Introduction
Autophagy is a process of cellular repair and
survival, during which cytoplasmic components are sequestered into
double-membrane vesicles. Previous research has revealed that
autophagy is stimulated in advanced atherosclerotic plaques by
oxidized lipids, inflammation and metabolic stress (1). Autophagy is beneficial in promoting
cellular recovery in adverse environments; it is also valuable in
inhibiting apoptosis. In advanced atherosclerosis (AS), macrophage
apoptosis, together with defective phagocytic clearance of the
apoptotic cells, promotes plaque necrosis, which results in acute
atherothrombotic cardiovascular events (2). Basal autophagy is beneficial in early
AS, however a detrimental effect is observed in advanced AS plaques
(3); autophagy is capable of
protecting cells from oxidative stress via the degradation of
damaged intracellular material; however, excessively stimulated
autophagic activity may result in significant destruction of the
cytosol (1). The latter scenario
leads to programmed cell death and may cause vascular endothelial
cell (VEC) death, which results in plaque destabilization.
Endothelial injury or death represents a predominant mechanism of
acute clinical events, due to the promotion of lesional thromboses
(4). Furthermore, insufficient
autophagic activity also reduces free cholesterol and
apolipoprotein A-I (5), and this
decreases the amount of high-density lipoprotein. Insufficient and
excessive autophagy of VECs are therefore both injurious, and the
regulation of autophagic homeostasis may be important in the
treatment of AS.
The molecular mechanism of autophagy
When Ashford and Porter (6) perfused rat livers with glucagon, they
discovered a large increase in the number of cellular lysosomes.
This phenomenon of self-feeding was referred to as autophagy. This
process involves the packaging of cytoplasmic proteins or
organelles into vesicles, followed by lysosomal fusion, to form an
autophagic lysosome; a cellular event which aids in metabolism and
in the renewal of organelles (7).
Autophagy is a primary catabolic survival process against various
types of stress. During macroautophagy, several substrates,
including lipids, pathogens, invading proteins, or even damaged
organelles may be sequestered into double-membrane vesicles known
as autophagosomes. In order to degrade their cargo, autophagosomes
must subsequently fuse with a lysosome, which contains various
hydrolases that are capable of degrading the sequestered substrates
(8). Autophagy responses are
triggered by stress; the elimination of damaged organelles and
subsequent release of energy substrates promotes cell survival
(9). Under physiological
conditions, autophagy is able to degrade dysfunctional organelles
and long-lived proteins; however, autophagic cell death will occur
if it is uncontrolled, due to the risk of excessive destruction of
organelles and important molecules (10). There is a manner of
organelle-selection during autophagy, with selective targeting of
lipid droplets, protein aggregates, endoplasmic reticulum,
peroxisomes, microorganisms, ribosomes and portions of the nucleus
or mitochondria (11). The basic
components obtained from cargo degradation are subsequently
released into the cytoplasm for recycling. Currently, three primary
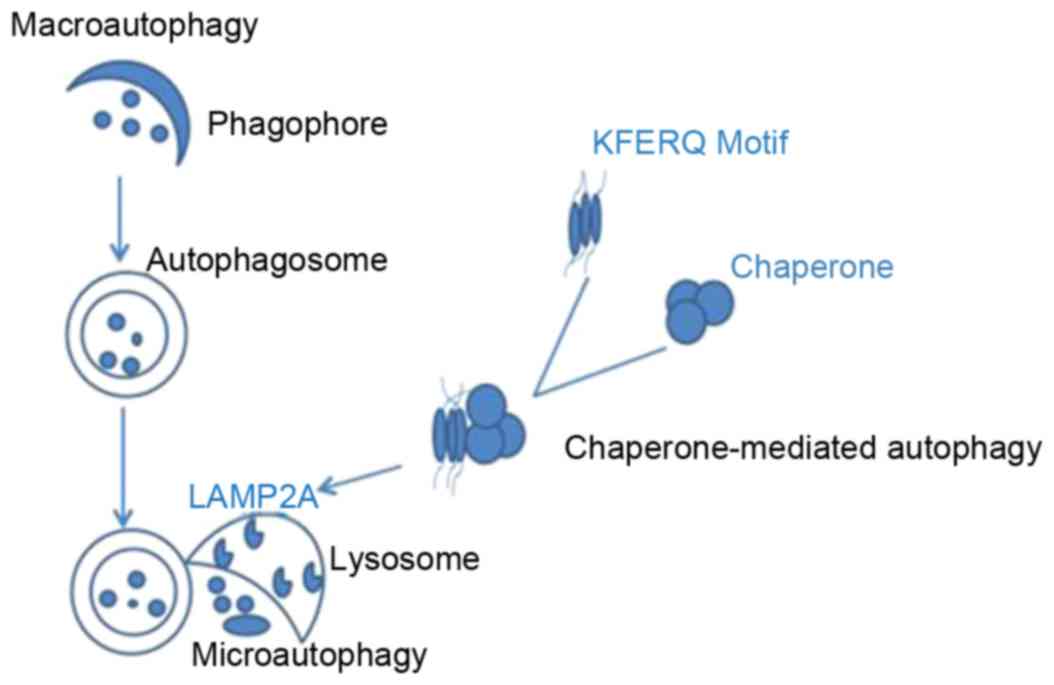
types of autophagy have been described (12): i) Microautophagy, which involves
direct engulfment of cytoplasmic material by lysosomes, via inward
