Introduction
Lung cancer is the most common cause of
cancer-associated fatality in men and the second most common in
women (1). The 5-year survival
rate following diagnosis of lung cancer is 15.6%, making it one of
the worst prognostic malignant tumors (2). The survival rate is lower compared
with breast, colon and prostate cancer (2). Cigarette smoking is responsible for
~90% of lung cancer incidences and leads to decreased survival
rates (3).
The major histological types of lung cancer include
adenocarcinoma, squamous cell carcinoma, large cell carcinoma and
small cell carcinoma. The incidence of lung adenocarcinoma (LAC)
increased gradually and this lung cancer has been the most
frequently occurring histological type in most parts of the world
in recent years (4).
Adenocarcinoma account for ~40% of all lung cancer cases (5). Smoking is a major cause of lung
adenocarcinoma (6). However, the
causes of the increase in adenocarcinomas are not clear.
Sequencing data from large-scale databases, such as
The Cancer Genome Atlas, have aided in identification of novel
factors and potentially targetable alterations in lung
adenocarcinomas (7). A number of
smoking-associated genes have been revealed in LAC, including the
cyclin D1 A870 G gene, and polymorphisms of this gene have been
indicated to modulate smoking-induced lung cancer risk (8). Estrogen receptor α promotes smoking
carcinogen-induced lung carcinogenesis via cytochrome P450 1B1
(9). The interactions between
smoking, polymorphisms of human 8-oxoguanine DNA glycosylase and
p53 are associated with the development of lung cancer (10). Interactions between smoking,
fragile histidine triad gene alterations (11) and excision repair
cross-complementation group 1 polymorphisms (12) have also been reported in lung
cancer. However, the recognized genetic changes in patients with
LAC who are smokers remain to be elucidated and further studies are
necessary to determine the underlying molecular mechanism of
smoking-induced LAC.
A recent study has aimed to identify
smoking-associated genes via the differential analysis of RNA
sequencing data (13). The study
analyzed two datasets with only two samples and identified 1,603
differentially expressed genes (DEGs). The authors also identified
that the possible alternative splicing of gene FCGBP may have an
impact on lung cancer. However, the small sample size could lead to
low reliability of the results.
In the present study, three gene expression datasets
of smokers and non-smokers with LAC (>50 samples/group) were
obtained and DEGs were identified using meta-analysis. A
protein-protein interaction (PPI) network of the DEGs was
constructed with the betweenness centrality (BC) analysis for the
selection of feature genes. Using the feature genes, a support
vector machine (SVM) classifier, which is able to distinguish
between samples from smokers and non-smokers with a high
classification accuracy, was constructed. The feature genes in the
SVM classifier were considered as the smoking-related genes in LAC
and enrichment analysis was conducted to identify significant
pathways.
Materials and methods
Gene expression data
To collect gene expression data from patients with
LAC who smoke or do not smoke, the Gene Expression Omnibus (GEO;
www.ncbi.nlm.nih.gov/geo/) database was
used and the key words ‘lung adenocarcinoma’, ‘Homo sapiens’
and ‘smoke’ were searched. The following inclusion criteria were
used to extract the corresponding datasets: i) They were gene
expression data; ii) they were from LAC samples; iii) information
concerning smoking was described; and iv) ≥50 samples were included
in each dataset. A total of three datasets were collected from the
GEO database, including GSE43458 (14), GSE10072 (15) and GSE12667 (16) (Table
I).
 | Table I.Data of the three collected gene
expression datasets. |
Table I.
Data of the three collected gene
expression datasets.
| Accession
number | Platform | Total samples
(n) | Non-smokers
(n) | Smokers (n) |
|---|
| GSE43458 |
HuGene-1_0-st-v1 | 110 | 40 | 40 |
| GSE10072 | HG-U133A | 107 | 16 | 42 |
| GSE12667 | HG-U133_Plus_2 | 75 | 8 | 43 |
Raw data in these three datasets were analyzed with
the affy package in R 3.2.1 (http://bioconductor.org/packages/release/bioc/html/affy.html)
(17). Probes were subsequently
mapped into genes. Probes corresponding to one gene were averaged
as the final expression value of the gene. Normalization was
performed with package limma (18)
of R to conduct the analysis of the datasets.
Screening of DEGs
Meta-analysis was used to enforce the analytical
reliability for gene expression data by combining data from
different datasets. DEGs associated with smoking in the three gene
expression datasets were screened via meta-analysis using the
MetaDE.ES package of R (19). The
method tested the heterogeneity of gene expression value from three
datasets with three statistic parameters: Tau2, Q-value
and Qpval. Subsequently, differential expression of genes between
smoking and non-smoking samples was assessed by determining the
P-value and false discovery rate (FDR). To determine the DEGs
associated with smoking, tau2=0, Qpval >0.05 and FDR
<0.05 were set as the cut-off points. Bidirectional clustering
analysis using the pheatmap package in R language (https://cran.r-project.org/web/packages/pheatmap/index.html),
which was based on the euclidean distance calculations for gene
expression values, was also conducted to examine whether the
selected DEGs were able to distinguish different samples, as
described previously (20).
Construction of PPI network
To investigate the interactions of DEGs, the DEGs
were mapped to the PPI database using the Human Protein Reference
Database (21). The interactions
of DEGs obtained were constructed into a PPI network with the
proteins that were connected with at least three DEGs. The network
was visualized with Cytoscape (22).
Calculation of BC
Feature genes that function as hub nodes in the PPI
network were screened using a BC algorithm (23). BC represented the degree of node in
the network and was calculated as follows:
CB(v)=∑t≠v≠u∈Vσst(v)σst
Where σst is the total number of shortest
paths from node s to node t; σst(ν) is the
number of shortest paths from s to t going through
v; BC scores were between 0 and 1, and a higher BC score
indicated a higher degree of the node.
Training and validation of SVM
classifier
SVM classifier comprises of feature genes that
distinguishes between different samples (24,25).
To construct the SVM classifier, one of the downloaded datasets,
GSE43458 (containing 40 non-smokers and 40 smokers) was selected as
the training dataset basing on the top 10, 20, 30, 40 and 50
feature genes ranked by BC scores. The feature genes in the SVM
classifier that could exactly distinguish between different samples
in GSE42458 were subjected to two-way clustering analysis using
pheatmap package in R 3.1.4 (https://cran.r-project.org/web/packages/pheatmap/index.html).
Sample similarity matrices were also obtained by computing the
Pearson's correlation coefficients of these genes using Cor package
in R 3.1.4 (https://stat.ethz.ch/R-manual/R-devel/library/stats/html/cor.html)
and top 50 genes were selected for further analysis. The clustering
and similarity matrices were visualized using heatmaps in pheatmap
package in R 3.1.4 (https://cran.r-project.org/web/packages/pheatmap/index.html).
The SVM classifier was validated with two
independent datasets, GSE10072 and GSE12667. Sensitivity (Se),
specificity (Sp), positive predictive value (PPV), negative
predictive value (NPV) and area under curve (AUC) were calculated
using the pROC package in R language (https://cran.r-project.org/web/packages/pROC/index.html)
to examine the classification accuracy of the SVM classifier as
described previously (26,27).
