Introduction
Normally, solid tumors rely on blood vessels for
growth and metastasis (1) because
blood vessels can carry oxygen and nutrients to solid tumors and
provide a pathway for tumor cell metastasis. Thus, inhibition of
blood vessel formation should block the vascular supply and reduce
patient mortality and morbidity due to these tumors (1,2).
Therapeutic approaches to block the vascular supply have been used
in clinical practice; however, these agents may cause hypoxia,
which may fuel tumor progression and treatment resistance (2,3). To
address this paradox, Jain et al (1), Carretero et al (4) and Peterson et al (5) used the emerging antiangiogenic agents
to normalize abnormal tumor vasculature, resulting in the more
efficient delivery of drugs and oxygen to targeted cancer cells and
the normalization of vascular endothelial cell (EC) connections to
reduce tumor cell migration into blood vessels. However the
mechanism remains unclear.
Normal and diseased ECs are divided into specialized
subtypes, which is the basis for vascular germination. The distal
branches of blood vessels play a navigational role but rarely
proliferate, while trailing stalk cells proliferate to extend the
vascular buds (6). Few reports
have demonstrated the metabolic pathways of ECs in vitro,
and even fewer reports have studied how EC metabolism regulates
angiogenesis in vivo (6).
ECs reportedly rely on glycolysis (7,8). One
of the rate-limiting checkpoints of glycolytic flux is the
conversion of fructose-6-phosphate (F6P) to fructose-1,6
bisphosphate (F1,6P2) through 6-phosphofructo-1-kinase (PFK-1)
(9,10).
Phosphofructokinase-2/fructose-2,6-bisphosphatase (PFKFB3) enzymes
synthesize fructose-2,6-bisphosphate (F2,6P2), an allosteric
activator of PFK-1 and the most potent stimulator of glycolysis.
Mechanistically, PFKFB3 not only regulates EC proliferation but
also controls the formation of filamentous pseudopods and squamous
cells and directed migration (6,11,12).
Among all PFKFB isozymes, PFKFB3 has the highest kinase activity,
which facilitates the production of intracellular F2, 6P2 levels
and controls its abundance (6,9,12).
Cyclooxygenase (COX) is an important rate-limiting
enzyme in vivo; it is composed of at least two subtypes,
COX-1 and COX-2 (13). COX-1 is a
normally expressed subtype in the human body (i.e., in the
physiological catalysis of arachidonic acid into prostaglandins,
which are involved in a variety of important physiological
functions). COX-2 is not expressed in most cells; however, it can
be induced through a variety of pathological factors, catalyzed by
the membrane phospholipid conversion of arachidonic acid produced
by prostaglandins and induced through the cascade of amplified
inflammatory processes mediated by pathophysiological processes
(14). Studies have shown that
COX-2 can be induced through inflammation, injury, carcinogens,
tumors, and cytokines in a variety of manners to promote tumor
progression (13,15). COX inhibitors, also known as
non-steroidal anti-inflammatory drugs (NSAIDs), reduce the
production of inflammatory mediator prostaglandins, produce
anti-inflammatory effects, and demonstrate analgesic functions
(15). A US epidemiological survey
reported that long-term use of low-dose aspirin can reduce the risk
of ovarian cancer in women, suggesting that aspirin may prevent
ovarian cancer (16); however,
aspirin and other COX-1 inhibitors have a wide range of effects.
Parecoxib and other selective COX-2 inhibitors not only demonstrate
anti-inflammatory and analgesic functions but also prevent the
occurrence of tumors; they inhibit tumor development, metastasis
and tumor cell proliferation, promote apoptosis, inhibit
angiogenesis, and demonstrate significant antitumor effects with
few adverse reactions. In addition, the biosafety of these
inhibitors is greatly improved relative to traditional treatments
(17,18).
Both COX-1 and COX-2 are expressed in ECs (19). However, the effect of COX
inhibitors on EC metabolism and tumor vessel normalization (TVN),
have been rarely reported. This study aimed to confirm that the
inhibitor parecoxib regulates the metabolism of EC glucose through
COX-2-VEGF-PFKFB3 signaling pathways, thereby affecting tumor
growth.
Materials and methods
Mice
C57BL/6 mice were obtained from the Animal Institute
of the Academy of Medical Science (Beijing, China). The mice were
kept under specific pathogen-free conditions at the Animal Center
of the Xinqiao Hospital, Third Military Medical University
(Chongqing, China). All animal experiments were approved by the
Ethics Committee of the Third Military Medical University.
Cell isolation and culture
ID8 cells (mouse ovarian epithelial papillary serous
adenocarcinoma) were obtained from the American Type Culture
Collection (ATCC). The cells were cultured in DMEM medium (Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with
10% heat-inactivated fetal bovine serum (FBS; Gibco), 2 mM fresh
L-glutamine and 100 units/ml penicillin/streptomycin (HyClone; GE
Healthcare Life Sciences, Logan, UT, USA) and then maintained in an
incubator at 37°C with 5% CO2.
Mouse ECs were isolated from perfused healthy livers
or livers infested with ID8 tumors from mice and their wild-type
(WT) littermates. Livers were dissected and placed into a 50 ml
conical tube containing 5 ml of the digestion buffer. The organs
were incubated in a water bath at 37°C for approximately 60 min,
with regular shaking of the tubes every 10 min. At the end of the
digestion process, the tissue was homogeneously dissociated, and
the reaction was stopped by adding 10 ml of an isolation buffer
containing PBS + 0.1% EDTA. Subsequently, the cell suspension was
filtered through a 100 µm cell strainer (Corning Costar, Inc.,
Corning, NY, USA), and the cells were washed twice with the
isolation buffer. Finally, the endothelial cells were isolated by
magnetic bead sorting using Dynabeads (CELLection™ Biotin Binder
kit; Thermo Fisher Scientific, Inc.) coated with anti-mouse CD31
(Anti-Mouse CD31 Clone 390; eBioscience; Thermo Fisher Scientific,
Inc.), according to the manufacturer's procedure. The purity of the
cultured ECs was confirmed by immunofluorescence analysis, showing
selective expression of EC-enriched markers.