invaginations of the lysosomal membrane; ii) macroautophagy,
characterized by formation of autophagosomes that fuse with
lysosomes; and iii) chaperone-mediated autophagy, which is
facilitated by a chaperone complex mediated by lysosomal-associated
membrane protein type 2A (LAMP2A), to degrade cytosolic proteins
with a specific targeting motif. Microautophagy and macroautophagy
may be selective or nonselective, and are observed in yeast and
higher eukaryotes; however, chaperone-mediated autophagy is a
selective process that has only been described in mammalian cells
(13). During chaperone-mediated
autophagy, specific protein substrates containing the amino acid
sequence KFERQ are recognized by chaperones (14), unfolded and translocated into the
lysosome, via LAMP2A. In microautophagy, uptake occurs at the
limiting membrane of the lysosome/vacuole, however this process
operates by directly sequestering the substrates via invagination
of the lysosome/vacuole membrane. Macroautophagy is the most
well-studied process out of the 3 types of autophagy (Fig. 1). Multiple autophagy-related genes
(ATG) and proteins have been described across the various stages of
autophagy (15). Macroautophagy
may be dissected into various steps based on the proteins involved:
Induction; nucleation of the autophagosome precursor; membrane
expansion and maturation of the autophagosome; fusion with the
lysosome/vacuole and recycling of the degraded cargo (16). Once autophagy is induced, assembly
of the phagophore is initiated by membrane nucleation. In both
yeast and mammals, the class III phosphatidylinositol 3-kinases
catalyze nucleation of the phagophore by producing
phosphatidylinositol 3-phosphate (PtdIns-3-P) and inducing the
recruitment of PtdIns-3-P binding proteins. Although membrane
nucleation has been established as a key step in the autophagic
process, the origin of the membrane that gives rise to the
phagophore, and subsequently the autophagosome, remains unclear.
Elongation and expansion of the phagophore membrane are key steps
in the autophagic process (17).
The autophagy related protein Atg12-Atg5-Atg16 and Atg8 conjugation
systems, which are inter-related ubiquitin-like conjugation
pathways, regulate this stage in both yeast and mammals (18). Previous research discovered that
functional cooperation occurs between the Beclin 1-binding proteins
Atg14L and Rubicon, and this regulates autophagy at different
stages. Atg14L is essential for autophagosome formation, and
deficiency of this protein results in defects of autophagic
degradation in mouse embryonic stem cells. Furthermore,
autophagosome maturation and endocytosis was enhanced and the
number of autophagosomes/autolysosomes was increased following
knockdown of Rubicon, indicating that Rubicon is negatively
involved in the formation of autophagosomes (19).
Autophagy has been described widely in the
cardiovascular system; autophagic activity is linked to
cardiovascular development, heart and vascular homeostasis and the
onset and progression of several cardiovascular diseases. Improved
understanding of the molecular mechanism involved may therefore
pave the way for novel therapeutic interventions in the treatment
of cardiovascular disease.
Autophagy in the vascular system
Previous research has indicated that basal autophagy
may have a significant impact on the functioning of the vasculature
(20). This is induced by
oxidative stress, which arises as a result of vascular disease and
cell death initiation, particularly during the early stages of AS.
