Introduction
Cerebral palsy (CP) is a group of permanent
disorders lead to lifelong movement disability and postural
dysfunction, due a non-progressive interference, lesion, or
abnormality in developing immature brain (1). Nearly half of these disabilities are
diagnosed in preterm born children, while full-term born children
are diagnosed with a prevalence of 2–2.5/1,000 live birth, this
number is several times higher in twins (2,3). The
role of genetic contribution to CP has been widely investigated.
Several CP candidate mutation genes have been identified by
whole-genome exome sequencing (4).
Rare copy number variants in 50 CP cases were detected and their
frequencies were determined (5).
However, the epigenome landscape of CP in monozygotic (MZ) twins
still remains largely unknown.
DNA methylation at the C-5 position of cytosine is a
key epigenetic modification that plays critical roles in regulating
gene expression during the development of embryos and can be
inherited by cell division (6–8).
Dysregulation of DNA methylation can lead to many complex diseases
including various types of cancer (9,10).
DNA methylation undergoes dynamic changes during multiple
biological processes and pathological changes (11–13).
The identification and functional annotation of different methylome
between MZ discordant twins is crucial for understanding the roles
of DNA methylation in the CP.
Reduced representation bisulfite sequencing (RRBS)
was first proposed in 2005 (14),
and has been proved as a powerful and cost-effective technology to
high-throughout generate the genome-scale DNA methylation profiling
at single nucleotide resolution in mammalian cells. Here, we
described the comprehensive DNA methylation profiles covering most
promoters, CpG islands of 4 MZ twin pairs discordant for CP by
using RRBS and generated the genomic CG, CHG and CHH contexts (H=A,
T or C) methylation patterns. We identified 190 differentially
methylated genes (DMGs) that exhibited significant different
methylation patterns between CP and normal healthy samples, these
DMGs may closely relate with the CP state. The methylome of 4 MZ
discordant twin pairs may be a remarkable resource to explore and
understand the epigenetic mechanism underlying the CP in MZ
twins.
Materials and methods
Patients and other family members signed an informed
consent form. This study was approved by the Jiamusi University
Hospital Ethics Committee.
Patients and DNA samples
The 4 pairs of MZ discordant twins in this study
were separately collected from Henan and Liaoning (the Third
Affiliated Hospital of Zhengzhou University, Shenyang Children's
Hospital Rehabilitation Department). Twin pairs met the following
criteria: Birth with no obstetric forceps and no brain trauma, or
medication-assisted treatment. The ages of MZ twins were 3 or 4
years at the time of enrollment. The CP diagnosis was confirmed by
a pediatric rehabilitation specialist using standard published
criteria relating to non-progressive disorders of movement control
and posture. The overall phenotypic, clinical characters of the
study cohort are shown in Table
I.
 | Table I.Clinical characters of 4 pairs of
monozygotic twins discordant for cerebral palsy. |
Table I.
Clinical characters of 4 pairs of
monozygotic twins discordant for cerebral palsy.
|
|
Sy02010f/Sy02011fcp |
Zz01040m/Zz01041mcp |
Zz01051f/Zz01050fcp |
Zz01060f/Zz01061fcp |
|---|
|
|
|
|
|
|
|---|
| Sex | F | F | M | M | F | F | F | F |
|---|
| Age (years) | 3 | 3 | 4 | 4 | 3 | 3 | 3 | 3 |
| Cerebral palsy
type | – | Spastic | – | Spastic | – | Spastic | – | Spastic |
| Gestational age
(weeks) | 31 | 31 | 28 | 28 | 32 | 32 | 39 | 39 |
| Birth weight
(kg) | 1.70 | 1.67 | 1.60 | 1.70 | 3.60 | 2.60 | 2.10 | 2.51 |
| Maternal age
(years) | 37 | 37 | 34 | 34 | 30 | 30 | 38 | 38 |
| Paternal age
(years) | 38 | 38 | 33 | 33 | 31 | 31 | 37 | 37 |
Blood samples were collected into tubes containing
EDTA by skilled nurses at the scene. One milliliter from collection
of peripheral blood samples was fixed on the FTA card (Whatman
patent product for DNA collection, transportation and storage at
room temperature), and submitted to zygosity appraisal institutions
to test 20 short tandem repeats (STRs). The pair was identified as
MZ twins if all the STRs were the same.
Genomic DNA was then isolated from a Qiagen DNeasy
Blood & Tissue kit (Qiagen Inc., Valencia, CA, USA) from
peripheral venous blood leukocytes from 4 pairs of MZ twins
discordant for CP, according to the manufacturer's instructions and
quantified with PicoGreen. Extracted DNA concentrations were
greater than 50 ng/ml, and OD260/280 value between 1.8 and 2.0. All
the prepared samples were immediately stored at −80°C.
Reduced representation bisulphite
sequencing and quality control
After DNA samples were extracted, the restriction
enzyme was used to cut the DNA into fixed length fragments. These
DNA fragments were added dA at 3′-end, equipped with adapters, then
size selected to 40–220 bp, treated with bisulfite, PCR amplified,
cloned and then pair-end sequenced with 49 bp read length using
Illumina high-throughput sequencing system. After a large amount of
raw data was generated, low quality reads with a ratio of N was
greater than 10% and the proportion of bases for which the quality
value was <20 was more than 50% of the entire read. Adapter
contamination, and duplication pairs were removed during the
process of quality control to get the clean reads.