Pathway enrichment analysis
Feature gene-related Kyoto Encyclopedia of Genes and
Genomes pathways (http://www.genome.jp/kegg/) were revealed using
Fisher's exact test as follows:
p=1–∑i=0x–1(Mi)(N–MK–i)(NK)
Where N represented the total number of
genes; M represented the number of genes in the pathway; and
K indicated the number of feature genes.
Results
DEGs
A total of 12,476 genes were in the three gene
expression datasets, and according to the set criteria, 347 DEGs
between smoking and non-smoking LAC samples were identified. The
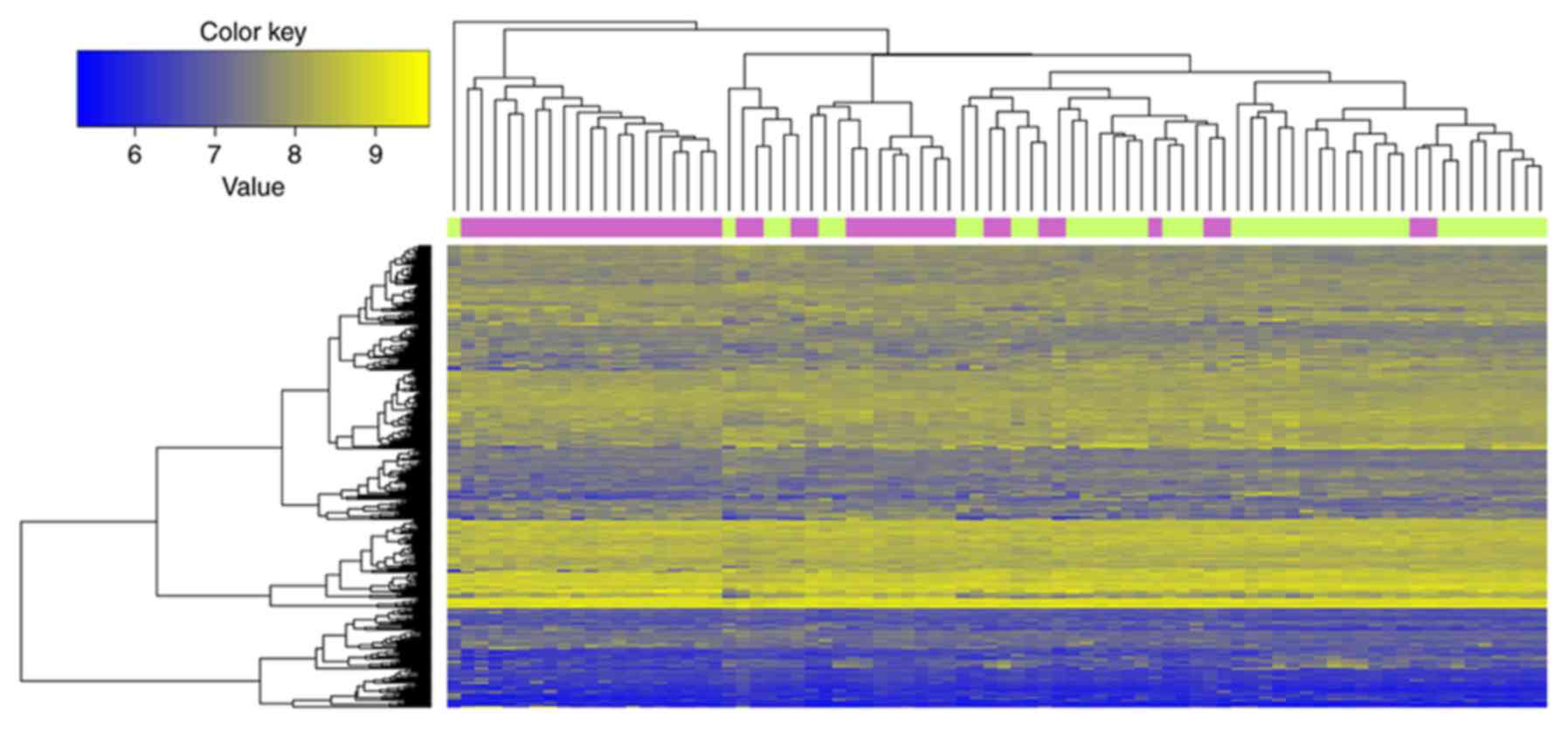
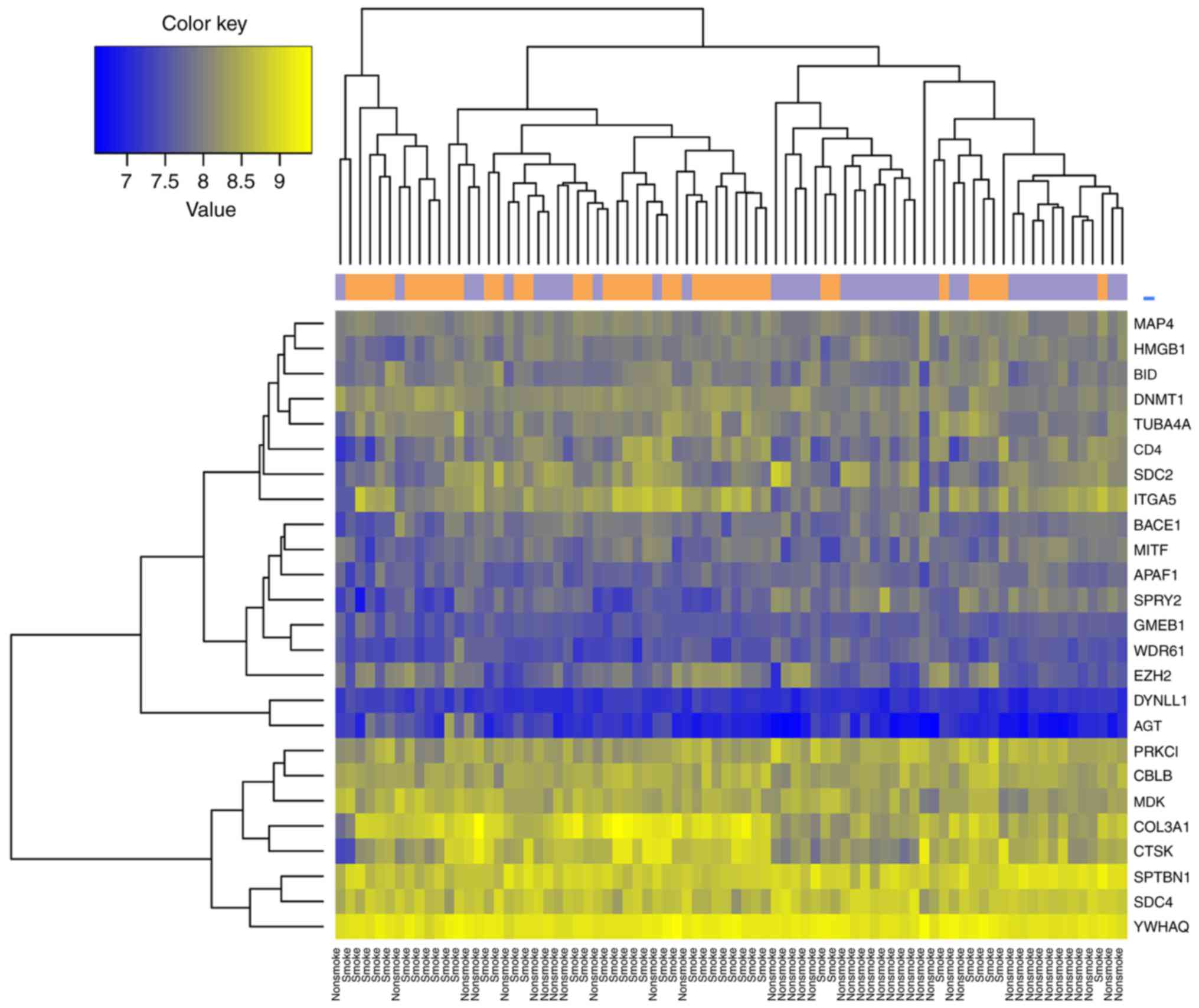
top 10 DEGs ranked by FDR are listed in Table II. As indicated in Fig. 1, the 347 DEGs distinguished the
samples of smokers from the non-smokers.