Autophagy is a cytoprotective mechanism in the normal vessel wall,
and there are several autophagy-signaling pathways involved in the
cardiovascular system, which have already been extensively reviewed
elsewhere (21), these pathways
include the following: i) Mammalian target of rapamycin (mTOR), a
highly conserved protein that can integrate several types of
extracellular signals, including nutritional signals and growth
factors, which are involved in gene transcription, protein
translation, ribosome synthesis, and the regulation of cell
apoptosis and autophagy. The mTOR protein accordingly serves an
important role in cell growth; ii) adenosine
monophosphate-activated protein kinase (AMPK) is a conserved
heterotrimeric protein kinase, which can stimulate autophagy when
adenosine monophosphate (AMP), the cell ‘starvation’ signal, is
increased. AMPK maintains the balance of adenosine triphosphate
generation and consumption in eukaryotic cells, via detection of
the cellular energy state. Furthermore, AMPK serves a key role in
regulating cell growth and proliferation, establishing and
stabilizing cell polarity; iii) inositol 1,4,5-trisphosphate and
inositol 1,4,5-trisphosphate receptor serve important roles in
regulating autophagy; iv) transcription factor tumor promoter p53
has contrasting roles in regulating autophagy, and these are
subcellular location-dependent; v) cyclic AMP (cAMP)-dependent
protein kinase A (PKA) is composed of two catalytic subunits and
two regulatory subunits, binding of cAMP to the regulatory subunit
results in a conformational change and subsequent release of the
catalytic subunit. Activation of the PKA catalytic subunits impacts
on the expression of related genes, and allows detection of the
nutritional and growth status of the cell; vi) histone
acetyltransferases and histone deacetylases (HDACs) are proteins
that regulate chromosome structure and gene expression. HDACs may
be targeted by small molecular inhibitors, and this may inhibit
pathological cardiac remodeling; vii) glycogen synthase kinase 3β
(GSK3β) is a multifunctional serine/threonine kinase, which is
commonly found in eukaryotic cells. GSK3β is involved in numerous
cellular signaling pathways, the predominant of which involves
regulation of glycogen metabolism, cell differentiation,
proliferation and gene expression; viii) nicotinamide adenine
dinucleotide is a rate-limiting enzyme, which is involved in
several physiological activities including cell metabolism, energy
synthesis, DNA repair, suppression of apoptosis and stimulation of
autophagic flux; ix) microRNAs are non-encoding RNAs that are
expressed in eukaryotic cells, and serve a regulatory role in cell
proliferation, differentiation and death (21).
Vascular endothelial cells (VECs)
VECs are located at the interior surface of the
vascular wall, and are an effective permeable barrier between
circulating blood and tissues. VECs also participate in the
regulation of cellular cholesterol, lipid homeostasis, signal
transduction, immunity, inflammation and hemostasis. Dysfunction of
the endothelium is a critical inducer for AS and other
cardiovascular diseases (20). A
recent study demonstrated that oxidized low-density lipoprotein
(oxLDL) may induce autophagy in VECs (22), resulting in increased expression of
microtubule associated protein 1 light chain 3 (LC3) and B cell
lymphoma/leukemia-1 (23).
Furthermore, serum starvation induces autophagy, however a
reduction in serum VECs induces apoptosis. The role of autophagy in
VECs has therefore received significant research attention. A
previous study demonstrated that human plasminogen Kringle 5 and
endostatin are inhibitors of angiogenesis; these molecules induce
apoptosis and inhibit cell proliferation, and can induce apoptosis
and promote autophagy in VECs (24).
Smooth muscle cells (SMCs)
SMCs demonstrate programmed cell death features in
the fibrous cap of advanced AS plaques (25). Features include the formation of
myelin figures, the aggregation of ubiquitin inclusion bodies in
the cytoplasm and significant vacuolization. Furthermore, myelin
figures are formed of phospholipids and plasma membrane fragments
arranged in concentric circles, and these reflect autophagic
degradation of the membranous cell structure. These autophagic
structures are not common in human atherosclerotic plaques;
however, they may be observed in cholesterol fed rabbit
atherosclerotic plaques (25).
Previous research has speculated that SMCs in the
fibrous cap are surrounded by basement membrane, indicating these
cells may undergo starvation-induced autophagy (26). However, in vitro studies
have suggested that there may be alternative pathways of induced
autophagy in the atherosclerotic plaque. For example, tumor
necrosis factor-α induces the expression of LC3 via the c-jun amino
terminal and protein kinase B pathway, and induces expression of
Beclin1 via the c-jun amino terminal pathway, thereby leading to
SMC death via autophagy (27).
Furthermore, mild oxidative stress also may activate autophagy,
thus promoting the removal of damaged organelles (28).
Macrophages
Macrophages demonstrate significant phagocytic
capabilities, and the cytoplasmic vesicles contained within these
cells may therefore result from autophagy, or from phagocytosis of
foreign bodies. Autophagic vesicles may be detected using an
antibody against a specific marker such as LC3. Cholesterol
acyltransferase (ACAT) is a sterol ester enzyme that regulates the
free cholesterol in cell membranes to prevent cell death.
Phytosterol is a substrate of ACAT, and a recent study revealed
that phytosterols may accumulate in macrophages in atherosclerotic
plaques, which may induce autophagic death, and necrosis of the
plaque (29). High levels of
phytosterols were also observed in patients with sitosterol
(30), and this may lead to the
premature development of a severe coronary AS thrombosis.