Mapping RRBS clean reads into
reference genome and methyl-cytosines analysis
These clean reads then were mapped into the human
reference genome (GRCh37) using BSMAP (15). Only reads uniquely aligned into
reference genome and of which 5′ restriction sites can be retained
to perform analysis. Furthermore, the rate of C-T conversion and
alignment were obtained from the BSMAP result. All cytosine sites
located on sex chromosomes were removed to eliminate the impact of
sex differences. The cytosine site in each uniquely mapped read was
reported as methylated or unmethylated status with different
patterns (CpG, CHG, CHH). And we calculated the methylation level
of each site by the formula, M/(M + UM), with M represents the
number of methylated reads and UM representing the number of
unmethylated reads.
Phylogenetic tree construction
The phylogenetic tree was constructed to assess the
4 pairs of discordant twins evolutionary patterns for DNA
methylation. DNA methylation euclidean distance matrices were
calculated using the common CpG sites with coverage greater or
equal to 4× in all the 8 samples. Based on the minimal evolution
algorithm, we applied the ‘fastme.bal’ function in the R package
‘ape’ to infer the phylogenetic tree (16).
Screening the DMGs
Among 4 pairs of MZ discordant twins, for each pair,
one was suffering from CP patient and the other was normal control.
The 8 samples were classified into two groups based on their
disease state. First, a Student's t-test was applied for each gene
to test whether the methylation of all CpG sites among the gene in
the two groups showed a significant difference. The P-values for
multiple-testing correction were further adjusted using the false
discovery rate (FDR) method. Second, the methylation level of each
gene was qualified by the mean value of its cytosines. For each
gene, we calculated the mean difference between every pairs of
twins. The gene which was regarded DMG satisfied the following two
conditions: The genes met strict controlled thresholds (P<0.05)
and the difference of its methylstion value was more than 0.2 in
three couples at least at the same direction would be retained for
the following analysis. All these analysis processes were
implemented in R language (v3.3.1).
Function enrichment analyses of gene
sets and genome regions
The DMGs were imported into Database for Annotation,
Visualization and Integrated Discovery (DAVID, v6.8) (17) to perform function enrichment
analysis using the default parameters. Annotated Gene Ontology (GO)
terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways
with P-value <0.05 were considered to be statistical
significant. The P-value was calculated by hypergeometric test or
Fisher's exact test. Moreover, GREAT (v.3.0.0) (18) was used to perform the function
enrichment analysis of the genome regions of these differentially
methylated gene sets. The annotated human phenotype and disease
ontology terms selected were found to be significant by
hypergeometric test (P<0.05).
Protein and protein interaction
network construction
The human comprehensive protein-protein network was
intergrated from 8 commonly used networks contained in the
Biomolecular Interaction Network Database (BIND), the Biological
General Repository for Interaction Datasets (BioGRID), the Database
of Interacting Proteins (DIP), the Human Protein Reference Database
(HPRD), intAct, the Molecular INTeraction database (MINT), the
Mammalian PPI Database of the Munich Information Center on Protein
Sequences (MIPS), PDZBase (a PPI database for PDZ-domains) and
reactome (19). The integrated
network was composed of 80,980 edges and 13,361 nodes. The DMGs
identified from the sequencing data were set as seed genes, and
then mapped into the background PPI network to extract sub PPI
network which composed of the seed genes and the genes directly
connecting to seed genes. The visualization of the sub-PPI network
was performed with cytoscape v3.2.0 (20).
Results
Description of the MZ twins discordant
for CP and RRBS sequencing
The genome-scale identification of the general
methylation pattern and different methylome is crucial for us to
understand the role of epigenetics in the inconsistent occurrence
of CP in MZ twins, as 4 pairs of MZ CP discordant twins were chosen
for RRBS sequencing (Table II).
Among the 4 sets of twins, three pairs were female and one was
male. The ages ranged from 3 to 4 years. The 8 samples were divided
into a disease group and a control group according to their status
of CP. DNA extracted from each sample was used for RRBS sequencing
to generate a global methylation pattern. The quality process was
performed for each sample (see the materials and methods section).
A total of 983 million clean reads were retained for these 8
samples. Subsequently, these clean reads were aligned to the
reference genome (GRCh37) using the alignment tool BSMAP. There
were 718 million reads only uniquely mapped into reference and of
which 5′ is restriction sites were obtained to perform the
subsequent analysis.
| Table II.Data description of reduced
representation bisulfite sequencing reads for 4 discordant twins
pairs. |
Table II.
Data description of reduced
representation bisulfite sequencing reads for 4 discordant twins
pairs.
|
| Sy02010f | Sy02011fcp | Zz01040m | Zz01041mcp | Zz01051f | Zz01050fcp | Zz01060f | Zz01061fcp |
|---|
| Clean reads (M) | 149,161,300 | 134,259,528 | 122,931,110 | 111,752,904 | 105,280,374 | 116,700,896 | 112,954,768 | 129,616,026 |
| Uniquely mapped reads
(M) | 108,295,641 | 99,082,196 | 89,070,401 | 79,623,368 | 75,125,797 | 87,301,722 | 82,058,642 | 97,377,446 |
| Uniquely mapped
rate (%) | 72.60 | 73.80 | 72.46 | 71.25 | 71.36 | 74.81 | 72.65 | 75.13 |
| Bisulfite
conversion rate (%) | 99.25 | 99.27 | 99.35 | 99.32 | 99.29 | 99.24 | 99.27 | 99.24 |
Comprehensive DNA methylation profile
of 4 discordant twins pairs
The number of clean reads ranged from 112 to 134
million in the disease group, and 105 to 149 million in the control
group. In total, 983 million clean reads from these 8 samples were
separately aligned to reference genome using BSMAP. The average
uniquely mapped rate between disease group and control group were
73.7 and 72.3% respectively. By pooling all clean reads together
from two groups, 718 million reads were uniquely mapped to the
reference genome with the average C-T conversion rate of 99.27%
(Table II). Non-conversion cases
were mostly caused by bisulfite treating or sequencing error. The
multiple metrics of RRBS sequencing of these 4 pairs of CP
discordant twins have already reached or exceeded anticipated
results.