| Table II.Top 10 candidate feature genes by
FDR. |
Table II.
Top 10 candidate feature genes by
FDR.
| ID | P-value | FDR |
tau2 | Qpval | Qval | Expression |
|---|
| ABCB11 |
1.52×10−05 |
5.59×10−04 | 0 |
9.27×10−01 |
8.28×10−03 | Up |
| ABCB6 |
2.27×10−03 |
2.23×10−02 | 0 |
9.63×10−01 |
2.18×10−03 | Up |
| ABCC2 |
2.11×10−03 |
2.11×10−02 | 0 |
9.51×10−01 |
3.83×10−03 | Up |
| ABCG5 |
4.01×10−06 |
2.10×10−04 | 0 |
9.00×10−01 |
1.58×10−02 | Up |
| ACD |
4.81×10−06 |
2.38×10−04 | 0 |
9.40×10−01 |
5.71×10−03 | Up |
| ADAMTS5 |
1.88×10−04 |
3.79×10−03 | 0 |
9.10×10−01 |
1.26×10−02 | Up |
| AGT |
2.89×10−03 |
2.64×10−02 | 0 |
8.47×10−01 |
3.71×10−02 | Up |
| AIM1L |
1.00×10−20 |
7.47×10−19 | 0 |
8.26×10−01 |
4.84×10−02 | Up |
| AKAP6 |
1.15×10−04 |
2.69×10−03 | 0 |
9.82×10−01 |
5.08×10−04 | Down |
| ALPL |
5.01×10−03 |
3.88×10−02 | 0 |
9.06×10−01 |
1.40×10−02 | Down |
PPI network
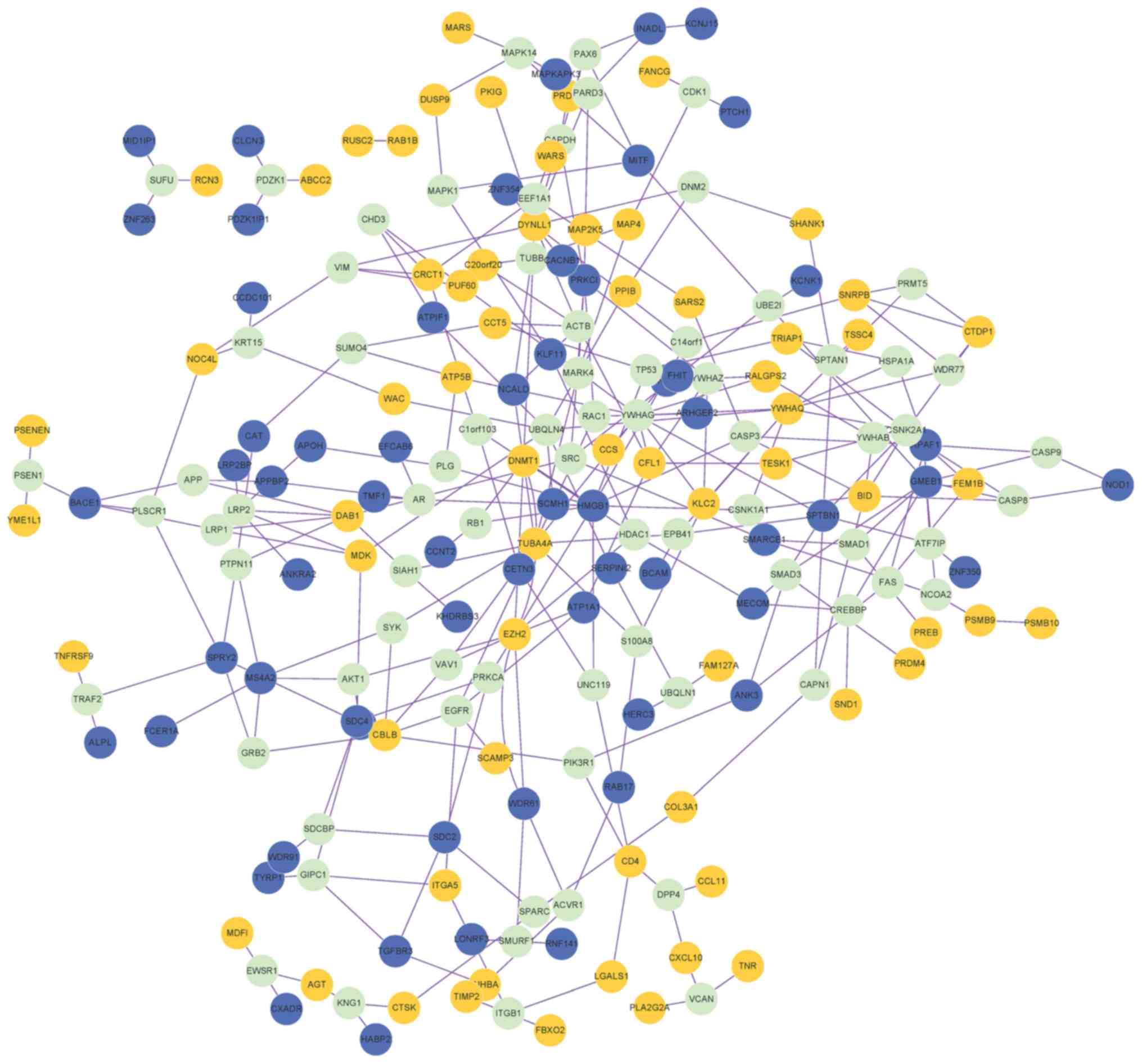
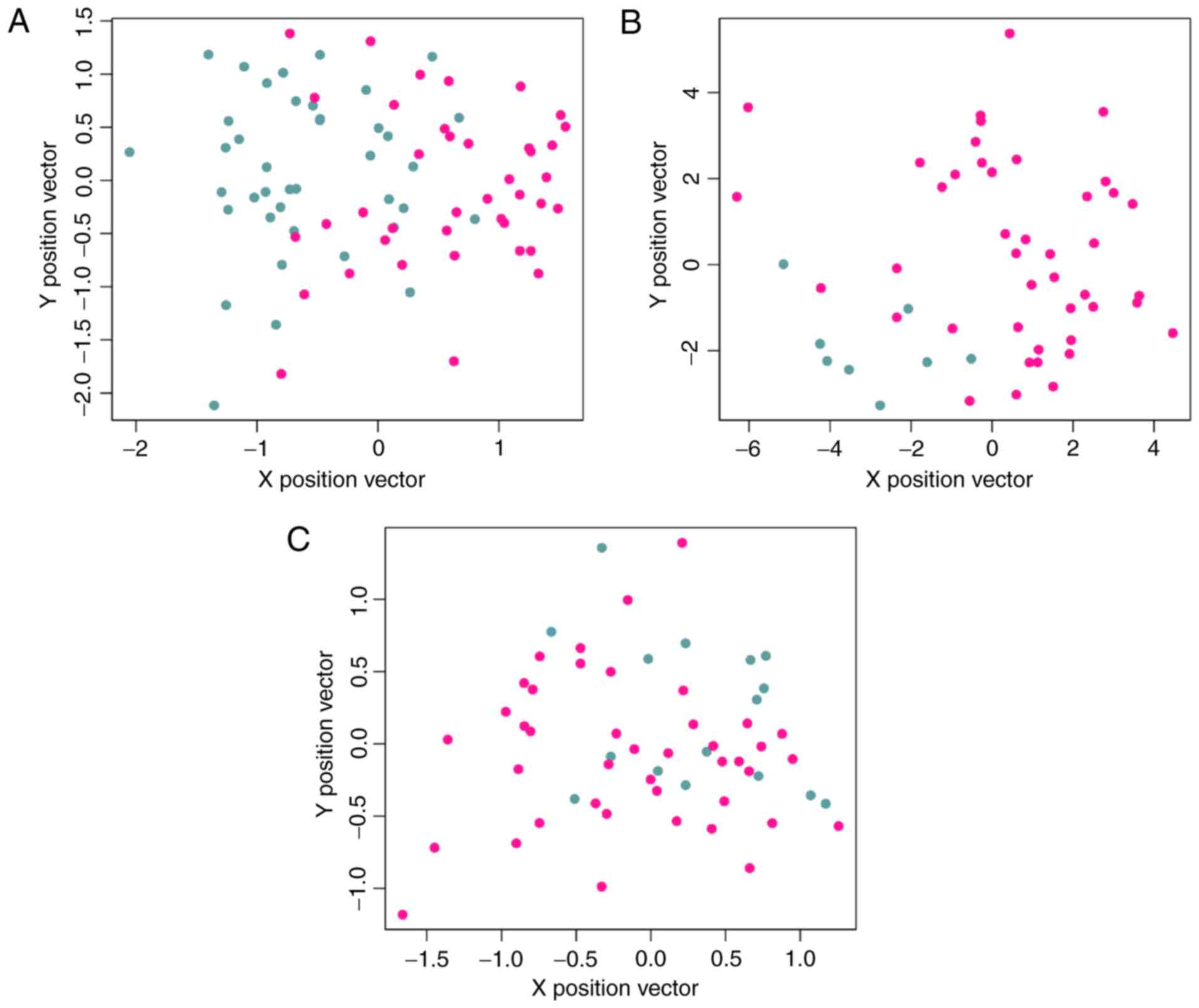
A PPI network containing 202 nodes (genes) and 300
edges (connection between nodes) was obtained (Fig. 2). The proteins that were connected
