Introduction
Chronic obstructive pulmonary disease (COPD) is one
of the most significant chronic conditions and is now the fourth
most common cause of death worldwide, which has an increasing
economic and social burden (1,2).
COPD affects >300 million people worldwide and accounts for 4
million deaths every year (3).
However, unlike heart disease and malignancy, mortality due to COPD
has increased progressively; without effective intervention, the
number of deaths caused by COPD is predicted to increase by >30%
between 2010 and 2020 (4).
Smoking, the major risk factor for COPD (5), is a powerful inducer of inflammatory
mediators, including oxidants and proteases (6). Emphysema is a characteristic of COPD,
which is caused by chronic inflammation in the lung that
contributes to lung tissue destruction and is mediated by a
multistep pathway, which involves an imbalance of metalloproteases,
including matrix metalloproteases (MMPs) and ADAM
metallopeptidases, and anti-metalloproteases, which include tissue
inhibitor of metalloproteinases (TIMPs) and α-2 macroglobulin. This
imbalance leads to MMP overproduction that is not sufficiently
counteracted by TIMPs (7,8). The imbalance of the metalloproteases
and anti-metalloproteases may be a potential target for the
treatment of the chronic inflammation and emphysema caused by
cigarette smoke (CS). The term ‘macrolide’ is used to describe
drugs with a macrocyclic lactone ring of ≥12 elements (9,10).
In addition to antimicrobial properties, other properties of
macrolides were suggested in the 1960s, particularly in chronic
inflammatory diseases, and these actions are observed only for
treatment with 14- and 15-membered macrolides, including
erythromycin (ERY), clarithromycin, roxithromycin and azithromycin
(11). The effects of macrolides
in patients with chronic inflammatory airway disease appear to be
independent of antimicrobial properties. A previous report
demonstrated the anti-inflammatory effect of macrolides in
CS-induced lung inflammation and emphysema, and also reported the
effect of ERY on the imbalance of
metalloproteases/anti-metalloproteases (12). However, the potential mechanism of
ERY in the inhibition of the inflammation in CS-induced emphysema
has not been clarified. Central to the inflammatory response
induced by CS is an increased level of chemotactic cytokines,
particularly interleukin (IL)-8. It is associated with an increase
in the activation of intracellular signaling molecules, including
mitogen-activated protein kinase (MAPK) and nuclear factor-κB
(NF-κB) (13,14). MAPKs, together with NF-κB
activation, are the most activated kinases in airway epithelial
cells and macrophages exposed to CS extracts, and these kinases are
important in inducing the expression of various pro-inflammatory
chemokines and cytokines, and inducing the activation of cytosolic
phospholipase A2 (15). The
present study evaluated the imbalance of
metalloproteases/anti-metalloproteases in CS-induced emphysema and
investigated the upstream signaling that altered
metalloprotease/anti-metalloprotease levels, via phosphorylation of
the MAPK family and NF-κB overactivation. The current study also
investigated whether ERY affects the
metalloprotease/anti-metalloprotease imbalance in a dose-dependent
manner, and if so, whether it does so by affecting signaling
pathways dependent on MAPKs or NF-κB.
Materials and methods
Animal model for cigarette smoke
exposure
All of the experiments were conducted in accordance
with the guidelines of the Research Ethics Committee of China
Medical University (Shenyang, China). Male Wistar rat weanlings
(6–7-weeks-old;170-210 g) were obtained from the Laboratory Animal
Center of China Medical University and were maintained in a
pathogen-free environment. They were fed laboratory rodent chow and
water ad libitum and were kept on a 12 h light/dark cycle.
Rats were randomly assigned into the following three groups:
control (n=12), CS exposure (n=12) and CS+ERY (n=12). Rats in the
CS group were exposed to smoke from 16 commercial cigarettes
(Marlboro; Philip Morris USA, New York, NY, USA; each cigarette
contains 0.9 mg nicotine, 14 mg carbon monoxide and 12 mg tar oil;
the smokescope v/V% was 8%) with the filters removed for 30 min
twice a day, 6 days per week for 12 weeks. The rats in the ERY
group were also exposed to CS for 12 weeks and treated with an
orally administered injection of ERY (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) at 100 mg/kg, once daily. The drug treatments
were performed 0.5 h prior to CS exposure from day 1of the 12-week
exposure period. The control group received a daily orally
administered injection of saline, which was the vehicle for ERY,
without any smoke exposure. At the end of week 12, rats were
sacrificed by exsanguination under anesthetic (40 mg/kg
pentobarbital intraperitoneally; Sigma-Aldrich; Merck KGaA) and
lungs were excised. The right upper lobes were taken and stored at
−80°C. The middle lobes of the right rat lungs, which were not
lavaged, were briefly washed in ice-cold saline containing 5 mM
CaCl2+1 mM MgCl2, fixed in ExCell Plus™ fixative
(American MasterTech Scientific, Inc., Lodi, CA, USA) at room
temperature for 24 h. Subsequently, the middle lobes of the right
rat lungs were embedded in paraffin blocks and sectioned (4 µm) for
conventional hematoxylin and eosin staining. The left lungs were
infused with 2 ml PBS four times. The bronchoalveolar lavage fluid
(BALF) was centrifuged for 10 min at 900 × g and 4°C. The cell-free
supernatants were stored at −80°C.
Morphologic and morphometric analyses
of lung tissue
Hematoxylin and eosin staining was performed on
paraffin-embedded lung tissue sections (4 µm). The measure of lung
tissue morphology was determined by light microscopy at a
magnification of ×100. The mean linear intercept (MLI) (16), a measure of inter-alveolar wall
distance, was defined by the total length of the cross-line/the
numbers of the alveolar walls intersecting the test lines. The mean
alveolar number (MAN) (16), an
indicator of alveolar density, was calculated by counting the
numbers of alveoli in each field. Five sections were analyzed per
animal, and four images were acquired from a randomly selected
location in each slide.