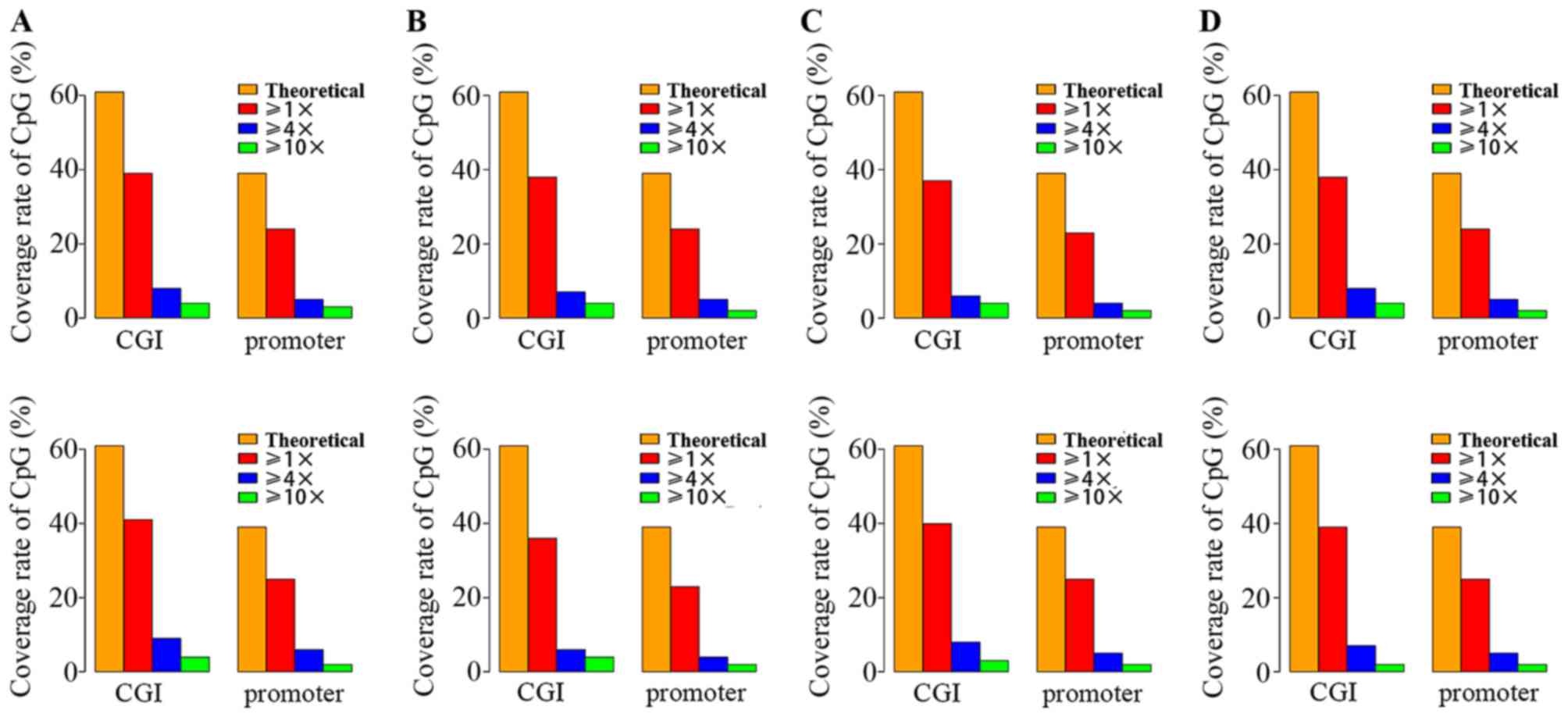
As shown in Fig. 1,
the coverage of single nucleotide cytosine of 4 twins at CpG
islands (CGI) was generally higher than promoter regions (Fig. 1). It also can be observed that
there was a difference between the actual single base coverage and
the theoretical value according to the RRBS technology. The number
of CpG sites was reduced with the increase of their coverage
(14). However, the CpG sites with
high coverage were more credible. Therefore, only the CpG sites
whose coverage ≥4 would be retained to carry out the following
analysis.
