetics.bwh.harvard.edu/pph2/index.shtml)
and scale-invariant feature transform (SIFT; www.sift.jcvi.org/), were integrated to predict
whether mutations affected protein function based on the structure
and function of the protein, and the conservation of amino acid
residues in different species sequences.
Gene functional enrichment
analysis
The gene sets screened were used for functional
annotation analysis by the Database for Annotation, Visualization
and Integrate Discovery software (25), which consists of the Kyoto
Encyclopedia of Genes and Genomes and Gene Ontology database. The
significance of gene groups enrichment was defined by a modified
Fisher's exact test and P<0.05 was considered to indicate a
statistically significant difference.
Results
Establishment of three-caller and HCC
data analysis
WES was analyzed in one HCC tumor and paired
adjacent tissues with the three-caller approach. The present study
acquired 96.30X and 79.18X coverages for the tumor and paired
adjacent tissues, respectively, in all of the targeted exonic
regions, with 93.4% of the base targeted at 20-fold and ≥99.1%
bases by a depth of at least two times. To identify the somatic
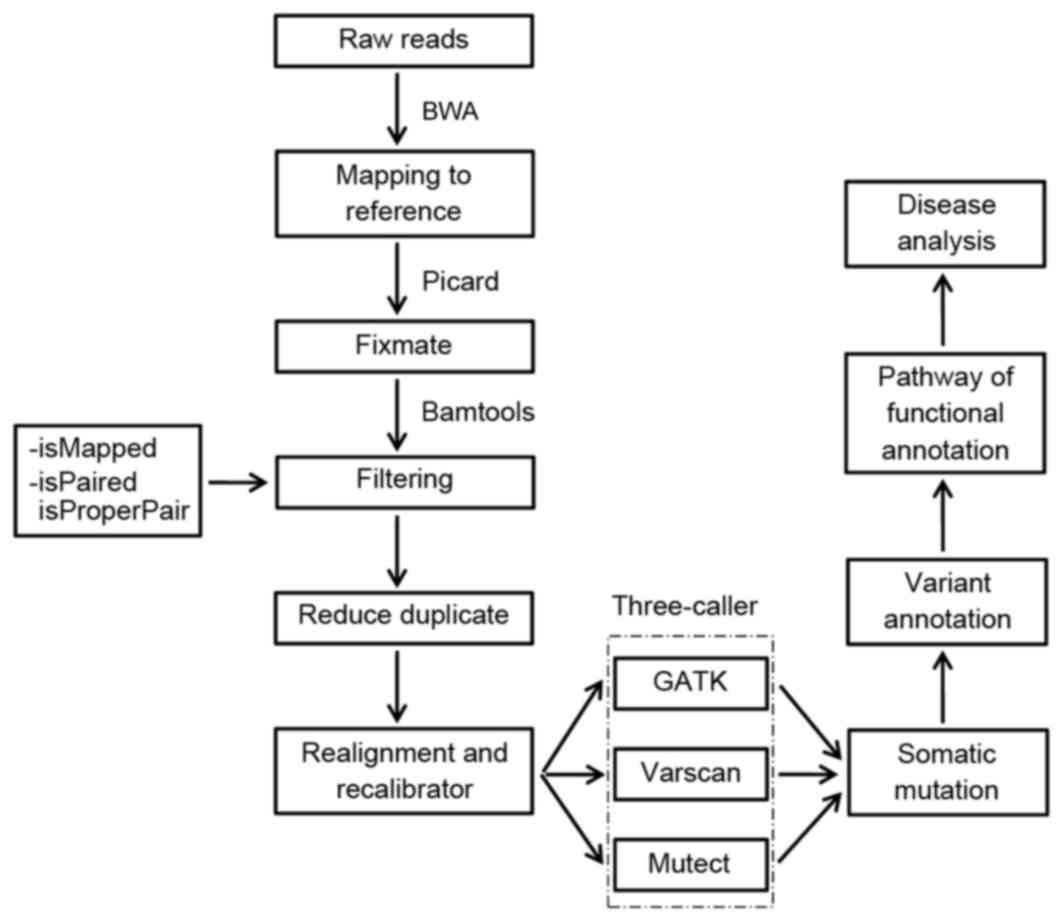
mutations, a flow chart was created with the following steps: i)
Quality evaluation of the raw reads; ii) reads map to a reference
genome; iii) somatic mutation identification with the three-caller
approach; iv) variant annotation; v) data visualization; and vi)
pathway analysis (Fig. 1).