Tumor challenge experiments
The ID8 cells were cultured until they reached 70%
confluence. Then, WT mice (females, 6–8 weeks old) were
subcutaneously injected in the right lateral flank with
5×105 ID8 cells in 100 µl PBS. At the same time, the
COX-2 inhibitor group was intraperitoneally injected with 200 µl
(250 µg/ml) of parixibox, and the control group was injected with
200 µl PBS. Tumor sizes were measured using calipers every three
days until the mice were sacrificed. The tumor volume was
calculated using the following formula: volume=0.5 × (width) 2 ×
length.
Immunofluorescence
Immunofluorescence analysis was performed on
cell-paved µ-slides. The abovementioned cells were fixed with
ice-cold 4% paraformaldehyde for 20 min at 37°C, blocked with
normal serum for 20 min at room temperature and then incubated
overnight in the dark at 4°C with specific antibodies against mouse
monoclonal CD34 (ab 8158; Abcam, Cambridge, UK) and mouse
monoclonal factor VIII (9035; Invitrogen™; Thermo Fisher
Scientific, Inc.). After three washes, the slides were stained with
Cy3-conjugated anti-mouse antibodies or anti-rabbit antibodies
(1:200; Abcam). Nuclei were counterstained with
4,6-diamidino-2-phenylindole (DAPI), and the stained sections were
then visualized with a confocal microscope (Olympus Corp., Tokyo,
Japan).
Western blot analysis
Proteins of ECs were extracted by RIPA [(1% Triton
X-100, 0.5% Na-deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 20
mmol/l Tris-HCl (pH 7.4)] with 1% PMSF (Beyotime Institute of
Biotechnology, Haimen, China). Then BCA kit (Beyotime Institute of
Biotechnology) was used to examine concentrations of protein. Equal
amount of protein samples were resolved on a 10% SDS-PAGE gel and
transferred onto a polyvinylidene difluoride (PVDF) membrane. After
blocking with blocking buffer (P0023B; Beyotime Institute of
Biotechnology), the membrane was probed with designated primary
overnight at 4°C: Anti-COX2/Cyclooxygenase 2 antibody (ab62331;
Abcam) and mouse anti-β Actin antibody (ab8226; Abcam). After
washed with 0.1% TBST, goat anti-rabbit secondary antibody
(1:5,000; Beyotime Institute of Biotechnology) and goat anti-mouse
secondary antibody (1:5,000; Beyotime Biotechnology) were used
respectively, followed by detection of the chemiluminescent signal
(Pierce; Thermo Fisher Scientific, Inc.).
Quantitative polymerase chain reaction
(qPCR)
Quantitative PCR was performed to validate the
enriched genes observed in microarray experiments. The individual
gene expression was analyzed with a PCR kit (Takara, Tokyo, Japan),
and carried out in triplicate with an ABI 7300 Prism Sequence
Detection System (Applied Biosystems, Foster City, CA, USA). The
PCR conditions were as follows: 95°C for 30 sec, followed by 40
cycles of 95°C for 5 sec, 60°C for 34 sec, and 72°C for 45 sec. The
relative gene expression levels were calculated using the
comparative Ct (ΔΔCt) method, with GAPDH as a reference gene. The
primers used for PCR.
PFKFB3 forward, CAA CTC CCC AAC CGT GAT TGT and
reverse, GAG GTA GCG AGT CAG CTT CTT; COX-2 forward, CTG CCG TCC
GAT TGA GAC C and reverse, CCC CTC CTT GTA CCA CTG TC; GAPDH
forward, AGG TCG GTG TGA ACG GAT TTG and reverse, TGT AGA CCA TGT
AGT TGA GGT CA
Statistical analysis
Statistical analysis data are expressed as mean ±
SEM. Significance was assessed by two-tailed unpaired Student's
t-test or other statistical method indicated in the test.
P<0.05, P<0.01 and P<0.001 were considered to indicate a
statistically significant difference. Statistical calculations were
performed using GraphPad Prism 7.0 (GraphPad Software, Inc., La
Jolla, CA, USA). All of the experiments were independently repeated
at least 3 times.
Results
Parecoxib may inhibit tumor
progression by downregulating VEGF
Previous studies have demonstrated growing evidence
for the involvement of COX-2-derived mediators in angiogenic
processes. Further demonstrating this role, NSAIDs that selectively
block COX-2 activity have both anti-angiogenic and
anti-carcinogenic actions (20,21).