There is an increasing body of research (30,31)
investigating the biological role of autophagy in the vascular wall
(15); it is suspected that
autophagic imbalance is associated with many vascular diseases,
such as pulmonary hypertension. Furthermore, there is recent
evidence that autophagy acts on a series of vascular processes,
ranging from angiogenesis to vascular wall calcification. Although
the autophagic mechanisms are different in endothelial cells and
SMCs, autophagosome formation is related to β-amyloid, which is
stimulated by oxidized lipids (32). Notably, the vasoactive substances
secreted by endothelial cells have important regulatory effects on
autophagy (15).
Autophagy in AS
Progressive stages of AS may be characterized by
distinctive molecular events. In early AS, cholesterol accumulates
to form foam cells, however early foam cell lesions are
non-occlusive and this is therefore asymptomatic. In advanced AS,
plaque rupture or erosion may cause acute clinical events. However,
in early AS with endothelial dysfunction (33), the repair of VEC injuries may
prevent the subsequent development of AS. Furthermore, in advanced
AS, apoptotic macrophages may be quickly cleaned-up by macrophages,
to prevent the progression of inflammation and plaque necrosis,
thereby delaying plaque development (29). Autophagy serves an important role
at each stage of AS; in early AS, autophagy may reduce lipid
accumulation and inhibit the formation of foam cells. As AS
progresses, autophagy can remove necrotic cells and delay
development of the plaque (34).
The specific autophagic mechanisms occurring at each stage are
currently unclear, and further research investigating these
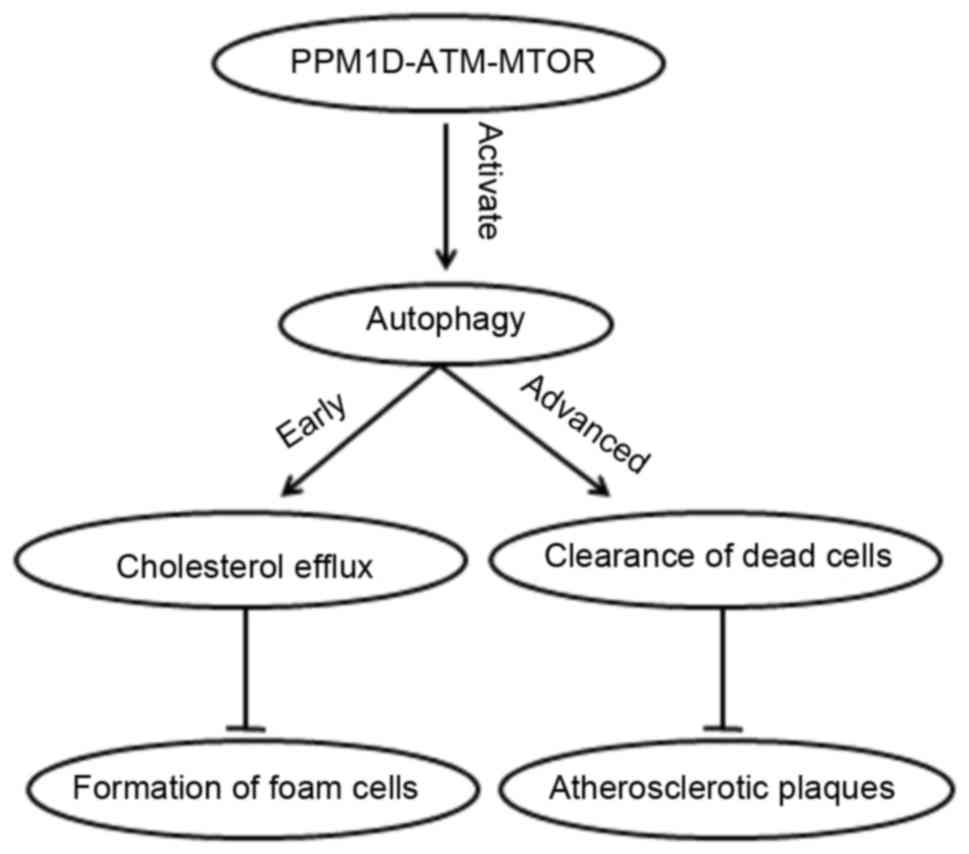
processes is warranted. In a recent study (35), protein phosphatase magnesium
dependent 1D (PPM1D) demonstrated a key role in the regulation of
autophagy during the development of AS. The conversion of
macrophages into foam cells is a major event in early AS, and PPM1D
deficiency inhibits the accumulation of lipid droplets in
macrophages, thus preventing the formation of foam cells and
delaying the development of atherosclerotic plaques. In early AS
stages, cholesterol predominantly accumulates in the cytoplasm as
lipid droplets; when macrophage PPM1D is deficient, autophagy is
activated and these cytosolic lipid droplets may be fused with
autophagosomes and lysosomes, to produce a free cholesterol efflux
(35).