Detecting SNVs in a HCC sample
Variant filtering was performed by GATK with the
following filter parameters: Low coverage (DP <5), low quality
(QUAL >30.0 and QUAL <5.0), very low quality (QUAL <30),
hard to validate [MQ0 ≥4 and MQ0/(1.0*DP)>0.1)] and
quality-by-depth (QD <1.5). The exome data from the samples were
calculated by running these parameters and reserved in a VCF file.
GATK was primarily used for identifying somatic mutations in the
sequencing data, including SNVs and indels.
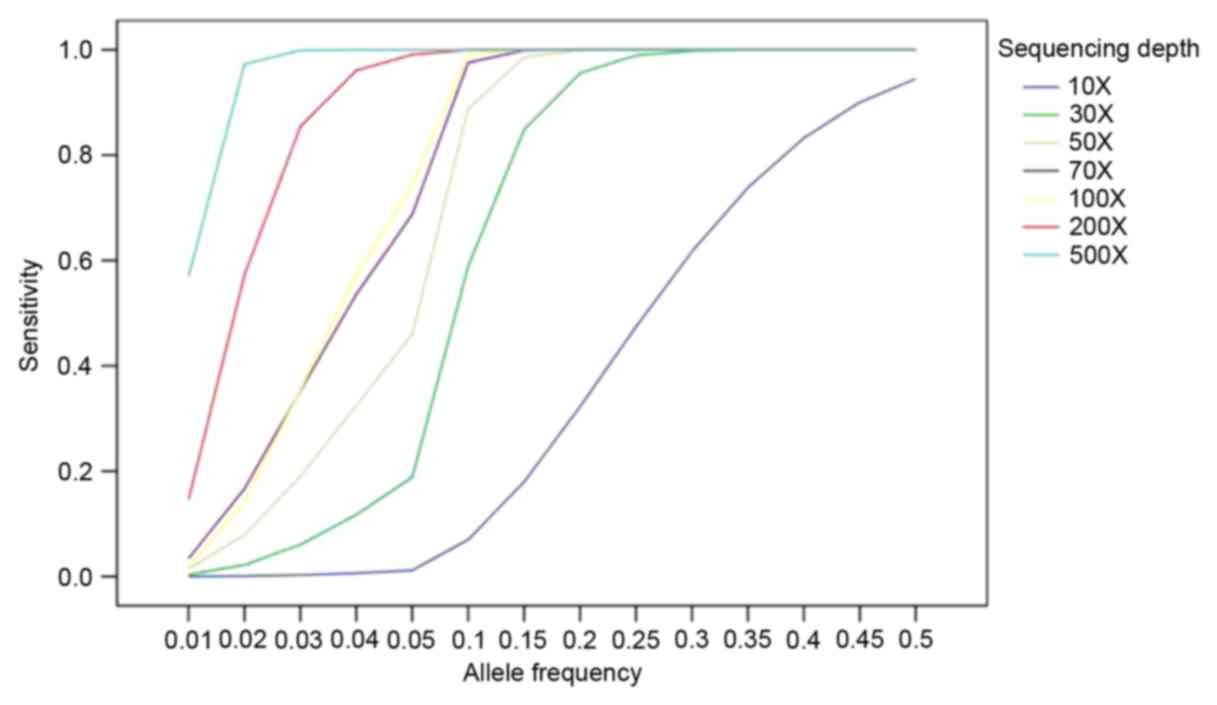
In order to identify the low allelic-fraction
mutations, MuTect was used to generate more performance in low
coverage (12). To illustrate how
high the sensitivity was based on allele fraction and sequencing
depth, a strategy was established based on the published data to
analyze the data (14). As shown
in Fig. 2, the sensitivity of
mutation was detected by MuTect approaching >90% at allele
frequency 10% with >80X sequencing depth and 80% at allele
frequency 5% with >80X.