To investigate whether COX-2 affects the formation and progression
of tumors, we used a tumor prevention model to confirm the effect
of a COX-2 inhibitor. We subcutaneously injected ID8 cells into WT
mice and divided them into two groups. Compared to the control
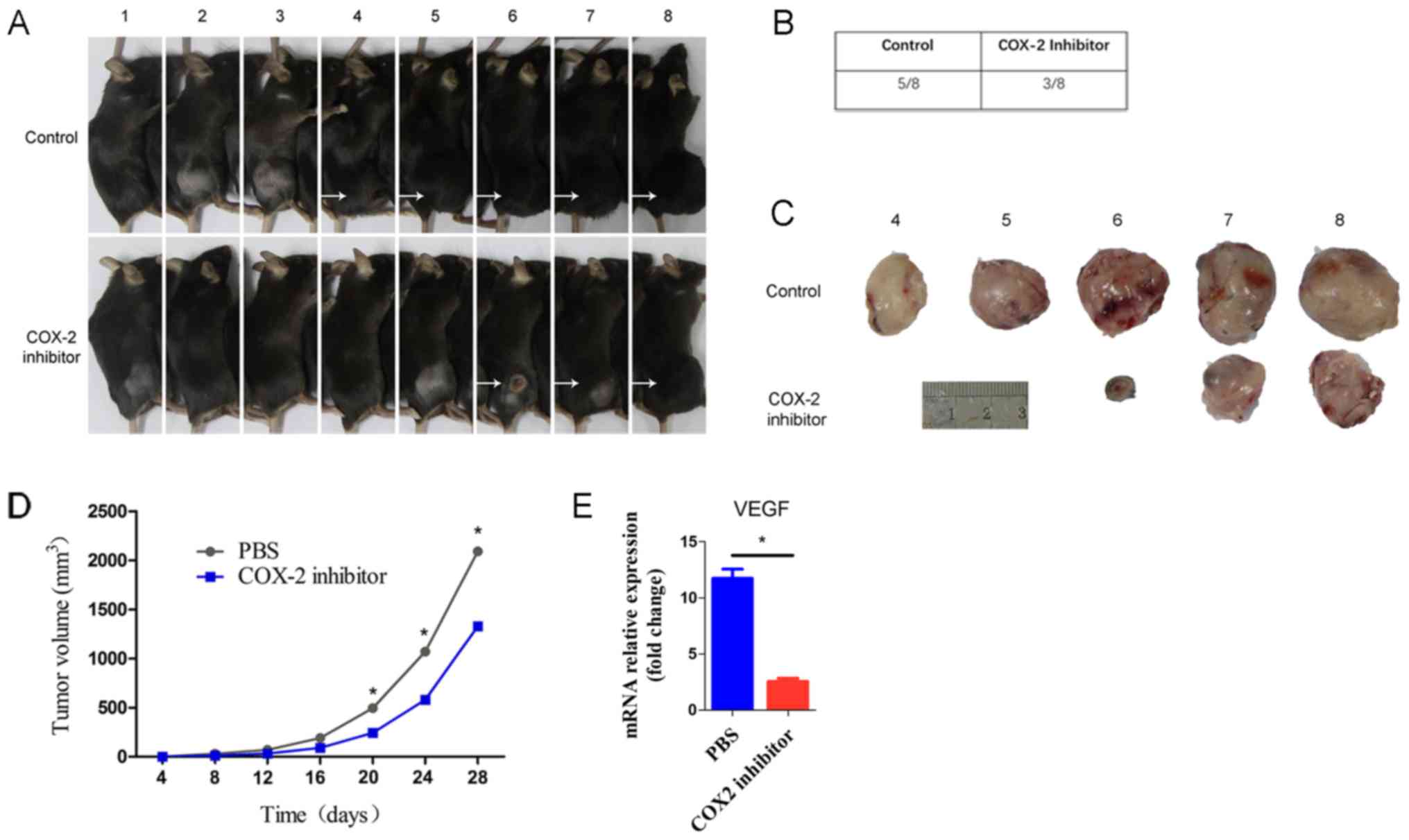
group, parecoxib completely inhibited tumor progression (Fig. 1A). The tumor incidence was 5/8 in
the control group and 3/8 in the COX-2 inhibitor group (Fig. 1B). As shown in the figure, the
volumes of excised tumors at termination (day 28) were markedly
smaller in the COX-2 inhibitor mice than in the control mice
(Fig. 1C). Additionally, the tumor
growth rate in the COX-2 inhibitor mice was slower than that in the
control mice (Fig. 1D). The above
data strongly suggest that parecoxib may inhibit tumor formation
and progression. Moreover, we found that the VEGF level in the
COX-2 inhibitor group was significantly lower than that in the
control group (Fig. 1E). The above
results are consistent with previously reported results (22,23).
These findings imply that the COX-2 inhibitor parecoxib regulates
VEGF expression in tumors, which then affects tumor progression.
Considering the relationship between VEGF and tumor angiogenesis,
we aimed to further clarify the regulation of tumor COX-2/VEGF on
tumor vascular ECs, particularly on glucose metabolism.
Expression of COX-2 in ovarian serous
cystadenocarcinoma (OV) correlates with VEGF and PFKFB3
expression
To gain more insight into the role of COX-2 in tumor
glucose metabolism, we analyzed RNA-seq expression data from The
Cancer Genome Atlas (TCGA; cancergenome.nih.gov). Pearson correlation analyses
were utilized to examine the correlations. The correlation analyses
of COX-2 (Symbol Report: PTGS2) with several genes related to
glucose metabolism are shown in Fig.
2. Most analyses demonstrated weak correlations; however, COX-2
and VEGF demonstrated a strong correlation (R=0.41) (Fig. 2A). The same result was observed
between VEGF and PFKFB3 (R=0.47, Fig.
2H). PFKFB3 acts as a glycolytic activator, and it is the
rate-limiting enzyme in glycolysis. The above results suggest that
the expression of COX-2 in tumors correlates with VEGF and PFKFB3
expression. However, these results are related to the entire tumor,
and the role in ECs is unknown.