Measurement of IL-8 and leukotriene B4
(LTB4)
The protein expression of IL-8 and LTB4-α in the
BALF supernatant of rats in each group was determined by ELISA kits
purchased from EnzoLife Sciences, Inc. (Farmingdale, NY, USA; cat
nos. ADI-900-074 and ADI-900-068, respectively), according to the
manufacturer's instructions.
Characterization of inflammatory cells
in BALF
The collected BALF samples from the left lung
tissues were centrifuged at 1,400 × g for 10 min at 4°C, and their
supernatants were stored at −80°Cuntil ELISA analysis. The pelleted
cells were resuspended in PBS, and 1×105 cells were
subjected to cytospin centrifugation on glass slides and fixed with
100% methanol for 15 min, followed by staining with
May-Grünwald-Giemsa solution for 15 min. A differential cell count
of BALF cell pellets was performed under a light microscope at a
magnification of ×200 according to morphological
characteristics.
Measurement of MMP-2, MMP-9 and
TIMP-1
The right upper lobes of lungs were lysed by RIPA
buffer (Cell Signaling Technology, Inc., Danvers, MA, USA) at a
ratio of 100 mg of tissue to 1 ml of lysis buffer. This was
followed by centrifugation at 1,500 × g for 20 min at 4°C. Total
protein concentration was determined using the Bradford assay
(Pierce; Thermo Fisher Scientific, Inc., Waltham, MA, USA), and
equal amounts of protein (30 µg) were separated by SDS-PAGE (4%
stacking gel and 10% separating gel). Protein was subsequently
transferred onto a nitrocellulose membrane by electroblotting. The
membrane was blocked for 1 h at room temperature with blocking
buffer [3% non-fat milk in TBS (pH 7.4) containing 0.1% Tween-20]
and was subsequently incubated overnight at 4°Cwith mouse
antibodies against MMP-2 (1:500; cat. no. ab7033;), MMP-9 (1:500;
cat. no. ab38898), TIMP-1 (1:500; cat. no. ab61224) and β-actin
(1:2,000; cat. no. ab6276; all from Abcam, Cambridge, UK). The
membrane was washed three times and incubated for 2 h at room
temperature with horseradish peroxidase-conjugated anti-mouse and
anti-rabbit immunoglobulin G (IgG; 1:5,000; cat. nos. sc-516102 and
sc-2357, respectively; Santa Cruz Biotechnology, Inc., Dallas, TX,
USA). The bound secondary antibody was detected using the Enhanced
Chemiluminescence Plus kit (GE Healthcare Life Sciences, Chalfont,
UK) and analyzed using the Las-3000 Luminescent Image Analyzer
(Fujifilm, Tokyo, Japan). The band intensities were quantified
using ImageJ software (National Institutes of Health, Bethesda, MD,
USA). The fold changes in protein expression were calculated
relative to the levels in the control group. The average values for
each group were determined by 3 independent experiments. Lung
tissues were sectioned (4 µm), cleared of paraffin and endogenous
peroxidases were blocked by incubation with 3%
H2O2 for 10 min at room temperature, followed
by three washes with PBS for 3 min. Tissue sections were
subsequently incubated with rabbit serum (Sigma-Aldrich; Merck
KGaA) for 10 min at ambient temperature. Sections were incubated
overnight with mouse MMP-2 (1:50; cat. no. ab7033; Abcam), mouse
MMP-9 (1:50; cat. no. ab38898; Abcam) or rabbit TIMP-1 (1:50; cat.
no. ab61224; Abcam) antibodies at 4°C, which was followed by the
addition of biotinylated rabbit anti-mouse (1:2,000; cat.
PA1-28567; Thermo Fisher Scientific, Inc.) or mouse anti-rabbit and
anti-mouse IgG secondary antibody (1:2,000; cat. nos. sc-2357 and
sc-516102; Santa Cruz Biotechnology, Inc.). To verify the binding
specificity for MMP-2, MMP-9 and TIMP-1, certain sections were
incubated with only a primary or secondary antibody. In these
situations, no positive staining was identified in the sections,
indicating that the immunoreactions were positive in all of the
experiments. Immunohistochemistry staining was performed in
accordance with the manufacturer's instructions and visualized by
the use of diaminobenzidine staining. Digital photos were analyzed
with Image-Pro Plus 4.1 (MediaCybernetics, Inc., Rockville, MD,
USA) by an observer who was blind to the group classification.
Measurement of phosphorylated (p)-MAPK
and NF-κB
Total protein was extracted from the upper right
lobes of the lungs by lysis with RIPA buffer (100 mg of tissue to 1
ml of lysis buffer). This was followed by centrifugation at 1,500 ×
g for 20 min at 4°C. The protein concentration was
determined using the Bradford assay and equal amounts of protein
(50 µg) were separated by SDS-PAGE (4% stacking gel and 10%
separating gel) for detection of extracellular signal-regulated
kinase (ERK)1/2, c-Jun NH2-terminal kinase (JNK) and p38. Following
electrophoresis and electrophoretic transfer of proteins to
nitrocellulose membranes, membranes were blocked with 3% non-fat
milk in TBS (pH 7.4) containing 0.1% Tween-20 for 1 h at room
temperature. The membranes were then rinsed three times in TBST and
incubated at room temperature with mouse monoclonal antibodies
against activated diphosphorylated ERK1/2 (1:500; cat. no.
sc-81492; Santa Cruz Biotechnology, Inc.), p-JNK (1:500; cat. no.
sc-293137; Santa Cruz Biotechnology, Inc.) and p-p38 (1:500; cat.
no. sc-17852-R; Santa Cruz Biotechnology, Inc.) for 1 h. β-actin
levels were also assessed as a loading control using a mouse
β-actin antibody (1:2,000; cat. no. ab6276; Abcam). The blots were
processed as described for the measurement of MMP-2, MMP-9 and
TIMP-1. To assess the activation of NF-κB, nuclear extracts were
analyzed by western blot analysis for the p65 fraction of NF-κB and
the NF-κB inhibitor α (IκBα). Nuclear extracts were isolated as
previously described (17). The
protein concentration was determined using the Bradford assay and
equal amounts of protein (30 µg) were separated by SDS-PAGE (4%
stacking gel and 10% separating gel). NF-κB was detected with NF-κB
p65 rabbit polyclonal antibody (1:500; cat. no. ab16502; Abcam) and
IκBα rabbit polyclonal antibody (1:500; cat. no. ab7217; Abcam).