The calling of SNVs by MuTect software was executed
through Java (version 1.6.0_45; www.oracle.com/technetwork/java/javase/downloads/java-archive-downloads-javase6-419409.html).
The default parameters of MuTect were kept to identify mutations.
Input database texts, including reference sequence hg19, dbsnp
v.135 and cosmic v54, were used for the MuTect algorithm. Somatic
point mutations were only identified by MuTect; GATK (version 1.5)
was used to analyze indels. SNVs located in exome regions were
screened with ≥20 coverage in the tumor, which was coupled with ≥4
alternate alleles and ≥4 allelic fraction of the altered base. The
paired normal sample also had 10X coverage at least in a certain
base. As many low coverage or low allelic fraction SNVs were
characterized by MuTect, SNVs with variants from low purity samples
not blindly rejected.
VarScan outperformed the other tools at higher
allelic fraction. A threshold of 6X for tumor and 8X for normal was
set, with ≥20% variation frequency. Subsequently, the present study
preferentially analyzed 20X coverage in the tumor, including
alternated variation accounting for 10X coverage, to eliminate
false positives.
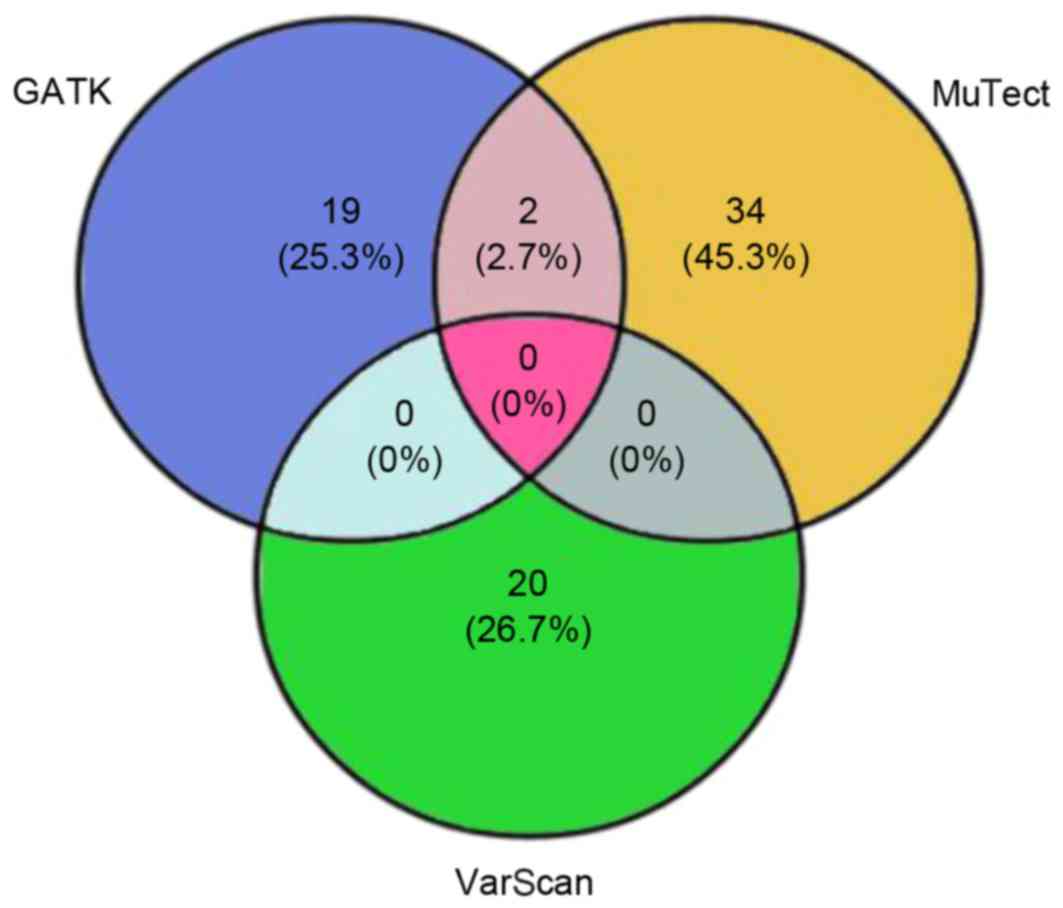
The present study proposed 75 candidate somatic
variants through the three-algorithm strategy (Fig. 3), including 50 nonsynonymous
mutations, 2 nonsense mutations, 20 synonymous mutations and 3
indels. The nonsynonymous to synonymous somatic SNV ratio was
2.5.