Macrophage apoptosis and defects in the removal of
apoptotic cells may promote plaque necrosis in advanced AS,
resulting in acute atherothrombotic cardiovascular events (36). Autophagy is activated by the
associated stimulation of AS. On the contrary, gathering numbers of
apoptotic macrophages inhibits autophagic activity, and this
promotes plaque necrosis. Furthermore, macrophage deficiency
resulted in an associated reduction in cholesterol efflux, a
process that served a significant protective role against the
development of AS. Previous research has demonstrated that PPM1D
(37), also known as wild-type
p53-induced phosphatase (WIP1), functions in a regulatory role in
AS development in mice. PPM1D knockdown decreases the lipid
accumulation of macrophages and inhibits the formation of foam
cells, thus inhibiting the progression of AS plaques. These events
are regulated by the ataxia telangiectasia mutated gene (ATM)-mTOR
signaling pathway, which controls the efflux of cholesterol, and
has a protective effect on early and advanced AS (24) (Fig.
2). A study involving WIP1 deficient mice allowed further
insight into the role of this protein in autophagy. In these mice,
lipid efflux was increased and autophagic flux was impaired,
thereby reducing the conversion of macrophages into foam cells, and
preventing the formation of AS. WIP1 regulates autophagy via ATM
and mTOR, which indicates that this protein may serve a protective
role in advanced AS; inactivation of mTOR may therefore provide a
novel therapeutic option in the treatment of AS (22). Other recent studies have
investigated the effect of small molecular weight compounds and
proteins on AS, including 3BDO, microtubule-associated protein 1
light chain 3 beta (MAP1LC3B) and lectin-like oxidized
low-density-lipoprotein receptor-1 (LOX-1) (38). 3BDO activation of mTOR serves to
protect endothelial cells and to inhibit the development of AS. The
mTOR protein is an evolutionarily conserved serine/threonine kinase
which has two forms, mTOR1 and mTOR2; mTOR1 predominantly regulates
cell growth, cell apoptosis, energy metabolism and autophagy and is
rapamycin-sensitive, whereas mTOR2 is associated with cell survival
and is not rapamycin-sensitive. Both mTOR forms are able to induce
autophagy. The 3BDO-induced inhibition of autophagy is mediated by
oxLDL in endothelial cells, and decreased ATG expression and
autophagy activity in apoE−/− mice. Conversely, 3BDO
barely impacts upon mTOR signaling in macrophages and smooth muscle
cells, and therefore 3BDO may selectively protect endothelial
cells, promoting atherosclerotic plaque stability via the mTOR
pathway (38). MAP1LC3B serves a
critical role in limiting the development of AS. Overexpression of
MAP1LC3B can stabilize plaque development in AS; the unfolded
protein response, which occurs under conditions of stress,
activates protective mechanisms by upregulating MAP1LC3B, and this
accomplishes a protective effect via the activation of basal
autophagy (39). OxLDL contributes
to endothelial cell dysfunction, which occurs via the induction of
oxidative stress and the subsequent initiation of apoptosis, and
this results in AS development and progression (26). Overexpression of the oxLDL receptor
1 (LOX-1) appears to serve a role in attenuating endothelial cell
protective autophagy. Previous research has demonstrated that early
changes in endothelial cell viability, following incubation with
oxLDL, are proceeded by impaired endothelial nitric oxide synthase
activity, and subsequent oxidative stress (40). This effect is associated with the
overexpression of the LOX-1 receptor, which mediates the
attenuation of protective autophagy, and leads to oxLDL-induced
bovine aortic endothelial cell death. These studies provided fresh
insight into the assessment of the pathophysiological mechanisms
underlying EC dysfunction, and allowed the identification of novel
therapeutic strategies that target the early stages of AS.