β-actin (1:2,000) served as the control. The blots were processed
as described for the measurement of MMP-2, MMP-9 and TIMP-1.
Statistical analysis
Statistical analyses were performed with SPSS 11.5
(SPSS, Inc., Chicago, IL, USA). All data were expressed as the mean
± standard deviation. Differences between multiple groups were
compared with a one-way analysis of variance. Differences between
two groups were tested for significance by the least significant
difference test. P<0.05 was considered to indicate a
statistically significant difference.
Results
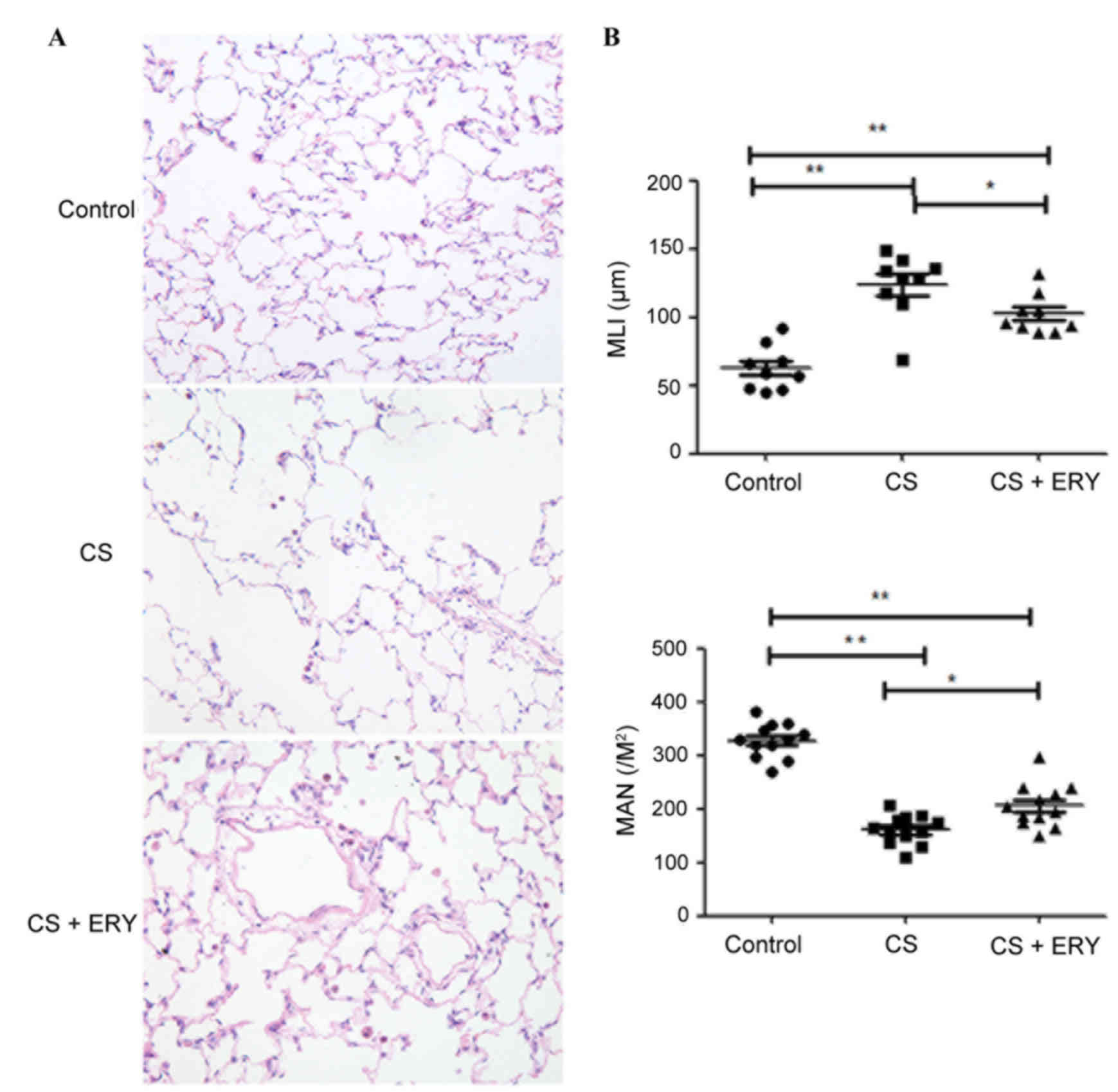
Effect of ERY on CS-induced lung
damage
Histopathological studies were performed to assess
the morphological damage caused by CS and the effect of ERY
treatment. To investigate the effect of ERY on the emphysema
induced by CS, MLI and MAN were measured. The control group
demonstrated no evident histological changes; the structure of the
alveoli was intact with regular ciliary arrangement in the control
group (Fig. 1A). Compared with the
morphology of the control group, CS induced significant
morphological changes characterized by conspicuous inflammation
accompanied with focal emphysema; the alveolar walls were broken
and merged, and the cavities of the alveoli were enlarged (Fig. 1A). MLI was significantly increased
(P<0.01; Fig. 1B), and MAN was
significantly lower (P<0.01; Fig.
1B) in the CS group compared with the control group. ERY
demonstrated a protective effect on emphysematous changes induced
by CS, with a significantly reduced MLI (P<0.05 vs. the CS
group; Fig. 1B) and a
significantly higher MAN (P<0.05 vs. the CS group; Fig. 1B) in the CS + ERY group compared
with the CS group. Compared with the morphological changes in the
CS group, the structural damage induced by CS was partially
recovered in the CS + ERY group (Fig.
1A); however, the MLI remained higher than in the control group
(P<0.01; Fig. 1B) and MAN
remained lower that the control group (P<0.01; Fig. 1B).