Analysis of somatic mutations
The predictive impact of amino acid substitution on
functional evidence was analyzed using PolyPhen-2/SIFT (Table I). The P94Q mutation was predicted
to affect the protein function of cell division cycle 7 protein,
which may be associated with neoplastic transformation of some
tumors and affect protein serine/threonine kinase activity. All of
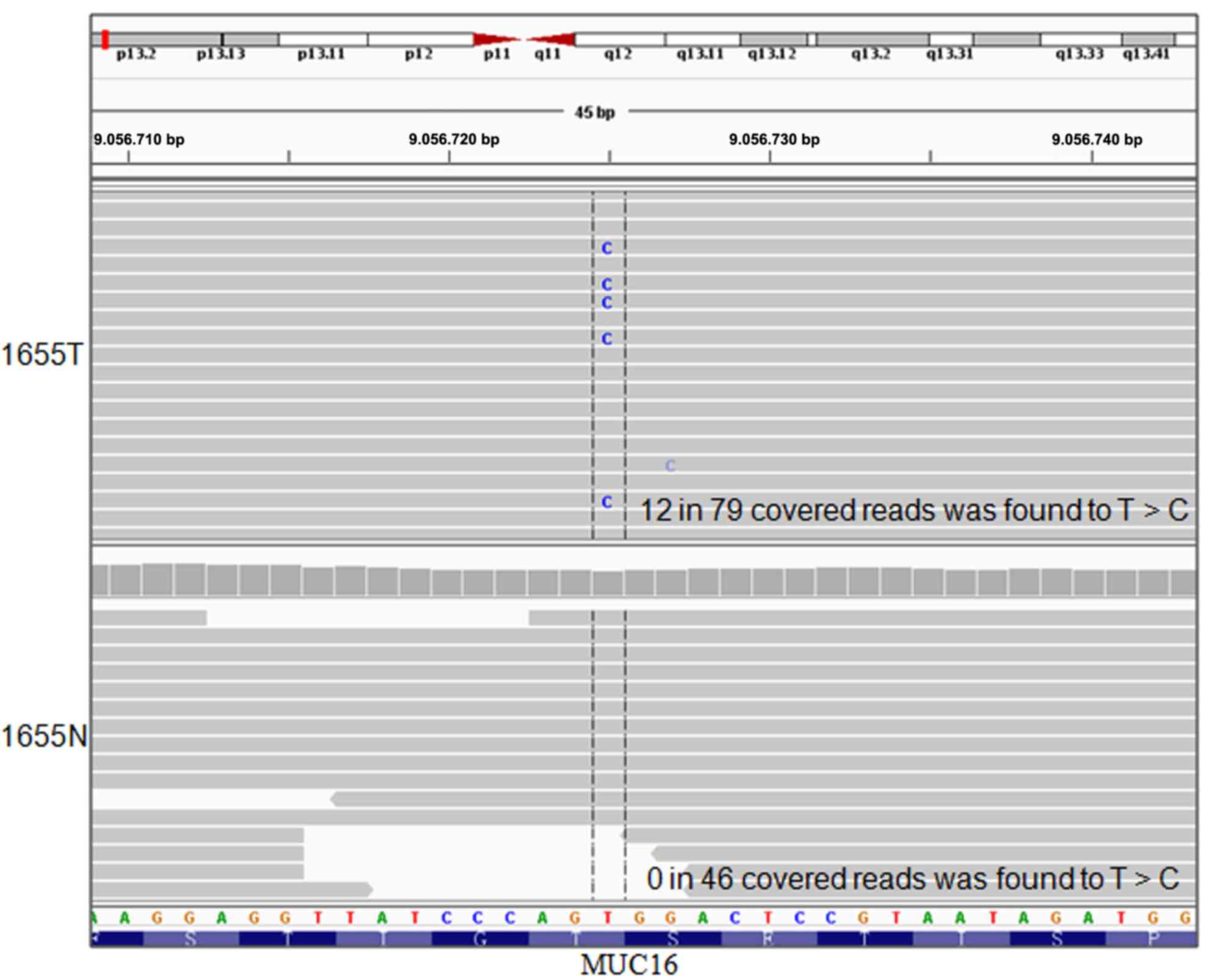
the putative somatic mutations were validated manually using IGV.