with ≥3 DEGs were also included in the PPI network. Degree
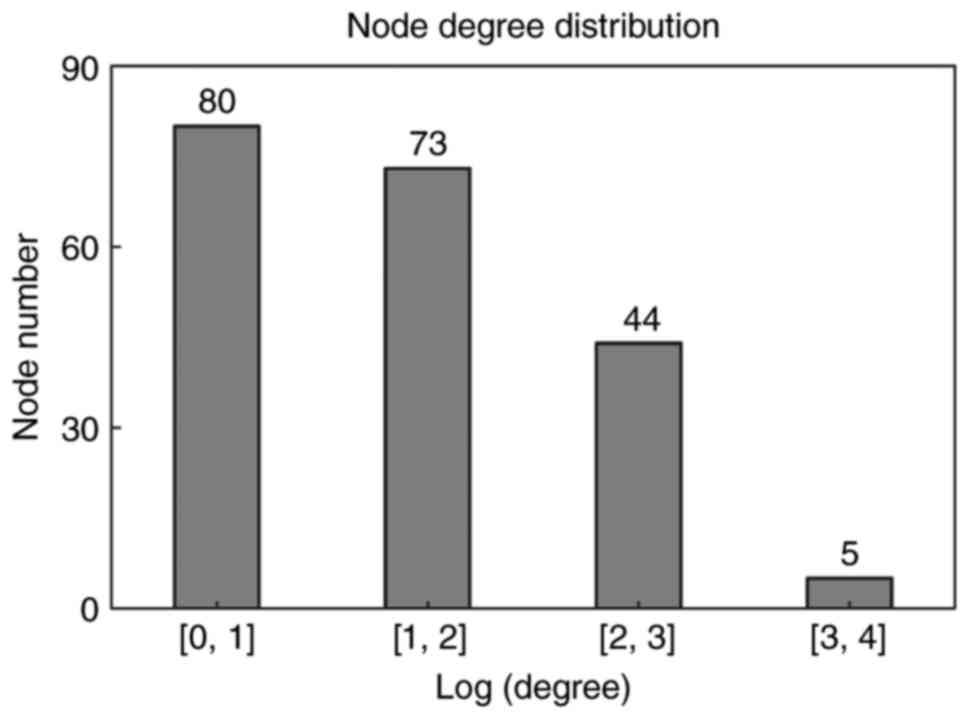
distribution of genes in the network is indicated in Fig. 3. Similar to biological networks,
the PPI network was scale-free, with the majority of genes (80
genes) exhibiting small degrees (Log transformed degree <1) and
few genes (only 5) exhibiting larger degrees (Log transformed
degree between 3 and 4). The genes with high degrees were hub
genes, indicating their roles in the development of
smoking-associated LAC.
Feature genes
BC was calculated for each node in the PPI network.
The top 10 genes by BC value were considered as the feature genes,
including high mobility group box 1 (HMGB1); dynein light chain
LC8-type 1; tubulin α 4a; 14-3-3 protein γ; tyrosine
3-monooxygenase; spectrin β, non-erythrocytic 1; ubiquilin 4; DNA
methyltransferase 1 (DNMT1); enhancer of zeste 2 polycomb
repressive complex 2 subunit (EZH2) and glucocorticoid modulatory
element binding protein 1 (Table
III).
| Table III.Top 10 genes ranked using BC. |
Table III.