ERY reduces the number of inflammatory
cells, and the levels of IL-8 and LTB4 present in the BALF
following CS exposure
As presented in Table
I, CS significantly increased the number of neutrophils
(P<0.01) and lymphocytes (P<0.01), and significantly
decreased the number of macrophages (P<0.01) compared with the
control group. Additionally, as presented in Fig. 2, rats in the CS group had
significantly increased levels of IL-8 (P<0.01) and LTB4
(P<0.01) compared with the control group. Compared with the CS
group, ERY treatment led to a significant reduction in the total
cell count (P<0.05; Table I),
the relative count of neutrophils (P<0.01; Table I) and lymphocytes (P<0.01;
Table I), and a significant
reduction in the levels of IL-8 (P<0.01) and LTB4 (P<0.01;
Fig. 2), and increased the
macrophage count (P<0.05; Table
I).
 | Table I.Cytological count in bronchoalveolar
lavage fluid of rats from different groups. |
Table I.
Cytological count in bronchoalveolar
lavage fluid of rats from different groups.
| Group | Total cell count
(x106/l) | Neutrophil (%) | Lymphocyte (%) | Macrophage (%) |
|---|
| Control | 4.20±2.41 | 3.02±1.96 | 3.96±2.02 | 86.52±12.49 |
| CS |
14.6±4.37b |
21.64±10.73b |
8.38±4.82b |
63.73±12.55b |
| ERY |
10.3±3.67b,c |
12.74±5.16b,d |
10.54±5.52a,c |
73.65±15.69a,c |
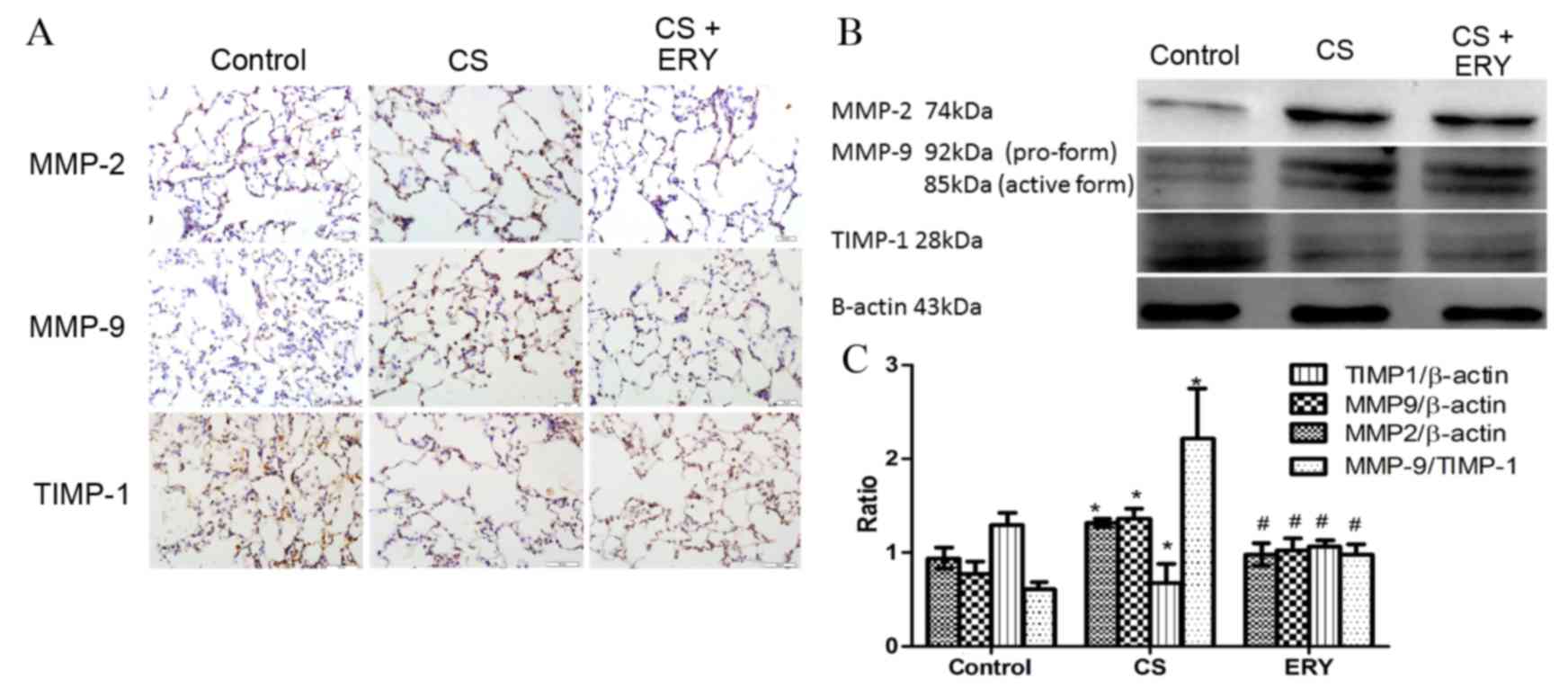
Effects of ERY on lung MMP-2, MMP-9
and TIMP-1 protein expression
To evaluate the effect of ERY on the regulation of
the imbalance of metalloproteases/anti-metalloproteases, the
present study performed immunohistochemical staining (Fig. 3A) and western blotting for MMP-2,
MMP-9 and TIMP-1 (Fig. 3B). The
protein expression levels of MMP-2 and MMP-9 in the CS group were
significantly increased compared with the control group (P<0.05
and P<0.05, respectively; Fig.
3B), while the level of TIMP-1 was significantly reduced
compared with the control group (P<0.05; Fig. 3B). While the expression of MMP-2
and MMP-9 were reduced significantly by ERY treatment compared with
the CS group (P<0.05 and P<0.05, respectively; Fig. 3B), the expression of TIMP-1 was
significantly increased by ERY treatment compared with the CS group
(P<0.05; Fig. 3B). The ratio of
MMP-9/TIMP-1 was increased significantly following CS exposure
compared with the control group (P<0.05; Fig. 3B) but significantly reduced in the
CS + ERY group compared with the CS group (P<0.05; Fig. 3B).
