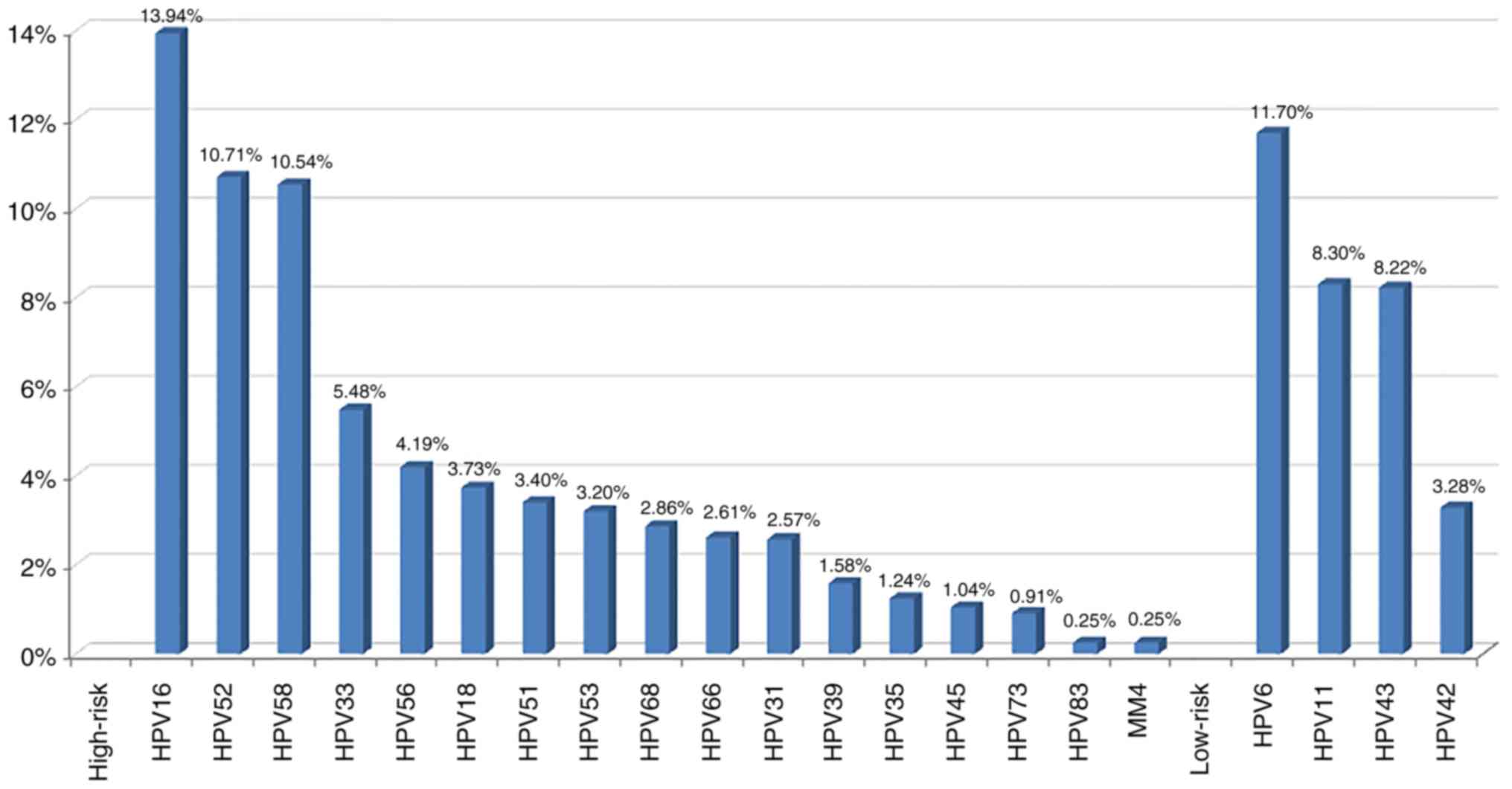
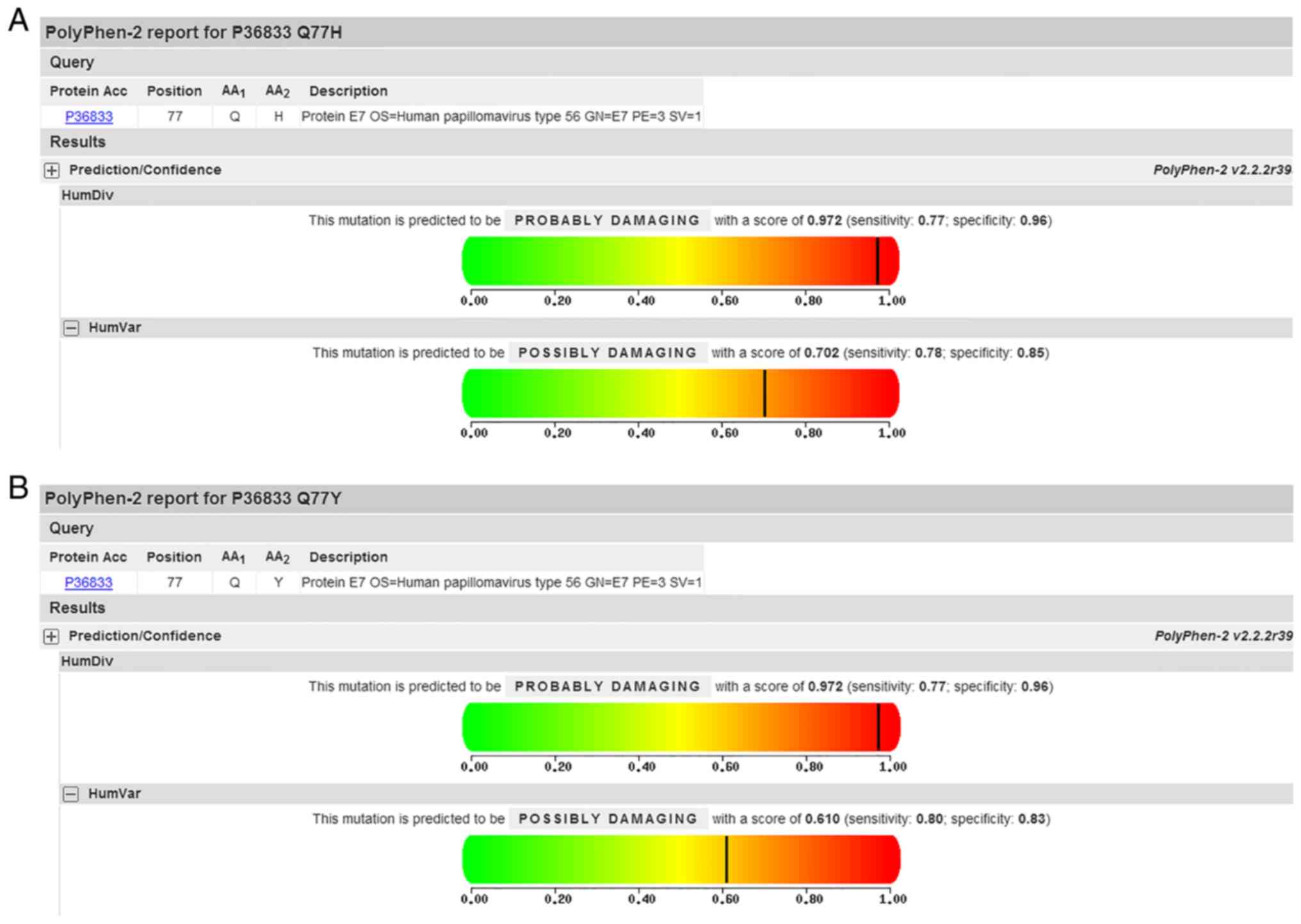
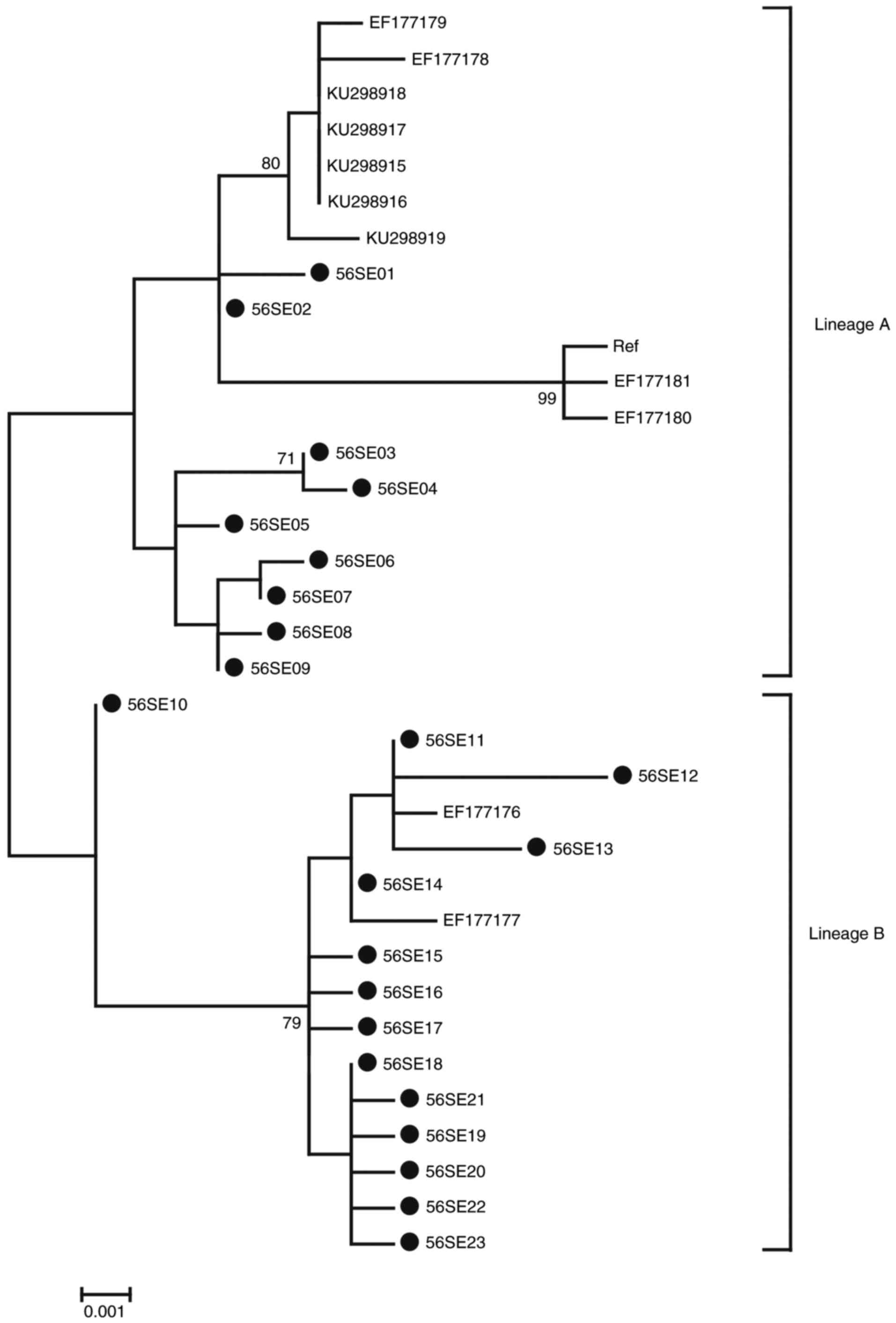
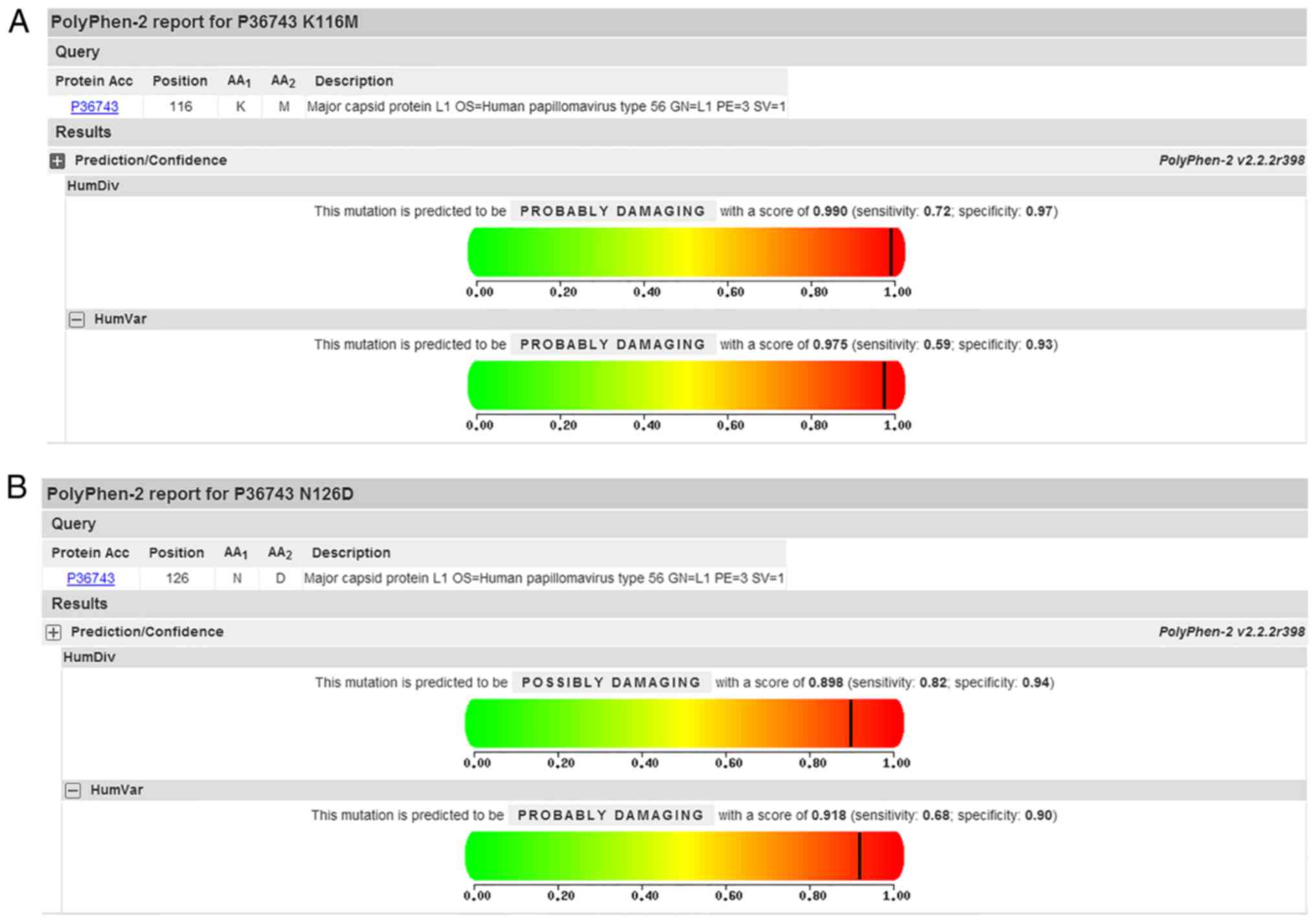
|
1
|
de Sanjose S, Quint WG, Alemany L, Geraets
DT, Klaustermeier JE, Lloveras B, Tous S, Felix A, Bravo LE, Shin
HR, et al: Human papillomavirus genotype attribution in invasive
cervical cancer: A retrospective cross-sectional worldwide study.
Lancet Oncol. 11:1048–1056. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ferlay J SI, Ervik M, Dikshit R, Eser S,
Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: GLOBOCAN 2012
v1.0Cancer Incidence and Mortality Worldwide: IARC CancerBase. No.