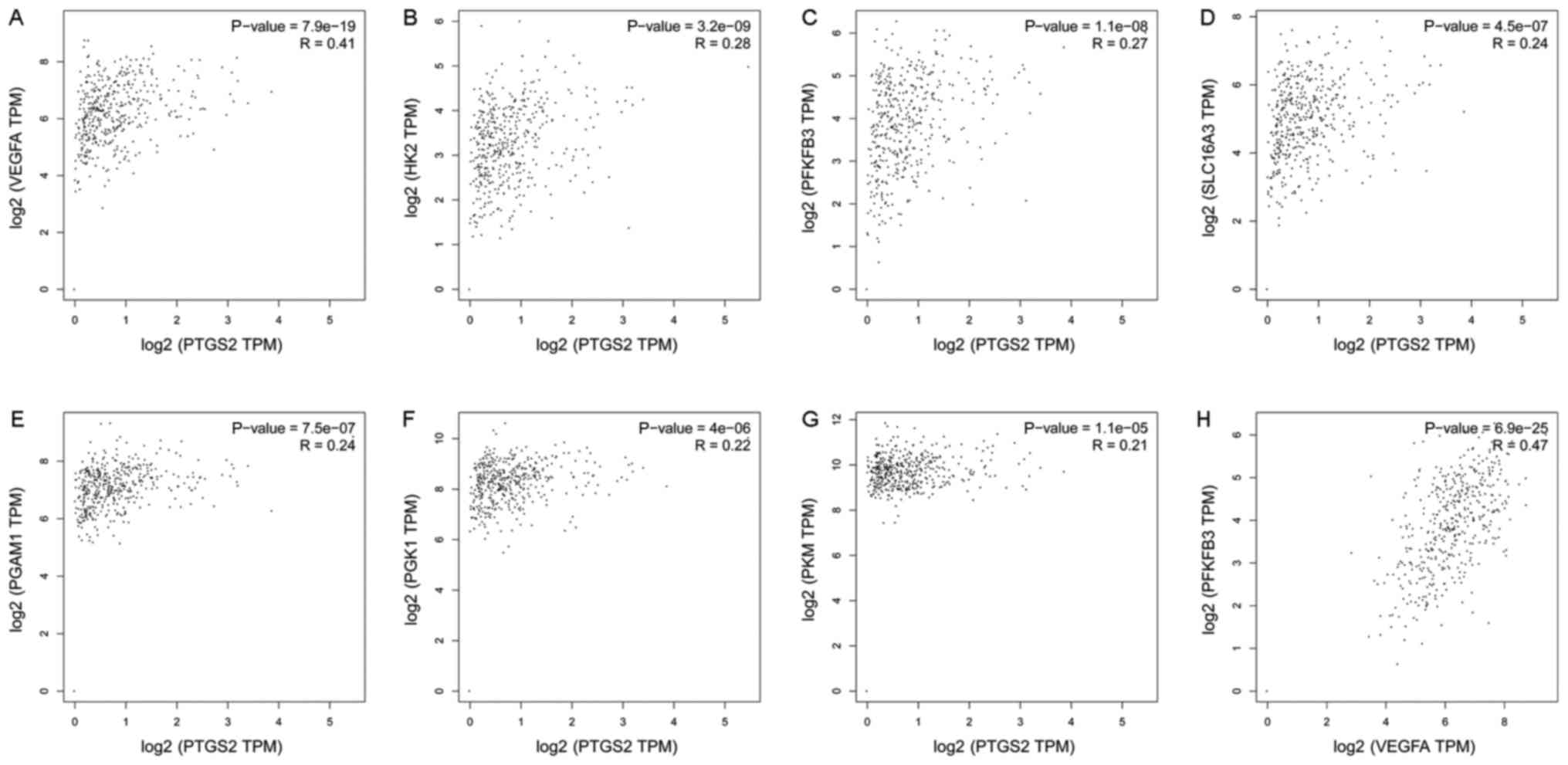 | Figure 2.Correlation analysis of glucose
metabolism-related genes. We analyzed RNA-seq expression data from
TCGA. Pearson's correlation analyses were utilized to examine the
correlations; we used the non-log scale for calculations and the
log-scale axis for visualization. The following correlations are
shown: (A) PTGS2 and VEGFA, (B) PTGS2 and HK2, (C) PTGS2 and
PFKFB3, (D) PTGS2 and SLC16A3, (E) PTGS2 and PGAM1, (F) PTGS2 and
PGK1, (G) PTGS2 and PKM, (H) VEGFA and PFKFB3. TCGA, The Cancer
Genome Atlas; VEGF, vascular endothelial growth factor; PFKFB3,
fructose-2,6-bisphosphatase. |
Characterization of glucose metabolism
in normal and tumor ECs
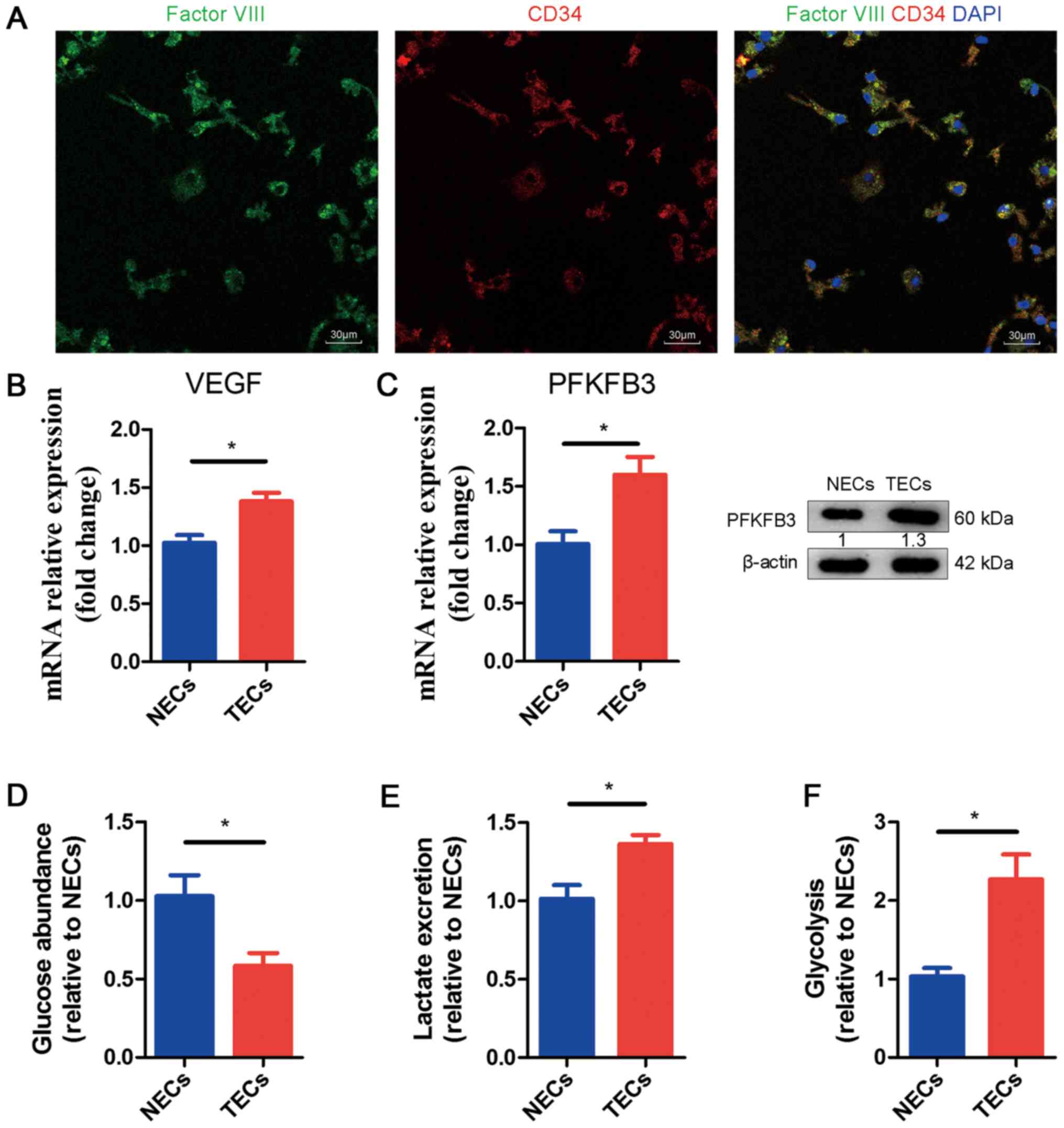
To further understand the mechanistic relationship
among COX-2, VEGF and PFKFB3 in ECs, we isolated tumor ECs (TECs)
and characterized their metabolic profiles. We injected ID8 cells
in the portal vein of WT mice to induce tumor growth in the liver.
After 15 days, we isolated ECs from the tumor-infested livers and
normal endothelial cells (NECs) from the livers of healthy mice (as
the control). First, we identified the TECs and NECs through
fluorescence. ECs can be stained to determine whether they are
factor VIII (green) positive and CD34 (red) positive; the merged
picture in Fig. 3A shows the
cytoplasm (yellow) and nucleus (blue). Subsequently, through
qRT-PCR, we examined the mRNA expression levels of VEGF and PFKFB3
in NECs and TECs. We noticed obviously increased mRNA expression in
TECs compared to NECs (Fig. 3B,
C). Meanwhile, we found that the protein levels of PFKFB3 were