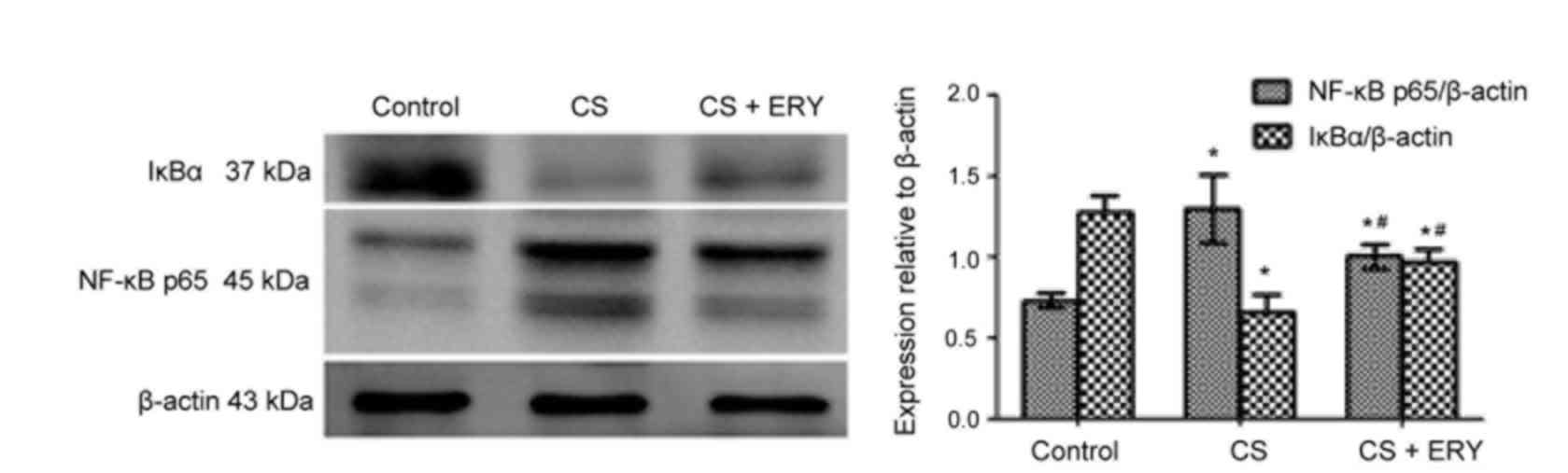
Effect of ERY on NF-κB activation
To investigate the mechanism of ERY-mediated
regulation of NF-κB in vivo, the present study investigated
the effect of ERY on IκBα (Fig.
4). Compared with the control group, the level of IκBα was
significantly decreased in the CS group (P<0.05; Fig. 4), while treatment with ERY
increased the expression of IκBα compared with the CS group
(P<0.05; Fig. 4). Thus, this
indicated that CS-induced IκBα degradation was significantly
blocked by treatment with ERY. Additionally, the activation of
NF-κB p65 was decreased significantly in the CS + ERY group
compared with the CS group (P<0.05; Fig. 4).
Effect of ERY on MAPK pathway
activation
The levels of phosphorylated MAPKs were measured by
western blot analysis (Fig. 5).
Total protein levels of MAPKs were used as controls for potential
fluctuations in the overall protein levels. ERK1/2 and p38
phosphorylation was significantly increased in the CS group
compared with the control group (P<0.05 and P<0.05,
respectively; Fig. 5), butp38 and
ERK1/2phosphorylationwas significantly reduced in the CS + ERY
group compared with the CS group (P<0.05; Fig. 5). No significant changes in JNK
phosphorylation levels in the CS group and CS + ERY group compared
with those in the control group (Fig.
5).
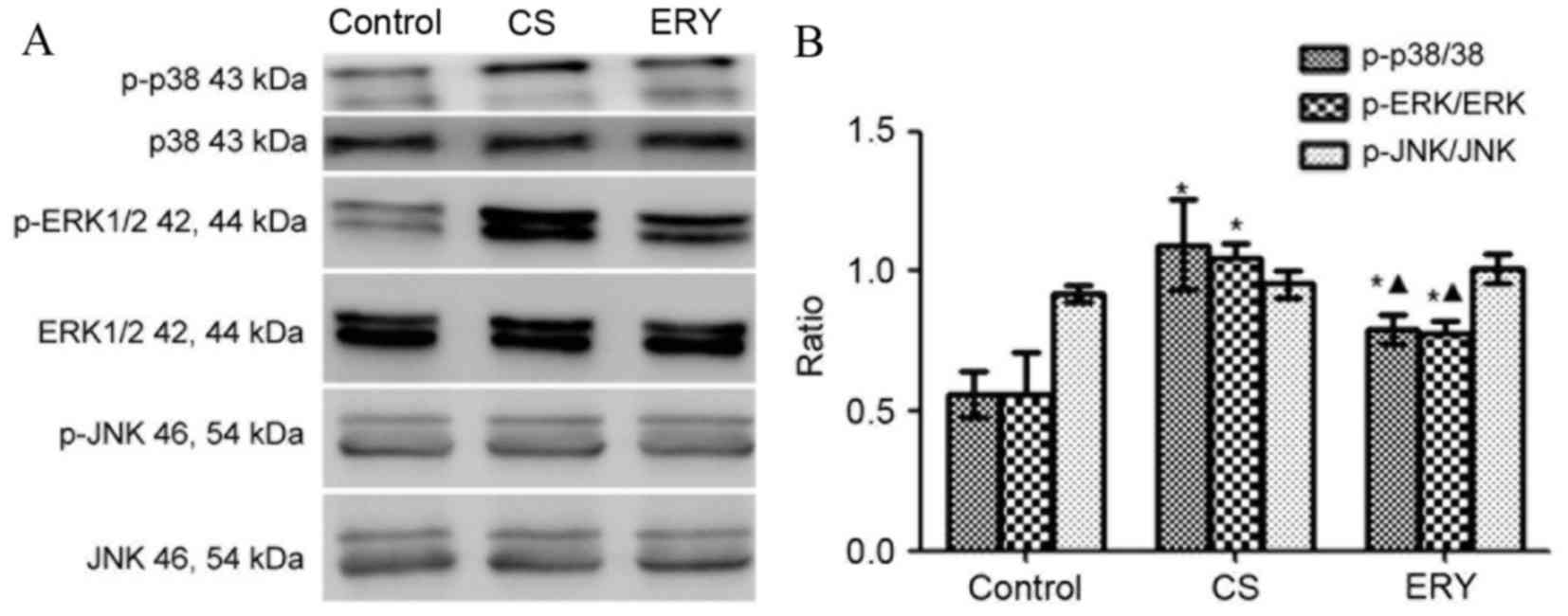 | Figure 5.Effects of ERY on the expression of
MAPK phosphorylation in CS-induced emphysema in rats by western
blotting. (A) The protein expression of MAPKs and p-MAPKs as
measured by western blotting. (B) For p-p38, p-ERK1/2, ERK1/2,
p-JNK and JNK, both bands were quantified in the densitometry.