Correlation of 4 twins discordant with
CP
It is an extremely rare phenomenon that one of MZ
twins is suffering from CP and the other is healthy in all 4 pairs
in this study. Naturally, it is worthy to explore whether the
methylation distribution exhibited significant differences between
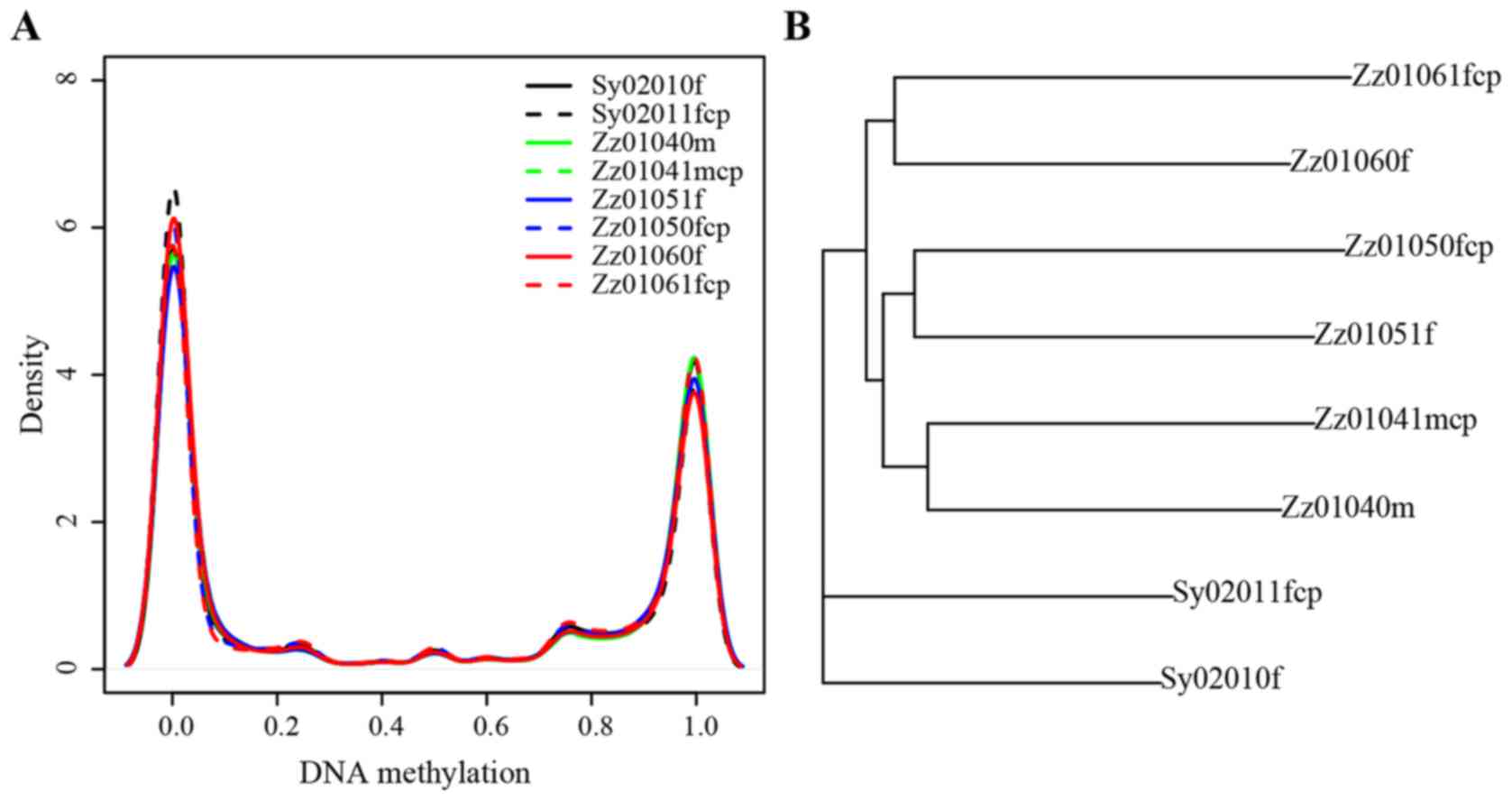
disease sample and normal sample of each twins. The bimodal
distribution of DNA methylation can be observed in each sample of 4
discordant twins pairs (Figs. 2
and 3A), which is consistent with
previous knowledge and observations (21). Moreover, the correlations of
methylation for each twin pair were ~0.98 (P<0.05) (Fig. 2) demonstrating that most of the
methylation patterns of MZ twins are consistent, and suggesting the
CP occurrence may be correlated with the DNA methylation alteration
on some certain genes/CpG stes.
Simultaneously, the overall methylation level for 8
samples was observed. The methylation level of all samples
illustrated the concordant bimodal distribution with the previous
studies (21) (Fig. 3A). The methylation level of most
CpG sites was 0 (completely unmethylated) or 1 (completely
methylated), with few sites showing intermediate methylation levels
which may due to the allele-specific methylation sequencing error
or the other reasons. Further inferred phylogenetic relationships
based on pairwise distances of common CpG sites among the 4 twins.
The phylogenetic tree topology is shown in Fig. 3B. Even though the twins experienced
different healthy status, all 4 twins had a closer evolutionary
relationship with their twin pair than other families. The
phenomenon that the MZ twins shared most of the CpG site
methylation level was expected, as not all but a few pathogenic
sites could drive the occurrence of CP. Herein, the study focused
on the excavation of the differentially methylated genes.