Conclusions
Recently, Han et al reported that curcumin
and resveratrol induced autophagy to protect VECs (41). Autophagy is an evolutionarily
conserved mechanism that serves a critical role in the regulation
of lipid metabolism in normal cells. Therefore, autophagy may
provide new therapeutic targets for lipid metabolism disorders,
including AS. Previous research has demonstrated that PPM1D may
inhibit the formation of foam cells by the ATM/mTOR signaling
pathway, via the regulation of autophagy-dependent cholesterol
efflux (42). Autophagy-mediated
inhibition of foam cell formation is induced by oxLDL, this results
in stimulation of transient receptor potential vanilloid type 1
(TRPV1) by capsaicin, and the AMPK signaling pathway is activated
by oxLDL to repair autophagic injury, with ultimate inhibition of
the formation of foam cells. Consequently, the role of autophagy
and TRPV1 in the formation of vascular smooth muscle cell foam
cells, may provide a novel therapeutic target for the treatment of
AS (43). Therefore, current
research indicates that atherosclerotic development is associated
with the dysfunction of autophagy, an event which results in
vascular oxidative stress, inflammation and plaque necrosis; this
knowledge may provide a novel mechanism through which the progress
of AS may be inhibited (44). This
disease may be treatable via an autophagic signal, and a few drugs
that can regulate autophagy have been identified. Autophagy is not
only protective, but it may also have an impaired impact on the
cell; however, there is limited clinical data to demonstrate its
curative role, and currently the treatment of cardiovascular
disease generally involves the downregulation of autophagy
(45).
In conclusion, macrophage apoptosis combined with
defective phagocytosis to clear the apoptotic cells promotes plaque
necrosis, thus leading to acute atherothrombotic cardiovascular
events in advanced AS. Inhibition of autophagy increases apoptosis
in macrophages, and treatment of macrophages with AS stimulators of
apoptosis resulted in the induction of autophagy (46). Defective macrophage autophagy
results in oxidative stress, plaque necrosis, macrophage apoptosis
and defective phagocytic clearance in AS (47). Previous research discovered that
the inhibition of autophagy, in vitro or in vivo,
resulted in increased apoptosis and reduced recognition of the
apoptotic cells by phagocytes (12). However, although autophagy is
beneficial in AS, excessive autophagy may promote plaque rupture
and the precipitation of acute clinical events, suggesting that
both insufficient and excessive autophagy of VECs is detrimental,
and the regulation of autophagic homeostasis is critical in the
treatment of AS. Currently these autophagic functions have not been
investigated in the context of vascular inflammation, and as these
studies progress more can be identified about whether autophagy is
a suitable target for therapeutic intervention in AS.
Acknowledgements
This work was supported by the National Natural
Science Foundation of China (grant no. 81670424), the National
Natural Science Foundation of Hunan Province (grant no.
2015JJ2118), the Key Scientific Research Fund of Hunan Provincial
Education Department (grant no. 15A166), the Scientific Research
Fund of the National Security Agency (grant no. hunan-0013-2016AQ),
the Scientific Research Fund of Hunan Provincial Health and Family
Planning Commission (grant no. B2016087), the Ph.D. Programs
Foundation of Ministry of Education of China (grant nos.
20114324120004 and 20124324110003), the Aid Program for Science and
Technology Innovative Research Team in Higher Educational
Institutions of Hunan Province (grant no. 2008-244), the Construct
Program of the Key Discipline in Human Province (grant no. 2011-76)
and the Postgraduate Innovation Project of Scientific Research of
University of South China (grant no. 2015XCX38).
References
|
1
|
Liao X, Sluimer JC, Wang Y, Subramanian M,
Brown K, Pattison JS, Robbins J, Martinez J and Tabas I: Macrophage
autophagy plays a protective role in advanced atherosclerosis. Cell
Metab. 15:545–553. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dong Z, Wang L, Xu J, Li Y, Zhang Y, Zhang
S and Miao J: Promotion of autophagy and inhibition of apoptosis by
low concentrations of cadmium in vascular endothelial cells.
Toxicol In Vitro. 23:105–110. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu H, Cao Y, Tong T, Shi J, Zhang Y, Yang
Y and Liu C: Autophagy in atherosclerosis: A phenomenon found in
human carotid atherosclerotic plaques. Chin Med J (Engl).
128:69–74. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang L, Li H, Zhang J, Lu W, Zhao J, Su L,
Zhao B, Zhang Y, Zhang S and Miao J: Phosphatidylethanolamine
binding protein 1 in vacular endothelial cell autophagy and
atherosclerosis. J Physiol. 591:5005–5015. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Le Guezennec X, Brichkina A, Huang YF,
Kostromina E, Han W and Bulavin DV: Wip1-dependent regulation of
autophagy, obesity, and atherosclerosis. Cell Metab. 16:68–80.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ashford TP and Porter KR: Cytoplasmic
components in hepatic cell lysosomes. J Cell Biol. 12:198–202.
1962. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
He C and Klionsky DJ: Regulation
Mechanisms and Signaling Pathways of autophagy. Annu Rev Genet.
43:67–93. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang YL, Cao YJ, Zhang X, Liu HH, Tong T,
Xiao GD, Yang YP and Liu CF: The autophagy-lysosome pathway: A
novel mechanism involved in the processing of oxidized LDL in human
vascular endothelial cells. Biochem Biophys Res Commun.