Densitometrically analyzed target p-MAPKs bands normalized to
MAPKs. Data are presented as the mean ± standard deviation. Control
rats were exposed to normal air and received daily saline
injections. CS rats were exposed to CS for 12 weeks. CS + ERY rats
were exposed to CS for 12 weeks and received daily injections of
100 mg/kg ERY. *P<0.05 vs. control group and
ΔP<0.05 vs. CS group; n=12 per group. MAPK,
mitogen-activated protein kinase; p-, phospho-; ERK, extracellular
signal-regulated kinase; JNK, c-Jun N-terminal kinase; CS,
cigarette smoke; ERY, erythromycin. |
Discussion
COPD is a lung disease characterized by progressive
airflow limitation due to the inflammation-driven destruction of
the alveolar walls (18). The
increased expression of MMPs is considered to be a key factor in
the development of COPD and emphysema (8,19).
Because CS exposure is the biggest risk factor for the development
of COPD, the present study aimed to investigate the effect of CS on
the imbalance of metalloproteases/anti-metalloproteases, and the
effect of ERY on the destruction of lung tissue and the inhibition
of the imbalance of metalloproteases/anti-metalloproteases. In the
present study, the expression of MMP-2 and MMP-9 was demonstrated
to be increased by stimulation with CS in a rat model of CS-induced
emphysema, while TIMP-1 expression was decreased. Previous studies
have demonstrated that the inhibition of MMPs by macrolides may
reduce the extracellular spread of inflammation (9,20).
In the current study, the imbalance of
metalloproteases/anti-metalloproteases was accompanied by altered
levels of inflammatory cells and cytokines in BALF, while ERY
reduced the imbalance of metalloproteases/anti-metalloproteases and
the changes in the inflammatory cells and cytokines in BALF to some
extent, which was also associated with the amelioration of the
emphysema. The potential signaling pathway of ERY in the inhibition
of the inflammation in CS-induced emphysema has not been previously
clarified. One previous study demonstrated the protective role of
ERY on CS-induced emphysema and its involvement in the reduction of
inflammation, the imbalance of MMP-9/TIMP-1 and the apoptosis of
lung structural cells (7). The
results of the present study provided novel evidence for the
protective effect of ERY on the development of emphysema induced by
CS via regulation of the expression of MMP-2, MMP-9 and TIMP-1, and
improvement of the inflammation of the lungs via MAPK/NF-κB
activation, thus indicating its potential use as a therapeutic
agent for the treatment of COPD. Similar studies on the potential
signaling pathway of the macrolides were performed in an asthma
model in vivo (21) or in
inflammation-stimulated cells in vitro (22). MAPKs, together with NF-κB
activation, are the kinases that are activated in airway epithelial
cells and macrophages exposed to CS extracts, which induces the
expression of various pro-inflammatory chemokines and cytokines
(17). A study on a
second-generation p38 MAPK inhibitor, SB681323, with the greatest
selectivity for p38α, demonstrated that a single oral dose
inhibited p-heat shock protein 27 and tumor necrosis factor-α
(TNF-α) production in whole blood obtained from patients with COPD
(23). In a 4-week treatment
regime using this p38 inhibitor in patients with COPD who were not
receiving inhaled corticosteroid therapy, a reduction in sputum
neutrophils and in serum fibrinogen, but not in serum C-reactive
protein, IL-8, IL-1β or IL-6 levels, was reported. This result was
associated with an improvement in forced vital capacity but not in
forced expiratory volume during the first second of a forced breath
(23). ERK1/2 and JNK inhibition
abrogated CS effects on heme oxygenase-1 expression and nuclear
factor erythroid 2 like 2/BTB domain and CNC homolog 1
translocation to the nucleus (24). Therefore, it is thought that MAPKs
are involved in the inflammatory responses induced by CS exposure,
endotoxins and oxidative stress through the activation and release
of pro-inflammatory cytokines/chemokines, post-translational
regulation of these genes and activation of inflammatory cell
migration (25). Inflammation in
COPD is amplified by increased oxidative stress, which activates
NF-κB, a transcription factor that orchestrates the expression of
multiple inflammatory genes, including TNF-α, C-X-C motif chemokine
ligand 8 (CXCL-8) and MMP-9 (26).
The current study demonstrated that CS induced MAPK
phosphorylation. The phosphorylation of p38 and ERK1/2 were
increased in the CS-induced emphysema model in vivo compared
with the control, while the oral administration of ERY suppressed
CS-induced emphysema by regulating inflammatory cytokines and the
MMP/anti-MMP imbalance that was associated with the changes in
MAPKs.
A number of studies have indicated that NF-κB is
regulated by macrolides (27–29).
For example, azithromycin inhibits pro-inflammatory mediators,
including CXCL-8, via inhibition of NF-κB in in vitro models
(28,29). Solithromycin, another macrolide,
inhibits activation of NF-κB under oxidative stress (30). Macrolides suppress the degradation
of IκBα, an inhibitor of NF-κB, and/or affect the downstream
dissociation from IκBα in the NF-κB signaling pathway (31). In the present study, CS activated
the expression of NF-κB p65 and lower expression of IκBα was
detected, while ERY inhibited the activation of NF-κB and promoted
the expression of IκBα.