The T>C transversion at position_9056725 in mucin 16 (MUC16) was
identified (Fig. 4), which was
then validated by Sanger sequencing.
 | Table I.Selected somatic mutations predicted
by Polyphen to affect protein function. |
Table I.
Selected somatic mutations predicted
by Polyphen to affect protein function.
| Hugo symbol | Amino acid
change | SIFT | SIFT score | Polyphen | Polyphen score |
|---|
| CSMD1 | Q2192R | Damaging | 0.04 | Probably
damaging | 0.973 |
| FREM1 | H822Q | Damaging | 0.01 | Probably
damaging | 0.972 |
| GP5 | I230N | Damaging | 0 | Probably
damaging | 0.997 |
| KCNA1 | E422K | Tolerated | 0.06 | Benign | 0.013 |
| CDC7 | P94Q | Damaging | 0 | Probably
damaging | 1 |
| DMBT1 | R2343W | Damaging | 0.02 | Probably
damaging | 0.998 |
| FAT2 | V3602I | Tolerated | 0.13 | Benign | 0.118 |
| C10orf90 | R188W | Tolerated | 0.08 | Benign | 0.015 |
Pathway analysis
The 75 genes with tumor-specific mutations
demonstrated significant functional enrichment of cell adhesion and
regulation of Ras GTPase activity (P<0.05; Table II). Notably, the genes encoding
cell adhesion demonstrated the most prevalent enrichment
(P=0.0089), indicating that the enriched mutations of cell adhesion
genes may serve pivotal roles in HCC development.
| Table II.Functional categories of the
tumor-specific mutation. |
Table II.
Functional categories of the
tumor-specific mutation.
| Biological
process | Count | P-value | Genes | Fold enrichment |
|---|
| Cell adhesion | 8 | 0.0089 | GP5, LGALS3BP, FREM1,
FAT2, FCGBP, COL5A3, PCDHGB4, MUC16 | 3.29 |
| Regulation of Ras
GTPase activity | 3 | 0.0487 | TBC1D3, AGAP3,
TBC1D3B, AGAP4 | 8.3 |
Discussion
WES technologies have provided extensive profiles of
genomic mutations in cancers, however, how to process the generated
dataset effectively for downstream analyses, remains a problem.
Currently the accuracy of variant calling is still influenced by a
number of factors. Firstly, low specificity and sensitivity of the
existing high-throughput sequencing may prevent the generation of
accurate mutation profiles (26).
Secondly, the BWA algorithms may produce incorrect base alignment.
Finally, the three algorithm tools, MuTect, VarScan and GATK, used
for identifying variants, present their respective limitations.
GATK is a semi-automated algorithm that calculates somatic
variants. VarScan identifies the most high-quality SNVs
preferentially, while MuTect outperforms in low-quality ones. Some
true SNVs are hard to differentiate due to a number of factors
including clonal heterogeneity, strand bias, low allele
frequencies, tumor contamination, high GC content of genomic
regions, sequencing errors and non-specificities in short read
mapping (12).