394:377–382. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Schrijvers DM, De Meyer GR, Herman AG and
Martinet W: Phagocytosis in atherosclerosis: Molecular mechanisms
and implications for plaque progression and stability. Cardiovasc
Res. 73:470–480. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ren J and Taegtmeyer H: Too much or not
enough of a good thing-the janus faces of autophagy in cardiac fuel
and protein homeostasis. J Mol Cell Cardiol. 84:223–226. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xie Y, You SJ, Zhang YL, Han Q, Cao YJ, Xu
XS, Yang YP, Li J and Liu CF: Protective role of autophagy in
AGE-induced early injury of human vascular endothelial cells. Mol
Med Rep. 4:459–464. 2011.PubMed/NCBI
|
|
12
|
Vindis C: Autophagy: An emerging
therapeutic target in vascular diseases. Br J Pharmacol.
172:2167–2178. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sato M, Seki T, Konno A, Hirai H, Kurauchi
Y, Hisatsune A and Katsuki H: Fluorescent-based evaluation of
chaperone-mediated autophagy and microautophagy activities in
cultured cells. Genes Cells. 21:861–873. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bonhoure A, Vallentin A, Martin M,
Senff-Ribeiro A, Amson R, Telerman A and Vidal M: Acetylation of
translationally controlled tumor protein promotes its degradation
through chaperone-mediated autophagy. Eur J Cell Biol pii.
S0171–9335. 2017.(Epub ahead of print).
|
|
15
|
Nussenzweig SC, Verma S and Finkel T: The
role of autophagy in vascular biology. Circ Res. 116:480–488. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mizushima N: Autophagy: Process and
function. Genes Dev. 21:2861–2873. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tang TX, Jo A, Deng J, Ellena JF, Lazar
IM, Davis RM and Capelluto DG: Structural, thermodynamic, and
phosphatidylinositol 3-phosphate binding properties of Phafin2.
Protein Sci. 2017.(Epub ahead of print). View Article : Google Scholar :
|
|
18
|
Gatica D, Chiong M, Lavandero S and
Klionsky DJ: Molecular mechanisms of autophagy in the
cardiovascular system. Circ Res. 116:456–467. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Matsunaga K, Saitoh T, Tabata K, Omori H,
Satoh T, Kurotori N, Maejima I, Shirahama-Noda K, Ichimura T, Isobe
T, et al: Two Beclin 1-binding proteins, Atg14L and Rubicon,
reciprocally regulate autophagy at different stages. Nat Cell Biol.
11:385–396. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
De Meyer GR, Grootaert MO, Michiels CF,
Kurdi A, Schrijvers DM and Martinet W: Autophagy in vascular
disease. Circ Res. 116:468–479. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lavandero S, Troncoso R, Rothermel BA,
Martinet W, Sadoshima J and Hill JA: Cardiovascular autophagy:
Concepts, controversies, and perspectives. Autophagy. 9:1455–1466.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Perrotta I and Aquila S: The role of
oxidative stress and autophagy in atherosclerosis. Oxid Med Cell
Longev. 2015:1303152015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ding Z, Wang X, Khaidakov M, Liu S, Dai Y
and Mehta JL: Degradation of heparan sulfate proteoglycans enhances
oxidized-LDL-mediated autophagy and apoptosis in human endothelial
cells. Biochem Biophys Res Commun. 426:106–111. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ramakrishnan S, Nguyen TM, Subramanian IV
and Kelekar A: Autophagy and angiogenesis inhibition. Autophagy.
3:512–515. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kockx MM, De Meyer GR, Buyssens N, Knaapen
MW, Bult H and Herman AG: Cell composition, replication, and
apoptosis in atherosclerotic plaques after 6 months of cholesterol
withdrawal. Circ Res. 83:378–387. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Salabei JK and Hill BG: Autophagic
regulation of smooth muscle cell biology. Redox Biol. 4:97–1033.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jia G, Cheng G, Gangahar DM and Agrawal
DK: Insulin-like growth factor-1 and TNF-alpha regulate autophagy
through c-jun N-terminal kinase and Akt pathways in human
atherosclerotic vascular smooth cells. Immunol Cell Biol.
84:448–454. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
KIFFIN R, Bandyopadhyay U and Cuervo M:
Oxidative stress and autophagy. Antioxidants Redox Signaling.