The results of the present study do not conclusively
demonstrate whether MAPK and NF-κB are upstream or downstream of
the signaling pathway. However, the changes identified in the
present study indicated that there may be an association between
the amelioration of emphysema when the emphysema model was treated
with ERY and the changes in MAPK and NF-κB. In a previous study of
acute and chronic CS exposure, the activation and expression of p38
MAPK in the lungs were compared between the following two mouse
strains: C57BL/6 emphysema-susceptible mice and NZW
emphysema-resistant mice: the selective p38 MAPK inhibitor,
SB203580, ameliorated CS-induced lung inflammation and injury
(32). ERY and clarithromycin
inhibited CXCL-8 with inactivation of NF-κB and/or activator
protein-1 in human bronchial epithelial cells in vitro
(33,34). In one in vitro experiment,
CS extract significantly induced ERK1/2 phosphorylation. PD98059, a
specific inhibitor of ERK-MAPK, significantly blocked the effect of
CS extract on ERK1/2 phosphorylation. Furthermore, PD98059
significantly inhibited the effect of CS extract on MMP-1
production and mRNA expression (35). These results indicate that CS may
stimulate the production of and potentially activates MMP-1 through
activation of the ERK1/2 signal transduction pathway. By inducing
MMP-1, CS may result in excessive tissue destruction and may
contribute to the development of emphysema (35).
In conclusion, CS may induce emphysema by
simultaneously disturbing the balance of
metalloproteases/anti-metalloproteases, increasing airway
inflammation and upregulating MAPKs together with increased NF-κB
activation signaling. Treatment with ERY reduced the development of
emphysema via inhibition of the phosphorylation of MAPKs and the
activation of NF-κB. These results revealed the anti-inflammatory
role of ERY in CS-induced emphysema and airway inflammation,
indicating that macrolides may have therapeutic potential for
chronic airway inflammation and the associated emphysema caused by
exposure to CS.
Acknowledgements
This research was supported by project 2012BAI05B01
from the Ministry of Science and Technology of the People's
Republic of China, project 2014021031 from the Education Department
of Liaoning Province and project 201409 from the Department of
Liaoning Province.
Glossary
Abbreviations
Abbreviations:
|
MAPK
|
mitogen-activated protein kinase
|
|
ERK1/2
|
extracellular signal-regulated
kinase-1/2
|
|
JNK
|
c-Jun NH2-terminal kinase
|
|
CS
|
cigarette smoke
|
|
ERY
|
erythromycin
|
|
MMP
|
matrix metalloprotease
|
|
TIMP
|
tissue inhibitor of
metalloprotease
|
References
|
1
|
Celli BR and MacNee W: ATS/ERS Task Force:
Standards for the diagnosis and treatment of patients with COPD: A
summary of the ATS/ERS position paper. Eur Respir J. 23:932–946.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nowak D, Berger K, Lippert B, Kilgert K,
Caeser M and Sandtmann R: Epidemiology and health economics of COPD
across Europe: A critical analysis. Treat Respir Med. 4:381–395.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fragoso CA: Epidemiology of chronic
obstructive pulmonary disease (COPD) in aging populations. COPD.
13:125–129. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chapman KR, Mannino DM, Soriano JB,
Vermeire PA, Buist AS, Thun MJ, Connell C, Jemal A, Lee TA,
Miravitlles M, et al: Epidemiology and costs of chronic obstructive
pulmonary disease. Eur Respir J. 27:188–207. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Stockley RA, O'Brien C, Pye A and Hill SL:
Relationship of sputum color to nature and outpatient management of
acute exacerbations of COPD. Chest. 117:1638–1645. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Spurzem JR and Rennard SI: Pathogenesis of
COPD. Semin Respir Crit Care Med. 26:142–153. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kwiatkowska S, Noweta K, Zieba M, Nowak D
and Bialasiewicz P: Enhanced exhalation of matrix
metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 in
patients with COPD exacerbation: A prospective study. Respiration.
84:231–241. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mocchegiani E, Giacconi R and Costarelli
L: Metalloproteases/anti-metalloproteases imbalance in chronic
obstructive pulmonary disease: Genetic factors and treatment
implications. Curr Opin Pulm Med. 17:(Suppl 1). S11–S19. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kanoh S and Rubin BK: Mechanisms of action
and clinical application of macrolides as immunomodulatory
medications. Clin Microbiol Rev. 23:590–615. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mazzei T, Mini E, Novelli A and Periti P:
Chemistry and mode of action of macrolides. J Antimicrob Chemother.
31:(Suppl C). S1–S9. 1993. View Article : Google Scholar
|
|
11
|
Perry DK, Hand WL, Edmondson DE and
Lambeth JD: Role of phospholipase D-derived diradylglycerol in the
activation of the human neutrophil respiratory burst oxidase.