Comparisons between SNVs calls analyzed with GATK,
MuTect and VarScan, revealed that only a few of the SNVs were
called by more than one of the tools (Fig. 3), thus it was difficult to select
candidate SNVs for further validation. The disagreement was
partially due to prior assumptions underlying each algorithm and
different error models. Therefore, further development of more
significant and accurate calling algorithms was required (27), however, combining MuTect/GATK with
VarScan produced more accurate SNVs. In light of these limitations
in genomic studies, the three-caller strategy was designed to
obtain accurate mutation information for clinical assessment.
The present study integrated different software
programs to form a modular pipeline for processing somatic SNVs and
indels. A series of software was used to perform data alignment,
data filtering, reducing duplicate and realignment, as well as
recalibrating through java. In the study of HCC, WES analysis
started with the acquisition of raw data to select several
candidate genes, which alluded to the potential effect of
cancer-associated somatic mutations on tumor progression. The
mutation set-based analysis revealed a number of potential somatic
events in HCC, including in CUB and sushi multiple domains 1,
FRAS1-related extracellular matrix 1 and MUC16 genes. The mutations
at different base positions of the same gene or different genes may
lead to disparate functions such as activation and inactivation
mutations. This may influence their physicochemical properties and
structure in comparison with wild-type proteins. Functional
enrichment analysis revealed the biological process enrichment of
cancer-specific mutations, including cell adhesion and regulation
of Ras GTPase activity. Experiments are required to validate the
variants which may affect interactions with other proteins and
disorder crucial signaling pathways (28).
In conclusion, the pipeline for HCC exome sequencing
data analysis demonstrated in the present study provided a
convenient strategy to identify the potentially functional
tumor-specific mutations, which may support our understanding of
the underlying mechanisms of HCC development.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant no. 31571434), the
National High Technology Research and Development Program of China
(grant no. 2012AA02A205) and the National Basic Research Program of
China (grant no. 2015CB553701).
References
|
1
|
Lengauer C, Kinzler KW and Vogelstein B:
Genetic instabilities in human cancers. Nature. 396:643–649. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bass AJ, Lawrence MS, Brace LE, Ramos AH,
Drier Y, Cibulskis K, Sougnez C, Voet D, Saksena G, Sivachenko A,
et al: Genomic sequencing of colorectal adenocarcinomas identifies
a recurrent VTI1A-TCF7L2 fusion. Nat Genet. 43:964–968. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chapman MA, Lawrence MS, Keats JJ,
Cibulskis K, Sougnez C, Schinzel AC, Harview CL, Brunet JP, Ahmann
GJ, Adli M, et al: Initial genome sequencing and analysis of
multiple myeloma. Nature. 471:467–472. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jia D, Dong R, Jing Y, Xu D, Wang Q, Chen
L, Li Q, Huang Y, Zhang Y, Zhang Z, et al: Exome sequencing of
hepatoblastoma reveals novel mutations and cancer genes in the Wnt
pathway and ubiquitin ligase complex. Hepatology. 60:1686–1696.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Biesecker LG and Green RC: Diagnostic
clinical genome and exome sequencing. N Engl J Med.
371:11702014.PubMed/NCBI
|
|
6
|
Cancer Genome Atlas Research Network.
Hammerman PS, Lawrence MS, Voet D, Jing R, Cibulskis K, Sivachenko
A, Stojanov P, McKenna A, Lander ES, et al: Comprehensive genomic
characterization of squamous cell lung cancers. Nature.
489:519–525. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cancer Genome Atlas Network. Muzny DM,
Bainbridge MN, Chang K, Dinh HH, Drummond JA, Fowler G, Kovar CL,
Lewis LR, Morgan MB, et al: Comprehensive molecular
characterization of human colon and rectal cancer. Nature.