8:152–162. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bao L, Li Y, Deng SX, Landry D and Tabas
I: Sitosterol-containing lipoproteins trigger free sterol-induced
caspase-independent death in ACAT-competent macrophages. J Biol
Chem. 281:33635–33649. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Moghadasian MH, Alsaif M, Le K, Gangadaran
S, Masisi K, Beta T and Shen GX: Combination effects of wild rice
and phytosterols on prevention of atherosclerosis in LDL receptor
knockout mice. J Nutr Biochem. 33:128–135. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kostin S, Pool L, Elsässer A, Hein S,
Drexler HC, Arnon E, Hayakawa Y, Zimmermann R, Bauer E, Klövekorn
WP and Schaper J: Myocytes die by multiple mechanisms in failing
human hearts. Circ Res. 92:715–724. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Deng M, Huang L, Ning B, Wang N, Zhang Q,
Zhu C and Fang Y: β-asarone improves learning and memory and
reduces Acetyl Cholinesterase and Beta-amyloid 42 levels in APP/PS1
transgenic mice by regulating Beclin-1-dependent autophagy. Brain
Res. 1652:188–194. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lin HH: In Vitro and in Vivo
Atheroprotective Effects of Gossypetin against Endothelial Cell
Injury by Induction of Autophagy. Chem Res Toxicol. 28:202–215.
2015.PubMed/NCBI
|
|
34
|
Martinet W and De Meyer GR: Autophagy in
atherosclerosis. Curr Atheroscler Rep. 10:216–223. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Torii S, Yoshida T, Arakawa S, Honda S,
Nakanishi A and Shimizu S: Identification of PPM1D as an essential
Ulk1 phosphatase for genotoxic stress-induced autophagy. EMBO Rep.
17:1552–1564. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Razani B, Feng C, Coleman T, Emanuel R,
Wen H, Hwang S, Ting JP, Virgin HW, Kastan MB and Semenkovich CF:
Autophagy links inflammasomes to atherosclerotic progression. Cell
Metab. 15:534–544. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Brichkina A and Bulavin DV: WIP-ing out
atherosclerosis with autophagy. Autophagy. 8:1545–1557. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Peng N, Meng N, Wang S, Zhao F, Zhao J, Su
L, Zhang S, Zhang Y, Zhao B and Miao J: An activator of mTOR
inhibits oxLDL-induced autophagy and apoptosis in vascular
endothelial cells and restricts atherosclerosis in apolipoprotein
E−/− mice. Sci Rep. 4:55192014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Swaminathan B, Goikuria H, Vega R,
Rodríguez-Antigüedad A, Medina López A, Mdel Freijo M, Vandenbroeck
K and Alloza I: Autophagic marker MAP1LC3B expression levels are
associated with carotid atherosclerosis symptomatology. PLoS One.
9:e1151762014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Mollace V, Gliozzi M, Musolino V, Carresi
C, Muscoli S, Mollace R, Tavernese A, Gratteri S, Palma E, Morabito
C, et al: Oxidized LDL attenuates protective autophagy and induces
apoptotic cell death of endothelial cells: Role of oxidative stress
and LOX-1 receptor expression. Int J Cardiol. 184:152–158. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Han J, Pan XY, Xu Y, Xiao Y, An Y, Tie L,
Pan Y and Li XJ: Curcumin induces autophagy to protect vascular
endothelial cell survival from oxidative stress damage. Autophagy.
8:812–825. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ouimet M: Autophagy in obesity and
atherosclerosis: Interrelationships between cholesterol
homeostasis, lipoprotein metabolism and autophagy in macrophages
and other systems. Biochim Biophys Acta. 1831:1124–1133. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Li BH, Yin YW, Liu Y, Pi Y, Guo L, Cao XJ,
Gao CY, Zhang LL and Li JC: TRPV1 activation impedes foam cell
formation by inducing autophagy in oxLDL-treated vascular smooth
muscle cells. Cell Death Dis. 5:e11822014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Arora M and Kaul D: Coronary
atherosclerosis: Significance of autophagic armour. World J
Cardiol. 4:271–274. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chew LH, Lu S, Liu X, Li FK, Yu AY,
Klionsky DJ, Dong MQ and Yip CK: Molecular interactions of the
Saccharomyces cerevisiae Atg1 complex provide insights into
assembly and regulatory mechanisms. Autophagy. 11:891–905. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Martinet W, Verheye S and De Meyer GR:
Everolimus-induced mTOR inhibition selectively depletes macrophages
in atherosclerotic plaques by autophagy. Autophagy. 3:241–244.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Panda PK, Mukhopadhyay S, Das DN, Sinha N,
Naik PP and Bhutia SK: Mechanism of autophagic regulation in
carcinogenesis and cancer therapeutics. Semin Cell Dev Biol.
39:43–55. 2015. View Article : Google Scholar : PubMed/NCBI
|