upregulated (Fig. 3C). We
performed glycolysis experiments to determine whether the metabolic
activity was greater in TECs or NECs. Our results show that glucose
consumption and lactate excretion in the medium were increased in
TECs, which suggested hyperglycolysis (Fig. 3D, E). We also noticed an increased
glycolytic flux in TECs compared to NECs (Fig. 3F).
COX-2 inhibition induces glucose
metabolism normalization in TECs
Because glycolysis activity is greater in TECs than
NECs, COX-2, the glycolytic activator PFKFB3 and glycolysis may be
attractive targets in TECs. To investigate whether the TEC
phenotype could be directly regulated by COX-2, we isolated ECs
from the tumor-infested livers and NECs from the livers of healthy
mice (as controls); the TECs were incubated with parecoxib (10
µg/ml). We observed that VEGF and PFKFB3 mRNA expression was
obviously increased in TECs compared to NECs, while in TECs exposed
to parecoxib, the mRNA expression was obviously decreased compared
to that in the non-exposed TECs and NECs (Fig. 4A, B). In addition, the protein
levels of PFKFB3 were downregulated in the TEC+inhibitor group
compared to the TECs and NECs (Fig.
4B). To determine which group demonstrated greater glucose
metabolism, we also performed a glycolysis experiment. Similar to
the gene results, our results showed that the glucose consumption
and lactate excretion of the TECs were increased in the medium,
which suggested that TECs were hyperglycolytic, and the glycolysis
levels of the COX-2 inhibitor-induced TECs tended to be normal
(Fig. 4C, D).