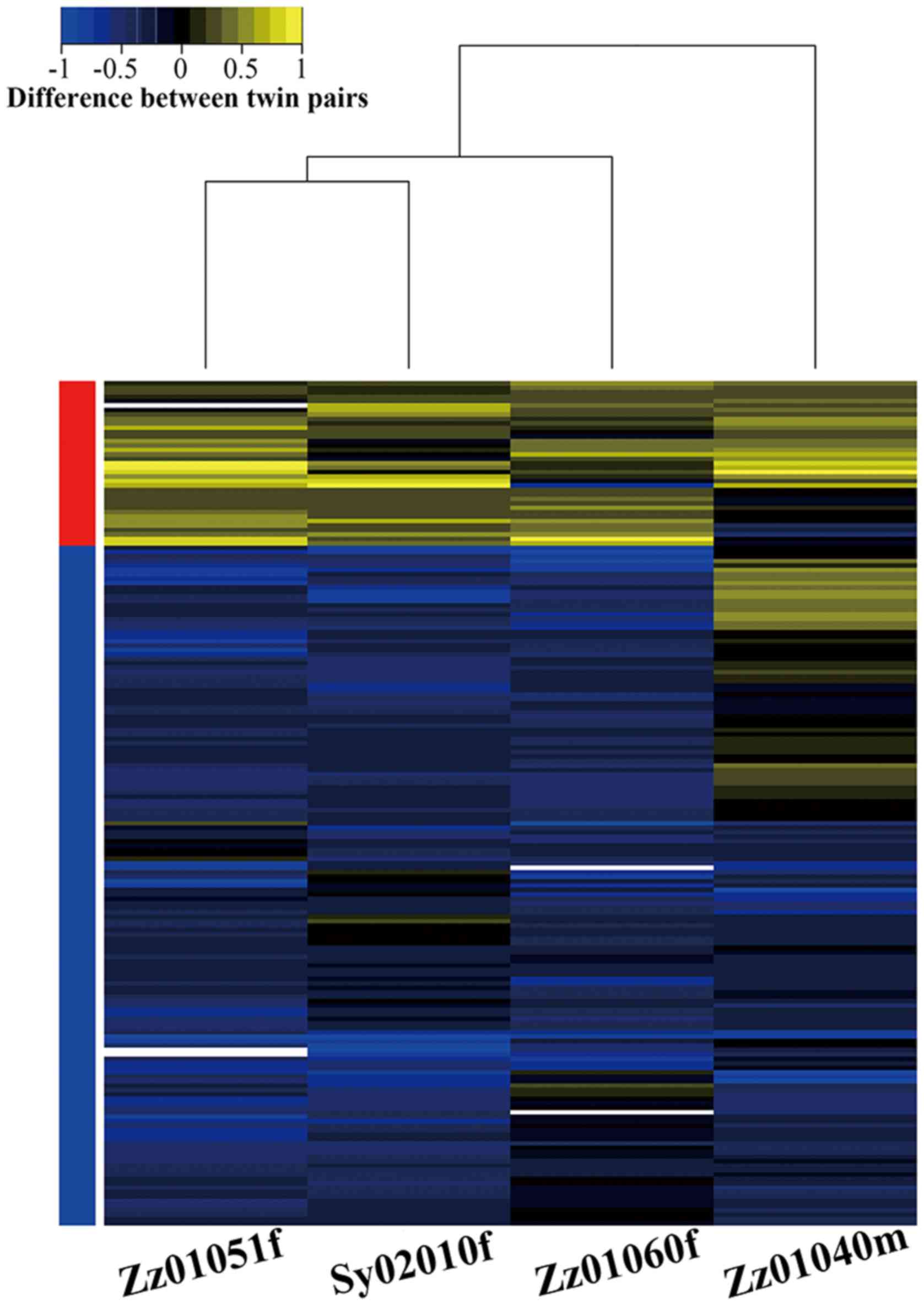
The identification of the DMGs
In this study, a total of 190 DMGs were identified
by Student's t-test (see Materials and methods). Among them, 37
DMGs showed hypermethylation in the disease group compared to the
normal group, and 153 DMGs presented hypomethylation among at least
three pairs of twins (Fig. 4).
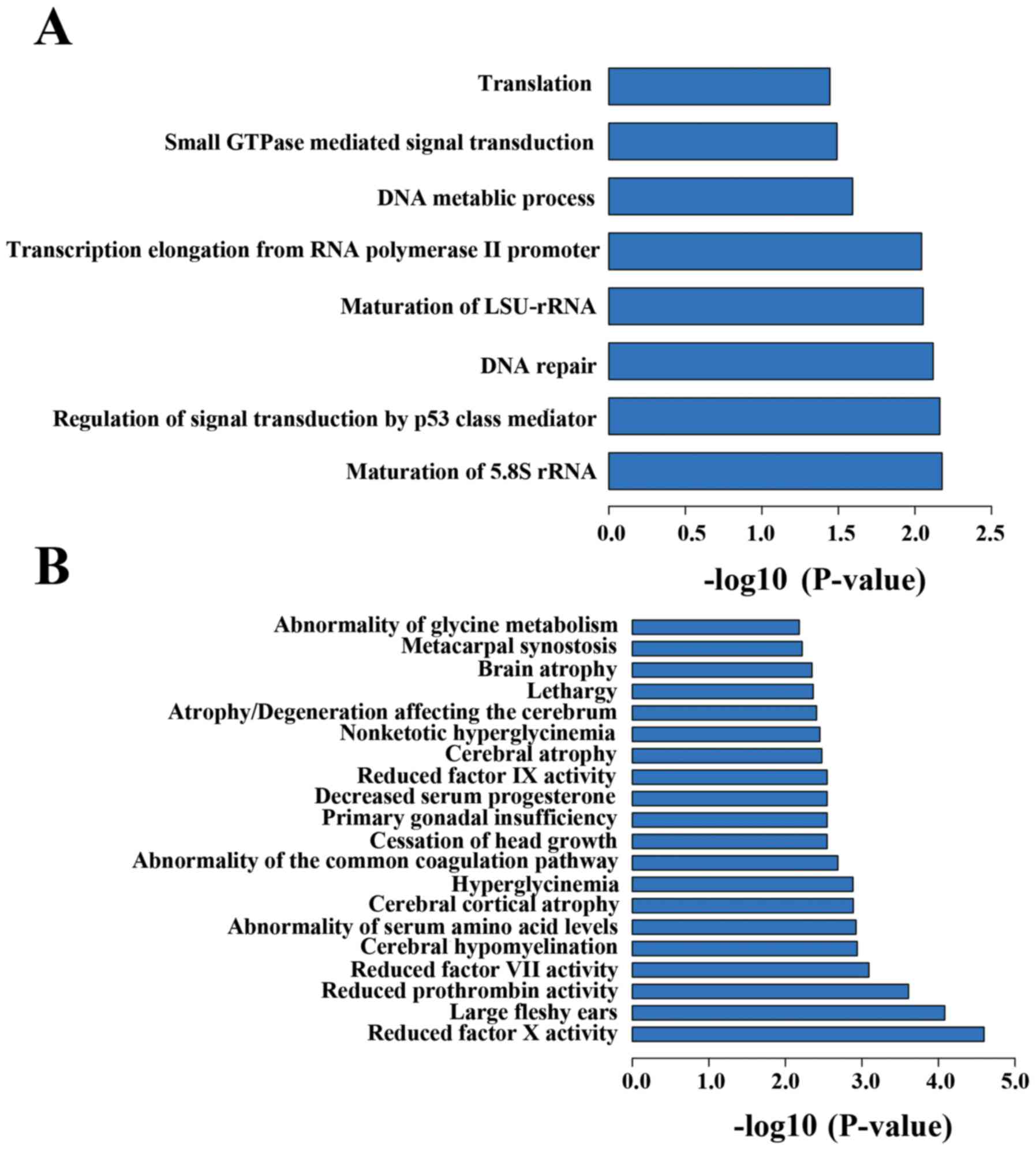
DAVID v6.8 (https://david.ncifcrf.gov/) was used for function
enrichment analysis of these 190 DMGs. These genes were mainly
enriched in maturation of 5.8S rRNA, regulation of signal
transduction by p53 class mediator and some basic biological
process like DNA repair, DNA metabolic process (Fig. 5A). These biological process (BP)
terms could be disturbed by the abnormal changes in DNA methylation
of some pathogenic genes. Moreover, these DMGs were also enriched
in biosynthesis of antibiotics, glycolysis/gluconeogenesis, and
propanoate metabolism KEGG pathways (Table III). Enrichment analysis for the
gene regions of DMGs by GREAT (Fig.