Top 10 genes ranked using BC.
| Gene | BC | Expression | Degree | P-value | FDR | Qpval | Qval |
|---|
| HMGB1 |
1.98×10−01 | Down | 11 |
1.63×10−03 |
1.76×10−02 |
8.95×10−01 |
1.74×10−02 |
| DYNLL1 |
1.77×10−01 | Up | 15 |
1.00×10−20 |
7.47×10−19 |
8.92×10−01 |
1.83×10−02 |
| TUBA4A |
1.37×10−01 | Up | 10 |
2.08×10−05 |
7.32×10−04 |
8.50×10−01 |
3.56×10−02 |
| YWHAG |
1.20×10−01 | − | 11 |
9.86×10−01 |
9.95×10−01 |
5.38×10−06 | 2.07×10 |
| YWHAQ |
1.07×10−01 | Up | 10 |
2.40×10−04 |
4.55×10−03 |
8.46×10−01 |
3.77×10−02 |
| SPTBN1 |
1.04×10−01 | Down | 7 |
9.39×10−04 |
1.23×10−02 |
8.53×10−01 |
3.43×10−02 |
| UBQLN4 |
1.00×10−01 | − | 7 |
9.31×10−01 |
9.67×10−01 |
5.95×10−02 |
3.55×100 |
| DNMT1 |
9.98×10−02 | Up | 7 |
1.20×10−04 |
2.77×10−03 |
8.61×10−01 |
3.05×10−02 |
| EZH2 |
8.51×10−02 | Up | 8 |
2.65×10−03 |
2.48×10−02 |
8.44×10−01 |
3.89×10−02 |
| GMEB1 |
8.45×10−02 | Down | 8 |
2.31×10−04 |
4.40×10−03 |
9.32×10−01 |
7.20×10−03 |
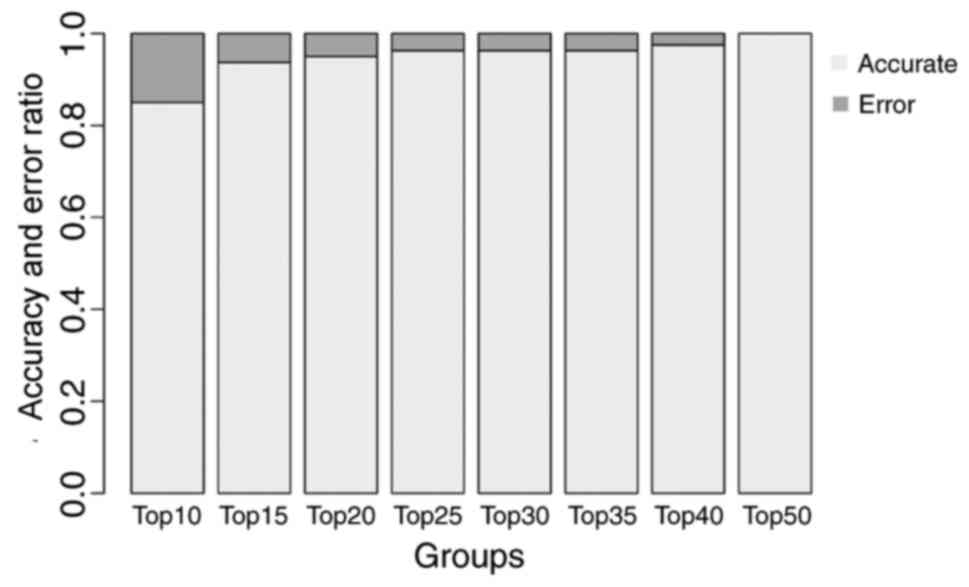
SVM classifier
Feature genes with the greatest BC values were used
to construct the SVM classifier basing on dataset GSE43458. There
were 8, 11, 14, 16, 18, 20, 22 and 26 feature genes in the top 10,
15, 20, 25, 30, 35, 40 and 50 genes, respectively. The training
process is indicated in Fig. 4.
The accuracy of the classifier reached 100% when the 26 feature
genes in the top 50 were included. Therefore, the classifier
comprised by these 26 feature genes were chosen as the final SVM
classifier. These feature genes included Cbl proto-oncogene B
(CBLB), DNMT1, EZH2, HMGB1, integrin α-5 (ITGA5), MDK, protein
kinase C ι (PRKCI) and sprouty receptor tyrosine kinase signaling
antagonist 2 (SPRY2).
Hierarchical clustering was performed for samples
from the training dataset using the 26 feature genes (Fig. 5). The classifier separated samples
of smokers from samples of non-smokers in dataset GSE43458
(Fig. 6A).
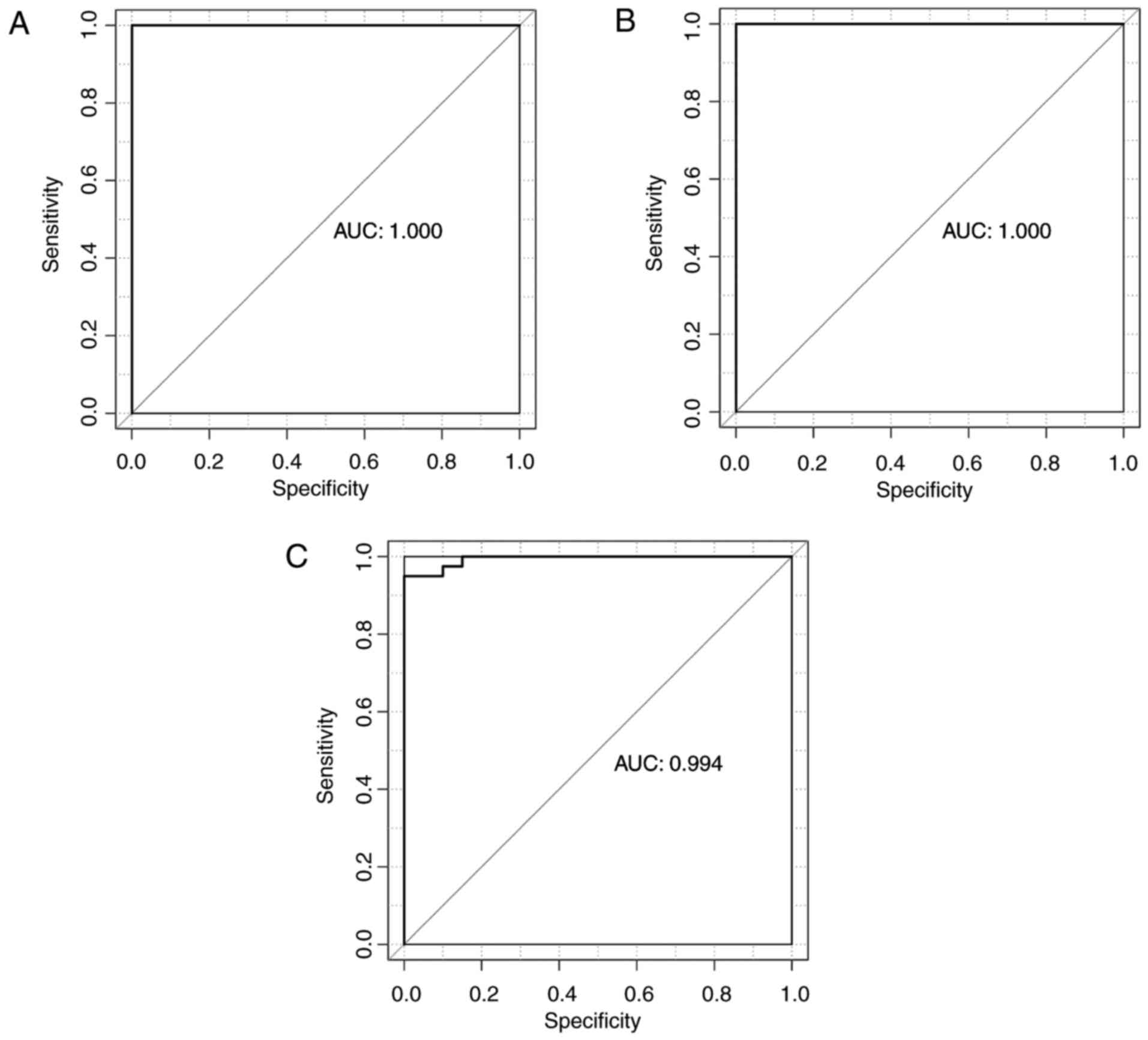
The SVM classifier was validated using dataset
GSE12667 and GSE10072. The classification accuracy in GSE12667 was
100% (Fig. 6B). In GSE10072, the
classifier identified 42 smokers (42/42, 100%) and 13 non-smokers
(13/16, 81.25%), and total accuracy was 94.83% (55/58) (Fig. 6C; Table IV). The classifier demonstrated
high accuracy of 100, 100 and 94.83% in GSE43458, GSE12667 and
GSE10072, respectively. Se, Sp, PPV, NPV and AUC results (Table IV) and receiver operating
characteristic curves were generated (Fig. 7).