11. IARC; Lyon: 2013, http://publications.iarc.fr/Databases/Iarc-Cancerbases/Globocan-2012-Estimated-Cancer-Incidence-Mortality-And-Prevalence-Worldwide-In-2012-V1-0-2012May
30–2012
|
|
3
|
Rampias T, Sasaki C and Psyrri A:
Molecular mechanisms of HPV induced carcinogenesis in head and
neck. Oral Oncol. 50:356–363. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bernard HU, Burk RD, Chen Z, van Doorslaer
K, zur Hausen H and de Villiers EM: Classification of
papillomaviruses (PVs) based on 189 PV types and proposal of
taxonomic amendments. Virology. 401:70–79. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bouvard V, Baan R, Straif K, Grosse Y,
Secretan B, El Ghissassi F, Benbrahim-Tallaa L, Guha N, Freeman C,
Galichet L, et al: A review of human carcinogens-Part B: Biological
agents. Lancet Oncol. 10:321–322. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Arbyn M, Tommasino M, Depuydt C and
Dillner J: Are 20 human papillomavirus types causing cervical
cancer? J Pathol. 234:431–435. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lörincz AT, Quinn AP, Goldsborough MD,
McAllister P and Temple GF: Human papillomavirus type 56: A new
virus detected in cervical cancers. J Gen Virol. 70:3099–3104.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhai L and Tumban E: Gardasil-9: A global
survey of projected efficacy. Antiviral Res. 130:101–109. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yang L, Yang H, Wu K, Shi X, Ma S and Sun
Q: Prevalence of HPV and variation of HPV 16/HPV 18 E6/E7 genes in
cervical cancer in women in South West China. J Med Virol.
86:1926–1936. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zeng Z, Yang H, Li Z, He X, Griffith CC,
Chen X, Guo X, Zheng B, Wu S and Zhao C: Prevalence and genotype
distribution of HPV infection in China: Analysis of 51,345 HPV
genotyping results from China's largest CAP certified laboratory. J
Cancer. 7:1037–1043. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang R, Guo XL, Wisman GB, Schuuring E,
Wang WF, Zeng ZY, Zhu H and Wu SW: Nationwide prevalence of human
papillomavirus infection and viral genotype distribution in 37
cities in China. BMC Infect Dis. 15:2572015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen Z, Wang Q, Ding X, Li Q, Zhong R and
Ren H: Characteristics of HPV prevalence in Sichuan Province,
China. Int J Gynecol Obstet. 131:277–280. 2015. View Article : Google Scholar
|
|
13
|
Lee K, Magalhaes I, Clavel C, Briolat J,
Birembaut P, Tommasino M and Zehbe I: Human papillomavirus 16 E6,
L1, L2 and E2 gene variants in cervical lesion progression. Virus
Res. 131:106–110. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cento V, Rahmatalla N, Ciccozzi M, Perno
CF and Ciotti M: Intratype variations of HPV 31 and 58 in Italian
women with abnormal cervical cytology. J Med Virol. 83:1752–1761.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xi LF, Schiffman M, Koutsky LA, Hughes JP,
Hulbert A, Shen Z, Galloway DA and Kiviat NB: Variant-specific
persistence of infections with human papillomavirus Types 31, 33,
45, 56 and 58 and risk of cervical intraepithelial neoplasia. Int J
Cancer. 139:1098–1105. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hubert WG: Variant upstream regulatory
region sequences differentially regulate human papillomavirus type
16 DNA replication throughout the viral life cycle. J Virol.
79:5914–5922. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Vande Pol SB and Klingelhutz AJ:
Papillomavirus E6 oncoproteins. Virology. 445:115–137. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Roman A and Munger K: The papillomavirus
E7 proteins. Virology. 445:138–168. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Buck CB, Day PM and Trus BL: The
papillomavirus major capsid protein L1. Virology. 445:169–174.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chan PK, Luk AC, Park JS, Smith-McCune KK,
Palefsky JM, Konno R, Giovannelli L, Coutlée F, Hibbitts S, Chu TY,
et al: Identification of human papillomavirus type 58 lineages and
the distribution worldwide. J Infect Dis. 203:1565–1573. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sun Z, Lu Z, Liu J, Wang G, Zhou W, Yang
L, Liu C and Ruan Q: Genomic polymorphism of human papillomavirus
type 52 in women from Northeast China. Int J Mol Sci.
13:14962–14972. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hang D, Yin Y, Han J, Jiang J, Ma H, Xie
S, Feng X, Zhang K, Hu Z, Shen H, et al: Analysis of human
papillomavirus 16 variants and risk for cervical cancer in Chinese
population. Virology. 488:156–161. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shen M, Ding X, Li T, Chen G and Zhou X:
Sequence variation analysis of HPV-18 Isolates in Southwest China.