Inhibition by phosphatidic acid phosphohydrolase inhibitors. J
Immunol. 149:2749–2758. 1992.PubMed/NCBI
|
|
12
|
Zhou Y, Tan X, Kuang W, Liu L and Wan L:
Erythromycin ameliorates cigarette-smoke-induced emphysema and
inflammation in rats. Transl Res. 159:464–472. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Birrell MA, Wong S, Catley MC and Belvisi
MG: Impact of tobacco-smoke on key signaling pathways in the innate
immune response in lung macrophages. J Cell Physiol. 214:27–37.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Moretto N, Facchinetti F, Southworth T,
Civelli M, Singh D and Patacchini R: alpha, beta-Unsaturated
aldehydes contained in cigarette smoke elicit IL-8 release in
pulmonary cells through mitogen-activated protein kinases. Am J
Physiol Lung Cell Mol Physiol. 296:L839–L848. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cheng SE, Luo SF, Jou MJ, Lin CC, Kou YR,
Lee IT, Hsieh HL and Yang CM: Cigarette smoke extract induces
cytosolic phospholipase A2 expression via NADPH oxidase, MAPKs,
AP-1, and NF-kappaB in human tracheal smooth muscle cells. Free
Radic Biol Med. 46:948–960. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zheng H, Liu Y, Huang T, Fang Z, Li G and
He S: Development and characterization of a rat model of chronic
obstructive pulmonary disease (COPD) induced by sidestream
cigarette smoke. Toxicol Lett. 189:225–234. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yin Y, Hou G, Li E, Wang Q and Kang J:
PPARγ agonists regulate tobacco smoke-induced Toll like receptor 4
expression in alveolar macrophages. Respir Res. 15:282014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rabe KF, Hurd S, Anzueto A, Barnes PJ,
Buist SA, Calverley P, Fukuchi Y, Jenkins C, Rodriguez-Roisin R,
van Weel C, et al: Global strategy for the diagnosis, management,
and prevention of chronic obstructive pulmonary disease: GOLD
executive summary. Am J Respir Crit Care Med. 176:532–555. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Atkinson JJ, Lutey BA, Suzuki Y, Toennies
HM, Kelley DG, Kobayashi DK, Ijem WG, Deslee G, Moore CH, Jacobs
ME, et al: The role of matrix metalloproteinase-9 in cigarette
smoke-induced emphysema. Am J Respir Crit Care Med. 183:876–884.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kanai K, Asano K, Hisamitsu T and Suzaki
H: Suppression of matrix metalloproteinase production from nasal
fibroblasts by macrolide antibiotics in vitro. Eur Respir J.
23:671–678. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ci X, Chu X, Xu X, Li H and Deng X:
Short-term roxithromycin treatment attenuates airway inflammation
via MAPK/NF-κB activation in a mouse model of allergic asthma.
Inflamm Res. 61:749–758. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Willems-Widyastuti A, Vanaudenaerde BM,
Vos R, Dilisen E, Verleden SE, De Vleeschauwer SI, Vaneylen A, Mooi
WJ, de Boer WI, Sharma HS and Verleden GM: Azithromycin attenuates
fibroblast growth factors induced vascular endothelial growth
factor via p38(MAPK) signaling in human airway smooth muscle cells.
Cell Biochem Biophys. 67:331–339. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Singh D, Smyth L, Borrill Z, Sweeney L and
Tal-Singer R: A randomized, placebo-controlled study of the effects
of the p38 MAPK inhibitor SB-681323 on blood biomarkers of
inflammation in COPD patients. J Clin Pharmacol. 50:94–100. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Goven D, Boutten A, Leçon-Malas V,
Boczkowski J and Bonay M: Prolonged cigarette smoke exposure
decreases heme oxygenase-1 and alters Nrf2 and Bach1 expression in
human macrophages: Roles of the MAP kinases ERK(1/2) and JNK. FEBS
Lett. 583:3508–3518. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chung KF: p38 mitogen-activated protein
kinase pathways in asthma and COPD. Chest. 139:1470–1479. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Barnes PJ and Adcock IM: Glucocorticoid
resistance in inflammatory diseases. Lancet. 373:1905–1917. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen X, Zeng S, Zou J, Chen Y, Yue Z, Gao
Y, Zhang L, Cao W and Liu P: Rapamycin attenuated cardiac
hypertrophy induced by isoproterenol and maintained energy
homeostasis via inhibiting NF-κB activation. Mediators Inflamm.
2014:8687532014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Aghai ZH, Kode A, Saslow JG, Nakhla T,
Farhath S, Stahl GE, Eydelman R, Strande L, Leone P and Rahman I:
Azithromycin suppresses activation of nuclear factor-kappa B and
synthesis of pro-inflammatory cytokines in tracheal aspirate cells
from premature infants. Pediatr Res. 62:483–488. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Matsumura Y, Mitani A, Suga T, Kamiya Y,
Kikuchi T, Tanaka S, Aino M and Noguchi T: Azithromycin may inhibit
interleukin-8 through suppression of Rac1 and a nuclear
factor-kappa B pathway in KB cells stimulated with
lipopolysaccharide. J Periodontol. 82:1623–1631. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kobayashi Y, Wada H, Rossios C, Takagi D,
Higaki M, Mikura S, Goto H, Barnes PJ and Ito K: A novel macrolide
solithromycin exerts superior anti-inflammatory effect via NF-κB
inhibition. J Pharmacol Exp Ther. 345:76–84. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Aghai ZH, Kode A, Saslow JG, Nakhla T,
Farhath S, Stahl GE, Eydelman R, Strande L, Leone P and Rahman I:
Azithromycin suppresses activation of nuclear factor-kappa B and
synthesis of pro-inflammatory cytokines in tracheal aspirate cells
from premature infants. Pediatr Res. 62:483–488. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Marumo S, Hoshino Y, Kiyokawa H, Tanabe N,
Sato A, Ogawa E, Muro S, Hirai T and Mishima M: p38
mitogen-activated protein kinase determines the susceptibility to
cigarette smoke-induced emphysema in mice. BMC Pulm Med. 14:792014.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Abe S, Nakamura H, Inoue S, Takeda H,
Saito H, Kato S, Mukaida N, Matsushima K and Tomoike H:
Interleukin-8 gene repression by clarithromycin is mediated by the
activator protein-1 binding site in human bronchial epithelial
cells. Am J Respir Cell Mol Biol. 22:51–60. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Desaki M, Takizawa H, Ohtoshi T, Kasama T,
Kobayashi K, Sunazuka T, Omura S, Yamamoto K and Ito K:
Erythromycin suppresses nuclear factor-kappaB and activator
protein-1 activation in human bronchial epithelial cells. Biochem
Biophys Res Commun. 267:124–128. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kim H, Liu X, Kohyama T, Kobayashi T,
Conner H, Abe S, Fang Q, Wen FQ and Rennard SI: Cigarette smoke
stimulates MMP-1 production by human lung fibroblasts through the
ERK1/2 pathway. COPD. 1:13–23. 2004. View Article : Google Scholar : PubMed/NCBI
|