| Table IV.Prediction results of the support
vector machine classifier in the three datasets. |
Table IV.
Prediction results of the support
vector machine classifier in the three datasets.
| Dataset | Samples (n) | Accuracy (%) | Se | Sp | PPV | NPV | AUC |
|---|
| GSE43458 | 80 | 100 | 1 | 1 | 1 | 1 | 1 |
| GSE12667 | 51 | 100 | 1 | 1 | 1 | 1 | 1 |
| GSE10072 | 58 | 94.83 | 1 | 0.813 | 0.933 | 1 | 0.994 |
Overrepresented biological
pathways
The 26 feature genes were indicated to be
significantly enriched in nine biological pathways (Table V): Extracellular matrix
(ECM)-receptor interaction, proteoglycans in cancer, cell adhesion
molecules, pathogenic Escherichia coli infection, p53
signaling pathway, microRNAs in cancer, bacterial invasion of
epithelial cells, apoptosis and hematopoietic cell lineage.
| Table V.A total of 9 biological pathways
significantly overrepresented by the 26 feature genes. |
Table V.
A total of 9 biological pathways
significantly overrepresented by the 26 feature genes.
| ID | Term | P-value | Genes |
|---|
| hsa04512 | Extracellular
matrix-receptor interaction |
5.68×10−03 | SDC4, ITGA5,
COL3A1 |
| hsa05205 | Proteoglycans in
cancer |
9.82×10−03 | SDC4, CBLB, ITGA5,
SDC2 |
| hsa04514 | Cell adhesion
molecules |
2.17×10−02 | SDC4, CD4,
SDC2 |
| hsa05130 | Pathogenic
Escherichia coli infection |
2.19×10−02 | YWHAQ, TUBA4A |
| hsa04115 | p53 signaling
pathway |
3.21×10−02 | APAF1, BID |
| hsa05206 | MicroRNAs in
cancer |
3.29×10−02 | EZH2, SPRY2, ITGA5,
DNMT1 |
| hsa05100 | Bacterial invasion
of epithelial cells |
3.91×10−02 | CBLB, ITGA5 |
| hsa04210 | Apoptosis |
4.86×10−02 | APAF1, BID |
| hsa04640 | Hematopoietic cell
lineage |
4.96×10−02 | CD4, ITGA5 |
Discussion
In the present study, three gene expression datasets
were obtained and a total of 347 DEGs were identified in samples
from smokers with LAC compared with non-smokers with LAC using
meta-analysis. A PPI network including 202 nodes and 300 edges was
constructed, from which 26 feature genes were identified. The SVM
classifier of these 26 genes separated smokers from non-smokers
with an accuracy >94% in all the three datasets. Pathway
enrichment analysis demonstrated that these feature genes were
primarily associated with cancer development- and
metastasis-associated pathways, including ECM-receptor interaction,
proteoglycans in cancer, cell adhesion molecules, p53 signaling
pathway, microRNAs in cancer and apoptosis.
Due to the generalization ability, SVM has been
widely used for analysis, including data classification and
function approximation (28–30).
SVM classifier has been demonstrated to distinguish whether one
cancer sample type possessed distinctive signatures of gene
expressions compared with other sample types (31). In the present study, an SVM
classifier with 26 feature genes successfully distinguished LAC
samples of smokers and non-smokers using bioinformatics analysis.
Yousef et al (32)
previously conducted a similar study for the identification of
biomarkers, by integrating interaction networks and an SVM
classifier, and subsequently obtained >90% accuracy in
classification of selected microarray datasets. Furthermore, a
previous study also demonstrated that the discriminant analysis
based on an SVM classifier achieved satisfactory results in the
classification of lung cancer samples (33).
Specific genes within the 26 feature genes have been
implicated in lung cancer or LAC. CBLB is a regulator of T-cell
response (34). It has been
reported that the single nucleotide polymorphisms of CBLB may
predict the definitive radiotherapy outcomes for non-small cell
lung cancer (NSCLC) (34). CBLB is
associated with icotinib-induced apoptosis and G1 phase arrest of
epidermal growth factor receptor mutation-positive NSCLC (35).
DNMT1 is responsible for maintaining methylation
patterns following DNA replication and has an important role in the
development of various types of cancer (36). DNA methylation alterations are
recognized as key epigenetic changes in cancer, influencing the
chromosomal instability through global hypomethylation and aberrant
gene expression via the alterations in methylation levels (37). The tobacco-specific carcinogen
nicotine-derived nitrosamine ketone induces the accumulation of
DNMT1 in patients with lung cancer (38). Furthermore, DNMT1 inhibits the
expression of, the tumor suppressor Wnt7a in NSCLC (39).
EZH2 is a member of the polycomb-group family, which
is associated with maintaining the transcriptional repressive state
of genes over successive cell generations (40). Yoon et al (41) previously suggested a correlation
between the genotype variants in EZH2 and reduced lung cancer risk.
Additionally, Zhang et al (42) determined that miR-138 inhibited
tumor growth through the repression of EZH2 in NSCLC. Notably, a
recent study indicated that EZH2 silencing with RNA interference
induced G2/M arrest in human lung cancer cells in vitro
(43), and Wang et al
(44) recently demonstrated that
EZH2 overexpression was associated with a poor prognosis for
patients with LAC. In the present study, it was indicated that EZH2
was upregulated in the samples of smokers and thus the present
findings suggest that EZH2 upregulation may result from
smoking.
HMGB1 has a role in tumor cell migration (45). Shen et al (46) indicated that the expression of
HMGB1 correlates with the progression of NSCLC. ITGA5 is considered
as a prognostic indicator in NSCLC (47).
MDK promotes cell growth, migration and
angiogenesis, in particular during tumorigenesis (48). A previous study indicated that MDK
protein overexpression is correlated with the malignant status and
prognosis of NSCLC (49).
Furthermore, MDK has been targeted as a therapeutic biomarker for
lung cancer (50).