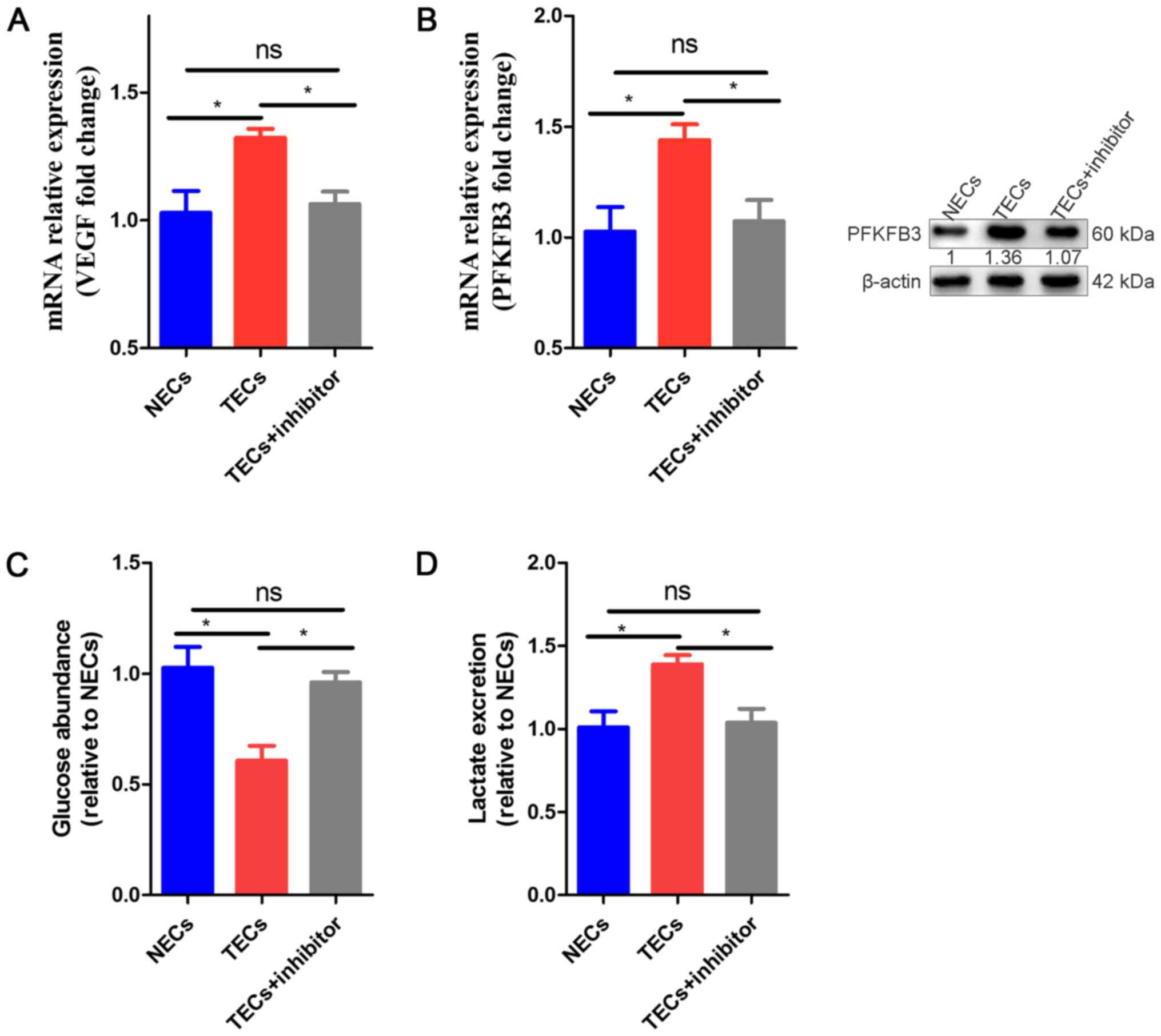 | Figure 4.Effect of COX-2 inhibition on TECS
Compared to NECs and TECs. (A) qRT-PCR analysis of mRNA expression
levels of VEGF in NECs, TECs and TECs + inhibitor. The values are
expressed relative to those of NECs (n=3). (B) qRT-PCR analysis of
mRNA expression and WB of protein expression levels of PFKFB3 in
NECs, TECs and TECs + inhibitor. The values are expressed relative
to those of NECs (n=3). (C) Glucose levels in the medium of TECs
and TECs + inhibitor, relative to those of NECs (n=3). (D) Lactate
levels in the medium of TECs and TECs + inhibitor, relative to
those of NECs (n=3). All data are shown as the means ± SEM.
*P<0.05. ECs, endothelial cells; TECs, tumor endothelial cells;
VEGF, vascular endothelial growth factor; PFKFB3,
fructose-2,6-bisphosphatase; NECs, normal endothelial cells. |
COX-2 inhibition regulates glycolysis
pathways in TECs
ECs rely on glycolysis instead of oxidative
metabolism for adenosine triphosphate (ATP) production (11,24).
The switch from a quiescent to an angiogenic phenotype, which
occurs in cancer, is metabolically demanding and mediated by
adaptations in EC metabolism. Lactate dehydrogenase B (LDH-B) is
upregulated in the tumor endothelium, and VEGF signaling increases
glycolytic flux by inducing GLUT1 and the glycolytic enzyme PFKFB3
(11,25). PFKFB3 catalyzes the synthesis of
fructose-2,6-bisphosphate (F2,6P2), which is an allosteric
activator of PFK-1. In our study, we used a COX-2 inhibitor to
treat TECs; it first inhibited the function of COX-2, resulting in
decreased VEGF production and PFKFB3 expression. The process of TEC
glycolysis is affected in this manner, and the products of
glycolysis and ATP are altered, resulting in a change in the
biological function of TECs (Fig.
5).
 | Figure 5.Metabolic pathways in TECs, and
parecoxib may regulate VEGF-PFKFB3 pathways. (A) TECs rely on
glycolysis instead of oxidative metabolism for ATP production and
upregulate PFKFB3 to increase the conversion of glucose into
lactate through glycolysis. However, VEGF induces PFKFB3 and
increases glycolytic flux, which is required for angiogenesis. (B)
Parecoxib inhibits COX-2 function, thereby reducing VEGF
production, which reduces PFKFB3 expression and, subsequently,
glycolytic flux. TECs, tumor endothelial cells; VEGF, vascular
endothelial growth factor; PFKFB3, fructose-2,6-bisphosphatase;
COX-2, cyclooxygenase-2; ATP, adenosine triphosphate. |
Discussion
ECs are arranged in the lumen of the blood vessel
and are, therefore, exposed to high concentrations of oxygen in the
blood. Thus, we suspected that these cells are better adapted to
oxidative metabolism. However, vascular germination is not
inhibited by mitochondrial respiration (6). In fact, ECs were found to be highly
glycolytic, resulting in up to an 85% increase in ATP production.
Even if glycolysis produces only two ATP molecules per molecule of
glucose, the glycolytic EC provides a wide variety of advantages
over the production of 34 molecules of ATP produced by the normal
oxidative metabolism of glucose (6). For example, anaerobic glucose
metabolism allows ECs to vascularize blood vessels without hypoxia.
In addition, glycolysis can produce more ATP molecules than
oxidants in a shorter time span than the normal metabolic pathway,
thereby rapidly providing ECs with the necessary energy to
germinate, form new blood vessels and rapidly evolve to restore the
oxygen supply of the surrounding tissue (26). In addition to the glycolytic
metabolites in the bypass pathway, the production of biomass
requires macromolecules and can also further regulate the function
of redox homeostasis, which is conducive to rapid vascular
germination.