487:330–337. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cancer Genome Atlas Network. Koboldt DC,
Fulton RS, McLellan MD, Schmidt H, Kalicki-Veizer J, McMichael JF,
Fulton LL, Dooling DJ, Ding L, et al: Comprehensive molecular
portraits of human breast tumours. Nature. 490:61–70. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Litchfield K, Summersgill B, Yost S,
Sultana R, Labreche K, Dudakia D, Renwick A, Seal S, Al-Saadi R,
Broderick P, et al: Whole-exome sequencing reveals the mutational
spectrum of testicular germ cell tumours. Nat Commun. 6:59732015.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang Z: Genomic landscape of liver
cancer. Nat Genet. 44:1075–1077. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kan Z, Zheng H, Liu X, Li S, Barber TD,
Gong Z, Gao H, Hao K, Willard MD, Xu J, et al: Whole-genome
sequencing identifies recurrent mutations in hepatocellular
carcinoma. Genome Res. 23:1422–1433. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang Q, Jia P, Li F, Chen H, Ji H, Hucks
D, Dahlman KB, Pao W and Zhao Z: Detecting somatic point mutations
in cancer genome sequencing data: A comparison of mutation callers.
Genome Med. 5:912013. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gerlinger M, Rowan AJ, Horswell S, Larkin
J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A,
Tarpey P, et al: Intratumor heterogeneity and branched evolution
revealed by multiregion sequencing. N Engl J Med. 366:883–892.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cibulskis K, Lawrence MS, Carter SL,
Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES
and Getz G: Sensitive detection of somatic point mutations in
impure and heterogeneous cancer samples. Nat Biotechnol.
31:213–219. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Koboldt DC, Zhang Q, Larson DE, Shen D,
McLellan MD, Lin L, Miller CA, Mardis ER, Ding L and Wilson RK:
Varscan 2: Somatic mutation and copy number alteration discovery in
cancer by exome sequencing. Genome Res. 22:568–576. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
McKenna A, Hanna M, Banks E, Sivachenko A,
Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly
M and DePristo MA: The genome analysis toolkit: A mapreduce
framework for analyzing next-generation DNA sequencing data. Genome
Res. 20:1297–1303. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mardis ER: Next-generation DNA sequencing
methods. Annu Rev Genomics Hum Genet. 9:387–402. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Metzker ML: Sequencing technologies-the
next generation. Nat Rev Genet. 11:31–46. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dohm JC, Lottaz C, Borodina T and
Himmelbauer H: Substantial biases inultra-short read data sets from
high-throughput DNA sequencing. Nucleic Acids Res. 36:e1052008.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nielsen R, Paul JS, Albrechtsen A and Song
YS: Genotype and SNP calling from next-generation sequencing data.
Nat Rev Genet. 12:443–451. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li H and Durbin R: Fast and accurate
long-read alignment with burrows-wheeler transform. Bioinformatics.
26:589–595. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li H, Handsaker B, Wysoker A, Fennell T,
Ruan J, Homer N, Marth G, Abecasis G and Durbin R: 1000 Genome
Project Data Processing Subgroup: The sequence alignment/map format
and SAMtools. Bioinformatics. 25:2078–2079. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ramos AH, Lichtenstein L, Gupta M,
Lawrence MS, Pugh TJ, Saksena G, Meyerson M and Getz G: Oncotator:
Cancer variant annotation tool. Hum Mutat. 36:E2423–E2429. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Robinson JT, Thorvaldsdóttir H, Winckler
W, Guttman M, Lander ES, Getz G and Mesirov JP: Integrative
genomics viewer. Nat Biotechnol. 29:24–26. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
da Huang W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Totoki Y, Tatsuno K, Yamamoto S, Arai Y,
Hosoda F, Ishikawa S, Tsutsumi S, Sonoda K, Totsuka H, Shirakihara
T, et al: High-resolution characterization of a hepatocellular
carcinoma genome. Nat Genet. 43:464–469. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lawrence MS, Stojanov P, Polak P, Kryukov
GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH,
Roberts SA, et al: Mutational heterogeneity in cancer and the
search for new cancer-associated genes. Nature. 499:214–218. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kwon SM, Cho H, Choi JH, Jee BA, Jo Y and
Woo HG: Perspectives of integrative cancer genomics in next
generation sequencing era. Genomics Inform. 10:69–73. 2012.
View Article : Google Scholar : PubMed/NCBI
|