PLoS One. 8:e566142013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Prado JC, Calleja-Macias IE, Bernard HU,
Kalantari M, Macay SA, Allan B, Williamson AL, Chung LP, Collins
RJ, Zuna RE, et al: Worldwide genomic diversity of the human
papillomaviruses-53, 56, and 66, a group of high-risk HPVs
unrelated to HPV-16 and HPV-18. Virology. 340:95–104. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wyant PS, Cerqueira DM, Moraes DS, Leite
JP, Martins CR, de Macedo Brígido M and Raiol T: Phylogeny and
polymorphism in the long control region, E6, and L1 of human
papillomavirus types 53, 56, and 66 in central Brazil. Int J
Gynecol Cancer. 21:222–229. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Clarke KR and Gorley RN: Primer v5: User
Manual\TutorialPRIMER-E. Plymouth: pp. 912001
|
|
27
|
Tamura K, Stecher G, Peterson D, Filipski
A and Kumar S: MEGA6: Molecular evolutionary genetics analysis
version 6.0. Mol Biol Evol. 30:2725–2729. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Buchan DW, Minneci F, Nugent TC, Bryson K
and Jones DT: Scalable web services for the PSIPRED protein
analysis workbench. Nucleic Acids Res. 41:W349–W357. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kel AE, Gössling E, Reuter I, Cheremushkin
E, Kel-Margoulis OV and Wingender E: MATCHTM: A tool for searching
transcription factor binding sites in DNA sequences. Nucleic Acids
Res. 31:3576–3579. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Nei M and Gojoborit T: Simple methods for
estimating the numbers of synonymous and nonsynonymous nucleotide
substitutions. Mol Biol Evol. 3:418–426. 1986.PubMed/NCBI
|
|
32
|
Burk RD, Harari A and Chen Z: Human
papillomavirus genome variants. Virology. 445:232–243. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Schiller JT and Lowy DR: Understanding and
learning from the success of prophylactic human papillomavirus
vaccines. Nat Rev Microbiol. 10:681–692. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pista A, Oliveira A, Barateiro A, Costa H,
Verdasca N and Paixao MT: Molecular variants of human
papillomavirus type 16 and 18 and risk for cervical neoplasia in
Portugal. J Med Virol. 79:1889–1897. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bernard HU: Regulatory elements in the
viral genome. Virology. 445:197–204. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Moody CA and Laimins LA: Human
papillomavirus oncoproteins: Pathways to transformation. Nat Rev
Cancer. 10:550–560. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Thomas M, Narayan N, Pim D, Tomaić V,
Massimi P, Nagasaka K, Kranjec C, Gammoh N and Banks L: Human
papillomaviruses, cervical cancer and cell polarity. Oncogene.
27:7018–7030. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chagas BS, Batista MV, Crovella S, Gurgel
AP, Silva Neto Jda C, Serra IG, Amaral CM, Balbino VQ, Muniz MT and
Freitas AC: Novel E6 and E7 oncogenes variants of human
papillomavirus type 31 in Brazilian women with abnormal cervical
cytology. Infect Genet Evol. 16:13–18. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yao Y, Huang W, Yang X, Sun W, Liu X, Cun
W and Ma Y: HPV-16 E6 and E7 protein T cell epitopes prediction
analysis based on distributions of HLA-A loci across populations:
An in silico approach. Vaccine. 31:2289–2294. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chan PK, Zhang C, Park JS, Smith-McCune
KK, Palefsky JM, Giovannelli L, Coutlée F, Hibbitts S, Konno R,
Settheetham-Ishida W, et al: Geographical distribution and
oncogenic risk association of human papillomavirus type 58 E6 and
E7 sequence variations. Int J Cancer. 132:2528–2536. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Fleury MJ, Touze A and Coursaget P: Human
papillomavirus type 16 pseudovirions with few point mutations in L1
major capsid protein FG loop could escape actual or future
vaccination for potential use in gene therapy. Mol Biotechnol.
56:479–486. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sapp M and Day PM: Structure, attachment
and entry of polyoma- and papillomaviruses. Virology. 384:400–409.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Pande S, Jain N, Prusty BK, Bhambhani S,
Gupta S, Sharma R, Batra S and Das BC: Human papillomavirus type 16
variant analysis of E6, E7, and L1 genes and long control region in
biopsy samples from cervical cancer patients in north India. J Clin
Microbiol. 46:1060–1066. 2008. View Article : Google Scholar : PubMed/NCBI
|