5B) found several cerebral abnormalities such as cerebral
cortical atrophy and cerebral atrophy were statistically
significant (P<0.05).
| Table III.The enrichment Kyoto Encyclopedia of
Genes and Genomes terms. |
Table III.
The enrichment Kyoto Encyclopedia of
Genes and Genomes terms.
| Term | Description | Count | P-value |
|---|
| hsa01130 | Biosynthesis of
antibiotics | 7 | 0.015 |
| hsa00010 |
Glycolysis/gluconeogenesis | 4 | 0.026 |
| hsa00640 | Propanoate
metabolism | 3 | 0.029 |
The subnetwork of DMGs from PPI
network
It is well known that proteins are involved in
almost all physiological processes. To some extent, the PPI network
can accurately describe the relationships between proteins. In
order to explore the potential functions of the DMGs, 190 DMGs were
mapped into a background network integrated from 8 PPI networks and
consisting of 80,980 edges and 13,361 nodes. The DMGs were set as
seed genes and the one step neighbors of the DMGs were extracted
from the background network. Duplicated relationships were removed,
leaving 1,068 nodes and 1,189 edges in the sub-PPI network
(Fig. 6). Each node represents a
gene and each edge represents the relationship between two nodes.
Not all DMGs were present in the sub-PPI network, with only 105
DMGs were retained. Among the network, hub genes were connected
with most genes and had higher degrees. These hub genes more tended
to perform important functions. Therefore, the top 1% hub genes in
the sub PPI network were identified (Table IV).
| Table IV.The hub differentially methylated
genes among protein-protein interaction network. |
Table IV.
The hub differentially methylated
genes among protein-protein interaction network.
| Ensembl gene
ID | Gene symbol | Degree | Direction |
|---|
|
ENSG00000069869 | NEDD4 | 107 | Down |
|
ENSG00000105401 | CDC37 | 74 | Down |
|
ENSG00000055332 | EIF2AK2 | 41 | Up |
|
ENSG00000114127 | XRN1 | 39 | Down |
|
ENSG00000196235 | SUPT5H | 38 | Down |
|
ENSG00000126581 | BECN1 | 35 | Up |
|
ENSG00000158019 | BRE | 34 | Up |
|
ENSG00000070061 | IKBKAP | 33 | Up |
|
ENSG00000152467 | ZSCAN1 | 32 | Down |
|
ENSG00000103423 | DNAJA3 | 31 | Down |
In order to find genes which may be truly influenced
by the DMGs, we analyzed the network through the network analyzer
in cytoscape v3.2.0 and obtained 160 non-differential methylation
genes which connected with at least two DMGs simultaneously. These
genes were used for performing the enrichment analysis. Amazingly,
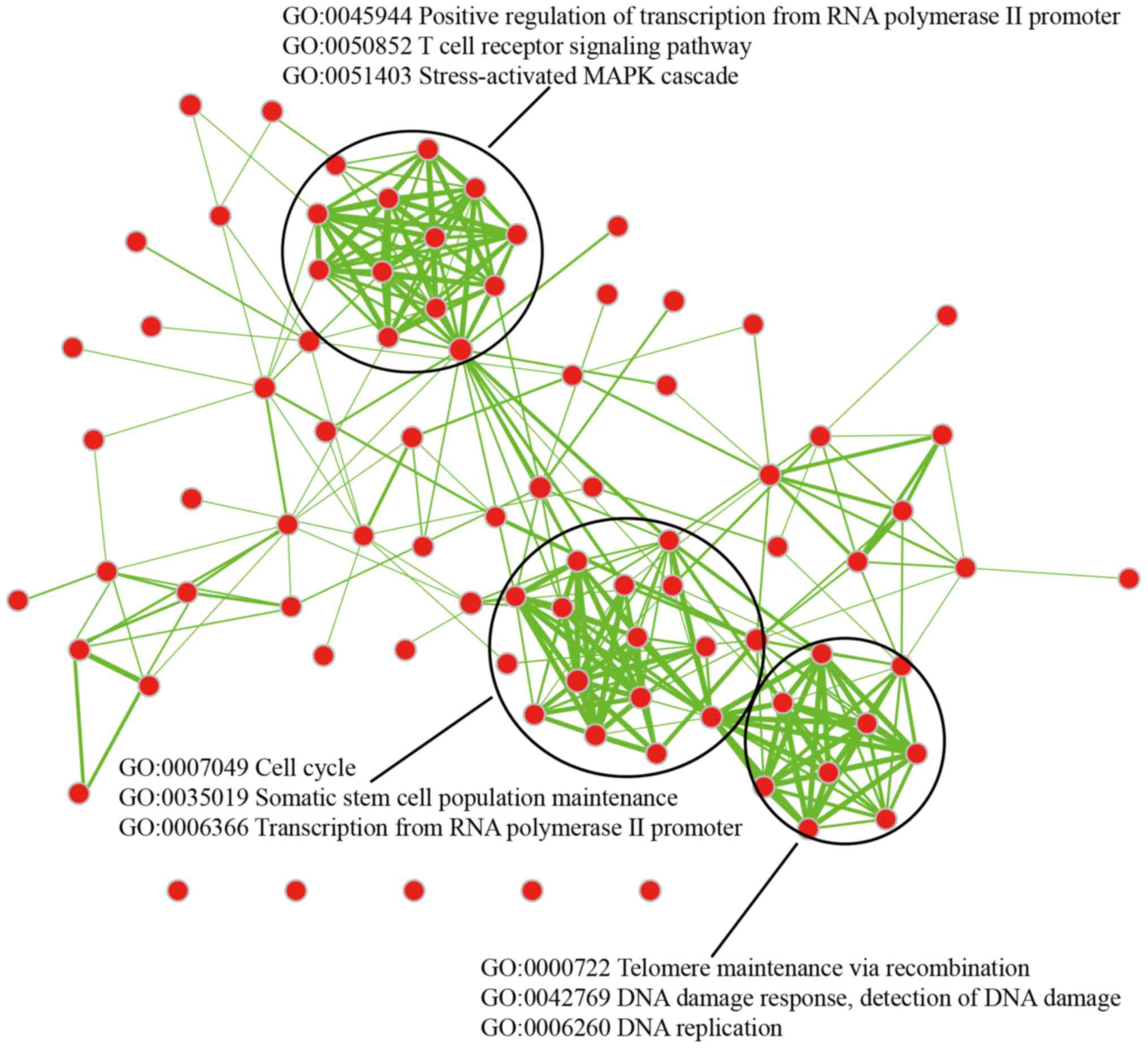
these genes were associated with many important GO terms (Fig. 7). It is suggested that these DMGs
may perform many important features by changing the connected genes
through PPI beyond our current understanding. We found that these
genes were also enriched in cellular response to nerve growth
factor stimulus and that this biological process was related with
nervous system which may cause the formation of CP. To a certain
extent, the methylation changes of these DMGs may directly or
indirectly lead the occurrence of diseases.
Discussion
Consistent with previous studies, the methylation
level in all the four discordant CP pairs exhibited the bimodal