VEGF increases the expression of PFKFB3 through
glycolysis. Reducing glycolysis by PFKFB3 gene inactivation in ECs
reduces the migration of tip cells and the proliferation of stem
cells to prevent vascular germination (6). In in vitro models, silencing
of PFKFB3 reduced EC movement, lamellipodia/filopodia formation and
directed migration and damaged EC spheroids (27). In addition, EC PFKFB3-deficient
mice showed impaired vascular growth and branch formation in the
neonatal retinal vasculature due to reduced stem cell proliferation
and damaged filamentous pseudopodia. Further analysis showed that
PFKFB3 was co-localized with F-actin and that other glycolytic
enzymes that had a related effect (12). Exposure to the glycolysis through
PFKFB3 overexpression boosts the tip cell phenotype during vascular
sprouting. The regulation of PFKFB3 levels (under- or
overexpression) did not alter the relative expression level of stem
cells, suggesting that the metabolic changes alone, particularly
PFKFB3-driven glycolysis, were sufficient to promote activity and
vascular germination.
EC dysfunction, which occurs when ECs are no longer
able to perform their physiological functions, may also contribute
to cardiovascular disease and diabetes (25). Thus, inhibition of pathological
vascular germination or prevention or reduction of EC dysfunction
is valuable for a wide variety of diseases. For the treatment of
overreaction, the most well-known and, to date, the only clinically
approved antiangiogenic strategy relies on growth factor signaling
(primarily VEGF signaling). This method is inadequate for cancer
treatment because of endogenous intolerance or resistance to the
secretion of angiogenic factors due to an increased compensatory
mechanism, which results in only limited benefits. The finding that
EC metabolism codetermines blood vessel formation may provide a
novel opportunity for antiangiogenic treatment based on an entirely
different mechanism. Indeed, PFKFB3 represents a target that
interferes with glycolysis and may counteract abnormal and
excessive formation of blood vessels in disease (28).
TVN is emerging as an anti-cancer treatment.
Previous TVN strategies have focused on targeting angiogenic
signals; however, targeting EC glucose metabolism as a method of
promoting TVN is still rare. Our data showed that COX-2 inhibition
reduces TEC glycolysis as follows: (1) TECs are hyperglycolytic, and
pharmacological blockade of COX-2 in TECs induces glycolysis
normalization. (2) In vivo,
the pharmacological blockade of COX-2 impairs tumor
progression.
COX-2 expression is associated with VEGF expression
in a variety of malignant tumors. Overexpression of COX-2 in tumor
tissue can promote angiogenesis, and VEGF may play a mediating
role. In Hodgkin's lymphoma, COX-2 overexpression was found to be
associated with angiogenesis, which induced Bcl-2 and VEGF
expression. COX-2 induces VEGF expression in the following manner:
(1) PGE2 upregulates the
expression of VEGF in gastric cancer cells by transactivating the
EGFR-MAPK signaling pathway, which is one of the basic mechanisms
of COX-2 that promotes tumor angiogenesis (13); (2)
COX-2 promotes tumor angiogenesis through the PGE2/HIF-1α/VEGF
signaling pathway (14); (3) COX-2, when overexpressed in
vivo, can activate the epidermal growth factor receptor (EGFR)
and increase the production of cyclic adenosine monophosphate
(cAMP) by producing PGE2 and activating the cAMP response element
binding protein (CREB).
COX inhibitors have become an important target for
the treatment of malignant tumors in recent years, and they have
been shown to have a significant inhibitory effect on some
malignant biological behaviors of tumor cells. Masferrer and others
have found that COX inhibitors can reduce the expression of
angiogenic factors and the downstream biological effects of COX-2.
Our results showed that pharmacological blockade of COX-2 in TECs
can reduce VEGF expression in the tumor microenvironment, and VEGF
can further influence the expression of PFKFB3, a key enzyme gene
of glycolysis. Pharmacological blockade of COX-2 in TECs restored
glycolysis in tumor vascular ECs, which may be an important basis
for the normalization of tumor blood vessels. COX-2 inhibitors have
been shown to play an important role in the treatment of a variety
of tumors as well as vascular abnormalities. Based on our findings,
we have reason to believe that COX-2 and TVN may become new targets
for tumor prevention and treatment.
References
|
1
|
Jain RK: Normalization of tumor
vasculature: An emerging concept in antiangiogenic therapy.
Science. 307:58–62. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Potente M, Gerhardt H and Carmeliet P:
Basic and therapeutic aspects of angiogenesis. Cell. 146:873–887.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jain RK: Antiangiogenesis strategies
revisited: From starving tumors to alleviating hypoxia. Cancer
Cell. 26:605–622. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Carretero R, Sektioglu IM, Garbi N,
Salgado OC, Beckhove P and Hämmerling GJ: Eosinophils orchestrate
cancer rejection by normalizing tumor vessels and enhancing
infiltration of CD8(+) T cells. Nat Immunol. 16:609–617. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Peterson TE, Kirkpatrick ND, Huang Y,
Farrar CT, Marijt KA, Kloepper J, Datta M, Amoozgar Z, Seano G,
Jung K, et al: Dual inhibition of Ang-2 and VEGF receptors
normalizes tumor vasculature and prolongs survival in glioblastoma
by altering macrophages. Proc Natl Acad Sci USA. 113:pp. 4470–4475.
2016; View Article : Google Scholar : PubMed/NCBI
|
|
6
|
De Bock K, Georgiadou M, Schoors S,
Kuchnio A, Wong BW, Cantelmo AR, Quaegebeur A, Ghesquière B,
Cauwenberghs S, Eelen G, et al: Role of PFKFB3-driven glycolysis in
vessel sprouting. Cell. 154:651–663. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Polet F and Feron O: Endothelial cell
metabolism and tumour angiogenesis: Glucose and glutamine as
essential fuels and lactate as the driving force. J Intern Med.