PRKCI is required for lung tumorigenesis as genetic
loss of PRKCI inhibits Kras-initiated hyperplasia and subsequent
lung tumor formation in vivo (51). SPRY2 inhibits cell migration and
proliferation in NSCLC (52). In
addition, a previous study has indicated that downregulation of
SPRY2 in NSCLC contributes to tumor malignancy (53).
Smoking can cause LAC and the incidence of this
disease increased in recent years (4). However, the reason for this increase
and the mechanism underlying smoking-associated development of LAC
remain to be elucidated. The present study identified genes
implicated in smoking-associated LAC, including CBLB, DNMT1, EZH2,
HMGB1, ITGA5, MDK, PRKCI and SPRY2. Most of these genes have been
reported in association with malignancy and certain were associated
with lung cancer. The identification of these characteristic genes
may aid in elucidating the mechanism underlying smoking
associated-lung adenocarcinoma. Although further experiments such
as validation the gene and protein expression level in the smoking
and non-smoking LAC samples were not performed limited by the LAC
samples available, these results may provide information to other
researchers in the field.
In conclusion, a number of key genes have been
revealed in smokers with LAC and some of these have been implicated
in lung cancer. However, the associations between the 26 feature
genes, smoking and LAC remain to be fully elucidated with further
studies.
Acknowledgements
The present study was supported by The Natural
Science Foundation of Heilongjiang Province (The Science Foundation
for Returned Overseas Students in Heilongjiang Province; grant nos.
LC2016036 and LC2015040) and Research Foundation of Harbin Medical
University Affiliated Second Hospital (grant nos. KYBS2015-10 and
KYBS2015-01).
References
|
1
|
Centers for Disease Control and Prevention
(CDC), . State-specific trends in lung cancer incidence and
smoking-United States, 1999–2008. MMWR Morb Mortal Wkly Rep.
60:1243–1247. 2011.PubMed/NCBI
|
|
2
|
Nanavaty P, Alvarez MS and Alberts WM:
Lung cancer screening: Advantages, controversies, and applications.
Cancer Control. 21:9–14. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bryant A and Cerfolio RJ: Differences in
epidemiology, histology, and survival between cigarette smokers and
never-smokers who develop non-small cell lung cancer. Chest.
132:185–192. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nakamura H and Saji H: Worldwide trend of
increasing primary adenocarcinoma of the lung. Surg Today.
44:1004–1012. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kong J, Xu F, He M, Chen K and Qian B: The
incidence of lung cancer by histological type: A population-based
study in Tianjin, China during 1981–2005. Respirology.
19:1222–1228. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cancer Genome Atlas Research Network, .
Comprehensive molecular profiling of lung adenocarcinoma. Nature.
511:543–550. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Devarakonda S, Morgensztern D and Govindan
R: Genomic alterations in lung adenocarcinoma. Lancet Oncol.
16:e342–e351. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gautschi O, Hugli B, Ziegler A, Bigosch C,
Bowers NL, Ratschiller D, Jermann M, Stahel RA, Heighway J and
Betticher DC: Cyclin D1 (CCND1) A870G gene polymorphism modulates
smoking-induced lung cancer risk and response to platinum-based
chemotherapy in non-small cell lung cancer (NSCLC) patients. Lung
Cancer. 51:303–311. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li MY, Liu Y, Liu LZ, Kong AW, Zhao Z, Wu
B, Long X, Wu J, Ng CS, Wan IY, et al: Estrogen receptor alpha
promotes smoking-carcinogen-induced lung carcinogenesis via
cytochrome P450 1B1. J Mol Med. 93:1221–1233. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cheng Z, Wang W, Song YN, Kang Y and Xia
J: hOGG1, p53 genes, and smoking interactions are associated with
the development of lung cancer. Asian Pac J Cancer Prev.
13:1803–1808. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang J, Chen D, Shen QM, Tian DL, Jiang
YH, Yin HN and Li HW: Association between cigarette smoking and
FHIT gene alterations in Chinese lung cancer. Lung Cancer.
29:2352000. View Article : Google Scholar
|
|
12
|
Zhou W, Liu G, Park S, Wang Z, Wain JC,
Lynch TJ, Su L and Christiani DC: Gene-smoking interaction
associations for the ERCC1 polymorphisms in the risk of lung
cancer. Cancer Epidemiol Biomarkers Prev. 14:491–496. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhou C, Chen H, Han L, Xue F, Wang A and
Liang YJ: Screening of genes related to lung cancer caused by
smoking with RNA-Seq. Eur Rev Med Pharmacol Sci. 18:117–125.
2014.PubMed/NCBI
|
|
14
|
Kabbout M, Garcia MM, Fujimoto J, Liu DD,
Woods D, Chow CW, Mendoza G, Momin AA, James BP, Solis L, et al:
ETS2 mediated tumor suppressive function and MET oncogene
inhibition in human non-small cell lung cancer. Clin Cancer Res.
19:3383–3395. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Landi MT, Dracheva T, Rotunno M, Figueroa
JD, Liu H, Dasgupta A, Mann FE, Fukuoka J, Hames M, Bergen AW, et
al: Gene expression signature of cigarette smoking and its role in
lung adenocarcinoma development and survival. PLoS One.
3:e16512008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ding L, Getz G, Wheeler DA, Mardis ER,
McLellan MD, Cibulskis K, Sougnez C, Greulich H, Muzny DM, Morgan
MB, et al: Somatic mutations affect key pathways in lung
adenocarcinoma. Nature. 455:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gautier L, Cope L, Bolstad BM and Irizarry
RA: Affy-analysis of Affymetrix GeneChip data at the probe level.
Bioinformatics. 20:307–315. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Smyth GK: Limma: Linear models for
microarray dataBioinformatics and Computational Biology Solutions
Using R and Bioconductor. Gentleman R, Carey V, Huber W, Irizarry R
and Dudoit S: Springer; New York: pp. 397–420. 2004
|
|
19
|
Wang X, Kang DD, Shen K, Song C, Lu S,
Chang LC, Liao SG, Huo Z, Tang S, Ding Y, et al: An R package suite
for microarray meta-analysis in quality control, differentially
expressed gene analysis and pathway enrichment detection.
Bioinformatics. 28:2534–2536. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang L, Cao C, Ma Q, Zeng Q, Wang H, Cheng
Z, Zhu G, Qi J, Ma H, Nian H and Wang Y: RNA-seq analyses of
multiple meristems of soybean: Novel and alternative transcripts,
evolutionary and functional implications. BMC Plant Biol.
14:1692014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Keshava Prasad TS, Goel R, Kandasamy K,
Keerthikumar S, Kumar S, Mathivanan S, Telikicherla D, Raju R,
Shafreen B, Venugopal A, et al: Human protein reference
database-2009 update. Nucleic Acids Res. 37(Database issue):
D767–D772. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Smoot ME, Ono K, Ruscheinski J, Wang PL
and Ideker T: Cytoscape 2.8: New features for data integration and
network visualization. Bioinformatics. 27:431–432. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Barthélemy M: Betweenness centrality in
large complex networks. Eur Phy J Conden Matter Com Sys.
38:163–168. 2004.
|
|
24
|
Zhang HH, Ahn J, Lin X and Park C: Gene
selection using support vector machines with non-convex penalty.