distribution in this study. We have observed that each pair of
twins exhibited similar methylome profiles, suggesting a stable
methylation pattern between MZ twins. We further constructed the
phylogenetic tree based on the methylome of these 4 CP pairs of
twins. The evolution relationship among the same twins was closer
than the others, which indicated the high stability of methylome of
MZ twins.
DNA methylation, as one of the most important
epigenetic modifications, undergoes dynamic changes during the
progress of lesions. A total of 190 DMGs were identified, of which
was 37 genes were hypermethylated in CP group and the others were
hypomethylated. Surprisingly, functional enrichment of these DMGs
has identified several statistically significant human phenotypes
closely related with cerebral abnormalities such as cerebral
cortical atrophy and cerebral atrophy. The non-DMGs close to the
identified DMGs in the comprehensive PPI network were extracted and
enriched in multiple basic biological pathways. These genes may be
indirectly involved in the occurrence and development of CP.
As twins are the matched controls for nearly all
genetic variants and many environmental factors, they provide an
opportunity to study DNA methylation (22). CP-discordant MZ twin pairs, who
shared an identical DNA sequence, provide an ideal model for
examining environmentally driven epigenetic factors in CP. The
clusters consist of DMGs and their neighbor non-DMGs in
sub-PPI-network identified in this study may provide us with
fascinating insights into novel CP susceptibility genes as well as
novel pathogenesis of CP for drug targeting.
However, there are also some limitations that should
be considered when interpreting these results. First, although the
study is a genome-wide study of DNA methylation variation in MZ
twin pairs discordant for CP to date, the sample size for each
subgroup (CP and unaffected CP) is small, partially sue to the
relative rarity of discordant MZ twins. Second, the DNA methylation
sequencing was implemented on DNA extracted from peripheral blood
rather sample from than the brain. Unfortunately, there is no
archived collection of post-mortem brain samples from CP-discordant
MZ twins. Nevertheless, recent research suggess that some
between-individual epigenetic variation is conserved across brain
and blood (23). Third, performing
longitudinal studies is the most conclusive approach to disentangle
potential cause vs. consequence of DNA methylation changes
associated with CP and is crucial to interpreting the epigenetic
effects. The main goal of longitudinal DNA methylation studies is
to identify whether DNA methylation changes occur prior to CP onset
and thus may be causal. When possible, longitudinal studies should
be a priority Finally, it is hard to conclude the causal
relationship for any DMGs associated with CP identified in this
study because of the lack of the corresponding RNA expression data.
Following the collection of more data from CP discordant MZ twins,
our future study is expected to combine RNA expression and DNA
methylation to better explain the aetiology of CP.