273:156–165. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cruys B, Wong BW, Kuchnio A, Verdegem D,
Cantelmo AR, Conradi LC, Vandekeere S, Bouché A, Cornelissen I,
Vinckier S, et al: Glycolytic regulation of cell rearrangement in
angiogenesis. Nat Commun. 7:122402016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xu Y, An X, Guo X, Habtetsion TG, Wang Y,
Xu X, Kandala S, Li Q, Li H, Zhang C, et al: Endothelial PFKFB3
plays a critical role in angiogenesis. Arterioscler Thromb Vasc
Biol. 34:1231–1239. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cantelmo AR, Conradi LC, Brajic A, Goveia
J, Kalucka J, Pircher A, Chaturvedi P, Hol J, Thienpont B, Teuwen
LA, et al: Inhibition of the glycolytic activator PFKFB3 in
endothelium induces tumor vessel normalization, impairs metastasis
and improves chemotherapy. Cancer Cell. 30:968–985. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
De Bock K, Georgiadou M and Carmeliet P:
Role of endothelial cell metabolism in vessel sprouting. Cell
Metab. 18:634–647. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Schoors S, De Bock K, Cantelmo AR,
Georgiadou M, Ghesquière B, Cauwenberghs S, Kuchnio A, Wong BW,
Quaegebeur A, Goveia J, et al: Partial and transient reduction of
glycolysis by PFKFB3 blockade reduces pathological angiogenesis.
Cell Metab. 19:37–48. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Di Francesco L, Dovizio M, Trenti A,
Marcantoni E, Moore A, O'Gaora P, McCarthy C, Tacconelli S, Bruno
A, Alberti S, et al: Dysregulated post-transcriptional control of
COX-2 gene expression in gestational diabetic endothelial cells. Br
J Pharmacol. Jul 3–2015.(Epub ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Leahy KM, Ornberg RL, Wang Y, Zweifel BS,
Koki AT and Masferrer JL: Cyclooxygenase-2 inhibition by celecoxib
reduces proliferation and induces apoptosis in angiogenic
endothelial cells in vivo. Cancer Res. 62:625–631. 2002.PubMed/NCBI
|
|
15
|
Sui W, Zhang Y, Wang Z, Wang Z, Jia Q, Wu
L and Zhang W: Antitumor effect of a selective COX-2 inhibitor,
celecoxib, may be attributed to angiogenesis inhibition through
modulating the PTEN/PI3K/Akt/HIF-1 pathway in an H22 murine
hepatocarcinoma model. Oncol Rep. 31:2252–2260. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gaul C: Aspirin for migraine in pregnancy.
This recommendation seems questionable. MMW Fortschr Med.
155:172013.(In German). View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Salvado MD, Alfranca A, Haeggström JZ and
Redondo JM: Prostanoids in tumor angiogenesis: Therapeutic
intervention beyond COX-2. Trends Mol Med. 18:233–243. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bijman MN, Hermelink CA, van Berkel MP,
Laan AC, Janmaat ML, Peters GJ and Boven E: Interaction between
celecoxib and docetaxel or cisplatin in human cell lines of ovarian
cancer and colon cancer is independent of COX-2 expression levels.
Biochem Pharmacol. 75:427–437. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Di Francesco L, Totani L, Dovizio M,
Piccoli A, Di Francesco A, Salvatore T, Pandolfi A, Evangelista V,
Dercho RA, Seta F and Patrignani P: Induction of prostacyclin by
steady laminar shear stress suppresses tumor necrosis factor-alpha
biosynthesis via heme oxygenase-1 in human endothelial cells. Circ
Res. 104:506–513. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
de Groot DJ, de Vries EG, Groen HJ and de
Jong S: Non-steroidal anti-inflammatory drugs to potentiate
chemotherapy effects: From lab to clinic. Crit Rev Oncol Hematol.
61:52–69. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Burleigh ME, Babaev VR, Yancey PG, Major
AS, McCaleb JL, Oates JA, Morrow JD, Fazio S and Linton MF:
Cyclooxygenase-2 promotes early atherosclerotic lesion formation in
ApoE-deficient and C57BL/6 mice. J Mol Cell Cardiol. 39:443–452.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yoshida S, Amano H, Hayashi I, Kitasato H,
Kamata M, Inukai M, Yoshimura H and Majima M: COX-2/VEGF-dependent
facilitation of tumor-associated angiogenesis and tumor growth in
vivo. Lab Invest. 83:1385–1394. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wu G, Luo J, Rana JS, Laham R, Sellke FW
and Li J: Involvement of COX-2 in VEGF-induced angiogenesis via P38
and JNK pathways in vascular endothelial cells. Cardiovasc Res.
69:512–519. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Vandekeere S, Dewerchin M and Carmeliet P:
Angiogenesis revisited: An overlooked role of endothelial cell
metabolism in vessel sprouting. Microcirculation. 22:509–517. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Goveia J, Stapor P and Carmeliet P:
Principles of targeting endothelial cell metabolism to treat
angiogenesis and endothelial cell dysfunction in disease. EMBO Mol
Med. 6:1105–1120. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Eelen G, de Zeeuw P, Simons M and
Carmeliet P: Endothelial cell metabolism in normal and diseased
vasculature. Circ Res. 116:1231–1244. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sun CM, Xiong DB, Yan Y, Geng J, Liu M and
Yao XD: Genetic alteration in phosphofructokinase family promotes
growth of muscle-invasive bladder cancer. Int J Biol Markers.
31:e286–e293. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Schoors S, Bruning U, Missiaen R, Queiroz
KC, Borgers G, Elia I, Zecchin A, Cantelmo AR, Christen S, Goveia
J, et al: Fatty acid carbon is essential for dNTP synthesis in
endothelial cells. Nature. 520:192–197. 2015. View Article : Google Scholar : PubMed/NCBI
|