Bioinformatics. 22:88–95. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Brown MP, Grundy WN, Lin D, Cristianini N,
Sugnet CW, Furey TS, Ares M Jr and Haussler D: Knowledge-based
analysis of microarray gene expression data using support vector
machines. Proc Natl Acad Sci USA. 97:pp. 262–267. 2000; View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Stojanović M, Andjelković Apostolović M,
Stojanović D, Milosević Z, Ignjatović A, Lakusić VM and Golubović
M: Understanding sensitivity, specificity and predictive values.
Vojnosanit Pregl. 71:1062–1065. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Parikh R, Mathai A, Parikh S, Chandra
Sekhar G and Thomas R: Understanding and using sensitivity,
specificity and predictive values. Indian J Ophthalmol. 56:45–50.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Orru G, Pettersson-Yeo W, Marquand AF,
Sartori G and Mechelli A: Using support vector machine to identify
imaging biomarkers of neurological and psychiatric disease: A
critical review. Neurosci Biobehav Rev. 36:1140–1152. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fan XJ, Wan XB, Huang Y, Cai HM, Fu XH,
Yang ZL, Chen DK, Song SX, Wu PH, Liu Q, et al:
Epithelial-mesenchymal transition biomarkers and support vector
machine guided model in preoperatively predicting regional lymph
node metastasis for rectal cancer. Br J Cancer. 106:1735–1741.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Han M, Dai J, Zhang Y, Lin Q, Jiang M, Xu
X, Liu Q and Jia J: Support vector machines coupled with proteomics
approaches for detecting biomarkers predicting chemotherapy
resistance in small cell lung cancer. Oncol Rep. 28:2233–2238.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Guyon I, Weston J and Barnhill S: Gene
selection for cancer classification using support vector machines.
Machine Learning. 46:389–422. 2002. View Article : Google Scholar
|
|
32
|
Yousef M, Ketany M, Manevitz L, Showe LC
and Showe MK: Classification and biomarker identification using
gene network modules and support vector machines. BMC
Bioinformatics. 10:3372009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Guan P, Huang D, He M and Zhou B: Lung
cancer gene expression database analysis incorporating prior
knowledge with support vector machine-based classification method.
J Exp Clin Cancer Res. 28:1032009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li P, Wang X, Liu Z, Liu H, Xu T, Wang H,
Gomez DR, Nguyen QN, Wang LE, Teng Y, et al: Single nucleotide
polymorphisms in CBLB, a regulator of T-cell response, predict
radiation pneumonitis and outcomes after definitive radiotherapy
for non-small-cell lung cancer. Clin Lung Cancer. 17:253–262. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mu X, Zhang Y, Qu X, Hou K, Kang J, Hu X
and Liu Y: Ubiquitin ligase Cbl-b is involved in icotinib
(BPI-2009H)-induced apoptosis and G1 phase arrest of EGFR
mutation-positive non-small-cell lung cancer. Biomed Res Int.
2013:7263752013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Rountree MR, Bachman KE, Herman JG and
Baylin SB: DNA methylation, chromatin inheritance, and cancer.
Oncogene. 20:3156–3165. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kerr KM, Galler JS, Hagen JA, Laird PW and
Laird-Offringa IA: The role of DNA methylation in the development
and progression of lung adenocarcinoma. Dis Markers. 23:5–30. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lin RK, Hsieh YS, Lin P, Hsu HS, Chen CY,
Tang YA, Lee CF and Wang YC: The tobacco-specific carcinogen NNK
induces DNA methyltransferase 1 accumulation and tumor suppressor
gene hypermethylation in mice and lung cancer patients. J Clin
Invest. 120:521–532. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tennis MA, Vanscoyk MM, Wilson LA, Kelley
N and Winn RA: Methylation of Wnt7a is modulated by DNMT1 and
cigarette smoke condensate in non-small cell lung cancer. PLoS One.
7:e329212012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
McCabe MT and Creasy CL: EZH2 as a
potential target in cancer therapy. Epigenomics. 6:341–351. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yoon KA, Gil HJ, Han J, Park J and Lee JS:
Genetic polymorphisms in the polycomb group gene EZH2 and the risk
of lung cancer. J Thorac Oncol. 5:10–16. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhang H, Zhang H, Zhao M, Lv Z, Zhang X,
Qin X, Wang H, Wang S, Su J, Lv X, et al: miR-138 inhibits tumor
growth through repression of EZH2 in non-small cell lung cancer.
Cell Physiol Biochem. 31:56–65. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Xia H, Zhang W, Li Y, Guo N and Yu C: EZH2
silencing with RNA interference induces G2/M arrest in human lung
cancer cells in vitro. Biomed Res Int. 2014:3487282014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wang X, Zhao H, Lv L, Bao L, Wang X and
Han S: Prognostic significance of EZH2 expression in non-small cell
lung cancer: A meta-analysis. Sci Rep. 6:192392016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhang C, Ge S, Hu C, Yang N and Zhang J:
miRNA-218, a new regulator of HMGB1, suppresses cell migration and
invasion in non-small cell lung cancer. Acta Biochim Biophys Sin
(Shanghai). 45:1055–1061. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Shen X, Hong L, Sun H, Shi M and Song Y:
The expression of high-mobility group protein box 1 correlates with
the progression of non-small cell lung cancer. Oncol Rep.
22:535–539. 2009.PubMed/NCBI
|
|
47
|
Zheng W, Jiang C and Li R: Integrin and
gene network analysis reveals that ITGA5 and ITGB1 are prognostic
in non-small-cell lung cancer. Onco Targets Ther. 9:2317–2327.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Jono H and Ando Y: Midkine: A novel
prognostic biomarker for cancer. Cancers. 2:624–641. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Yuan K, Chen Z, Li W, Gao CE, Li G, Guo G,
Yang Y, Ai Y, Wu L and Zhang M: MDK protein overexpression
correlates with the malignant status and prognosis of non-small
cell lung cancer. Arch Med Res. 46:635–641. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hao H, Maeda Y, Fukazawa T, Yamatsuji T,
Takaoka M, Bao XH, Matsuoka J, Okui T, Shimo T, Takigawa N, et al:
Inhibition of the growth factor MDK/midkine by a novel small
molecule compound to treat non-small cell lung cancer. PLoS One.
8:e710932013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Regala RP, Davis RK, Kunz A, Khoor A,
Leitges M and Fields AP: Atypical protein kinase C{iota} is
required for bronchioalveolar stem cell expansion and lung
tumorigenesis. Cancer Res. 69:7603–7611. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Sutterlüty H, Mayer CE, Attems J, Setinek
U, Mikula M, Mikulits W, Micksche M and Berger W: Inhibition of
cell migration and proliferation in non-small cell lung cancer
(NSCLC) by Sprouty 2 (Spry2) via K-Ras dependent and independent
pathways. Cancer Res. 66 Suppl 8:S349–S350. 2006.
|
|
53
|
Sutterlüty H, Mayer CE, Setinek U, Attems
J, Ovtcharov S, Mikula M, Mikulits W, Micksche M and Berger W:
Down-regulation of Sprouty2 in non-small cell lung cancer
contributes to tumor malignancy via extracellular signal-regulated
kinase pathway-dependent and -independent mechanisms. Mol Cancer
Res. 5:509–520. 2007. View Article : Google Scholar : PubMed/NCBI
|