In conclusion, the RRBS sequencing of 4 discordant
twin pairs at whole-genome scale presented in this study attempts
to reveal the methylation landscape of MZ twins discordant for CP.
It should be the first research on the internal links between CP
occurrence and DNA methylation alterations in twins, improving our
understanding in the role of methylation in the different disease
states in twins. The occurrence of CP is may not only be affected
by genetics (24), but also by
epigenetic factors.
Acknowledgements
This study was supported by National Natural Science
Foundation of China Project (grant no. 81273174), study on the
etiology and pathogenesis of CP based on the twin crowd; and by the
Heilongjiang Cerebral Palsy Treatment and Management Center.
References
|
1
|
Palisano R, Rosenbaum P, Walter S, Russell
D, Wood E and Galuppi B: Development and reliability of a system to
classify gross motor function in children with cerebral palsy. Dev
Med Child Neurol. 39:214–223. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mutch L, Alberman E, Hagberg B, Kodama K
and Perat MV: Cerebral palsy epidemiology: Where are we now and
where are we going. Dev Med Child Neurol. 34:547–551. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yokoyama Y, Shimizu T and Hayakawa K:
Prevalence of cerebral palsy in twins, triplets and quadruplets.
Int J Epidemiol. 24:943–948. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
McMichael G, Bainbridge MN, Haan E,
Corbett M, Gardner A, Thompson S, van Bon BW, van Eyk CL, Broadbent
J, Reynolds C, et al: Whole-exome sequencing points to considerable
genetic heterogeneity of cerebral palsy. Mol Psychiatry.
20:176–182. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
McMichael G, Girirajan S, Moreno-De-Luca
A, Gecz J, Shard C, Nguyen LS, Nicholl J, Gibson C, Haan E, Eichler
E, et al: Rare copy number variation in cerebral palsy. Eur J Hum
Genet. 22:40–45. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Smith ZD and Meissner A: DNA methylation:
Roles in mammalian development. Nat Rev Genet. 14:204–220. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bird A: DNA methylation patterns and
epigenetic memory. Genes Dev. 16:6–21. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wen Y, Wei Y, Zhang S, Li S, Liu H, Wang
F, Zhao Y, Zhang D and Zhang Y: Cell subpopulation deconvolution
reveals breast cancer heterogeneity based on DNA methylation
signature. Brief Bioinform. 18:426–440. 2017.PubMed/NCBI
|
|
9
|
Das PM and Singal R: DNA methylation and
cancer. J Clin Oncol. 22:4632–4642. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Timp W and Feinberg AP: Cancer as a
dysregulated epigenome allowing cellular growth advantage at the
expense of the host. Nat Rev Cancer. 13:497–510. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu H, Li S, Wang X, Zhu J, Wei Y, Wang Y,
Wen Y, Wang L, Huang Y, Zhang B, et al: DNA methylation dynamics:
Identification and functional annotation. Brief Funct Genomics.
15:470–484. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ziller MJ, Gu H, Müller F, Donaghey J,
Tsai LT, Kohlbacher O, De Jaqer PL, Rosen ED, Bennett DA, Bernstein
BE, et al: Charting a dynamic DNA methylation landscape of the
human genome. Nature. 500:477–481. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Varley KE, Gertz J, Bowling KM, Parker SL,
Reddy TE, Pauli-Behn F, Cross MK, Williams BA, Stamatoyannopoulos
JA, Crawford GE, et al: Dynamic DNA methylation across diverse
human cell lines and tissues. Genome Res. 23:555–567. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Meissner A, Gnirke A, Bell GW, Ramsahoye
B, Lander ES and Jaenisch R: Reduced representation bisulfite
sequencing for comparative high-resolution DNA methylation
analysis. Nucleic Acids Res. 33:5868–5877. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xi Y and Li W: BSMAP: Whole genome
bisulfite sequence MAPping program. BMC Bioinformatics. 10:2322009.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Desper R and Gascuel O: Fast and accurate
phylogeny reconstruction algorithms based on the minimum-evolution
principle. J Comput Biol. 9:687–705. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
National Institute of Allergy and
Infectious Diseases (NIAID), NIH, . Database for Annotation,
Visualization, and Integrated Discovery (DAVID). 2006.https://david.ncifcrf.gov/ease/ease.jsp
|
|
18
|
McLean CY, Bristor D, Hiller M, Clarke SL,
Schaar BT, Lowe CB, Wenger AM and Bejerano G: GREAT improves
functional interpretation of cis-regulatory regions. Nature
Biotechnol. 28:495–501. 2010. View
Article : Google Scholar
|
|
19
|
Zhang C, Zhao H, Li J, Liu H, Wang F, Wei
Y, Su J, Zhang D, Liu T and Zhang Y: The identification of specific
methylation patterns across different cancers. PLoS One.
10:e01203612015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Meissner A, Mikkelsen TS, Gu H, Wernig M,
Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB,
et al: Genome-scale DNA methylation maps of pluripotent and
differentiated cells. Nature. 454:766–770. 2008.PubMed/NCBI
|
|
22
|
Bell JT and Spector TD: DNA methylation
studies using twins: What are they telling us? Genome Biol.
13:1722012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wong CC, Meaburn EL, Ronald A, Price TS,
Jeffries AR, Schalkwyk LC, Plomin R and Mill J: Methylomic analysis
of monozygotic twins discordant for autism spectrum disorder and
related behavioural traits. Mol Psychiatry. 19:495–503. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nelson KB, Dambrosia JM, Iovannisci DM,
Cheng S, Grether JK and Lammer E: Genetic polymorphisms and
cerebral palsy in very preterm infants. Pediatr Res. 57:494–499.
2005. View Article : Google Scholar : PubMed/NCBI
|