Introduction
Arteriosclerotic cardiovascular disease is
life-threatening and has a high mortality rate in China (1). Atherosclerotic plaque erosion and
plaque rupture are the primary pathologies associated with acute
coronary syndrome, which may lead to formation of complete or
incomplete occlusive thrombi and myocardial ischemia (2,3). In
the treatment of myocardial ischemia, revascularization is
necessary to restore the blood flow (4,5).
However, following blood supply restoration, ischemic tissues
produce excess free radicals which cause further severe injury to
the ischemic tissues known as ischemia/reperfusion (I/R) injury
(6,7).
Statins down-regulate low density lipoprotein (LDL)
receptors by inhibiting 3-hydroxy-3-methyl-glutaryl-CoA reductase
and reduce cholesterol synthesis in hepatocytes (8,9).
Previous studies have indicated that statins can also serve a
variety of physiological functions, including improving endothelial
function, reducing inflammation and delaying hardening of the
arteries (10,11). Statins are also involved in the
synthesis of extracellular matrix proteins (12) It has also been indicated that
rosuvastatin (RS) stabilizes arterial plaques and exhibits a
stronger lipid-lowering capacity compared with other statins
(13,14). However, the function and mechanism
of action of RS in myocardial ischemia/reperfusion (MI/R) remain to
be elucidated.
Peroxisome proliferator-activated receptor-γ
(PPAR-γ) is a ligand-inducible transcription factor which can
regulate a number of biological processes associated with the
cardiovascular system (15).
PPAR-γ belongs to nuclear receptor superfamily which is primarily
expressed in adipose tissues (16). Previous studies have also indicated
that PPAR-γ agonists reduce inflammation and myocardial injury
caused by MI/R (17,18). Therefore, PPAR-γ is a novel
treatment target used to prevent heart disease complications
including heart failure (19).
However, the mechanism of PPAR-γ in MI/R needs to be extensively
studied.
Uncoupling proteins (UCPs) are located in the inner
mitochondrial membrane and act as anion carrier proteins (20). Previous studies have demonstrated
that activation of UCP3 could reduce ATP synthesis by uncoupling
oxidative phosphorylation, lowering proton gradient, decreasing ROS
generation, transporting fatty acid anions and reducing
peroxide-associated damage (21,22).
It has been reported that UCP3 is able to prevent mitochondrial
injury (23). It has also been
demonstrated that the expression levels of UCP1 increased in IR
myocardium and served a critical role in both cardioprotection
against MI/R injury and induction of ischemic preconditioning
(24). Recently, mitochondrial
uncoupling protein 2 (UCP2) has been reported to serve a role in
cardiac hypertrophy and myocardial injury (25,26).
Nevertheless, the precise role and mechanism of UCP2 in
cardioprotection remain unclear.
PPAR subtypes have been implicated in
transcriptional regulation of UCP, and PPARs are expressed in many
organs, such as the heart and pancreas (27). It has been demonstrated that PPAR-γ
could regulate the transcription of UCP2 in INS-1E cells (28). Based on the aforementioned data,
the authors of the present study hypothesized that PPAR-γ and UCP2
may be associated with cardioprotection and MI/R.
Therefore, the present study investigated the effect
of RS on oxidative stress in an in vivo model of MI/R. The
effects of RS and atractyloside (ATR) on myocardial infarct size
were also studied. It was further detected whether RS affected
cardiomyocyte viability, LDH activity and ROS content in
vitro following oxygen-glucose deprivation/reperfusion (OGD/R)
damage. Furthermore, the possible involvement of caspase-9,
cytochrome c (cyt c), PPAR-γ and UCP2 in the MI/R injury was also
verified.
Materials and methods
Ischemia-reperfusion (MI/R) model
A total of 48 healthy adult male New Zealand white
rabbits (weight, ~4.0–5.0 kg; age, 6 months) were purchased from
Guangdong Medical Laboratory Animal Center (Foshan, China). Rabbits
were randomly divided into 4 groups, with 12 rabbits in each group.
Rabbits had free access to food and water and were housed at 20°C
with 60–70% humidity and a 12 h light/dark cycle. Animals were
fasted 12 h prior to surgery. All experimental animals used in the
present study received ethical approval for experimental research.
The project protocol was approved by the Institutional Review Board
of Fujian Province Medical Association. Prior to surgery,
anesthesia was induced by intramuscular injection of ketamine
(25–40 mg/kg) and acepromazine (1–2 mg/kg). In the sham group, a
suture was placed around the coronary artery, without induction of
MI/R. In the MI/R group, rabbits were subjected to ischemia for 30
min followed by 120 min of reperfusion. In the RS group, RS
(CRESTOR®; AstraZeneca, Cambridge, UK) was administrated
once at a dose of 5 mg/kg 12 h prior to MI/R. In the RS + ATR
group, RS was administrated once at a dose of 5 mg/kg 12 h prior to
MI/R, and ATR (Chengdu Herbpurify Co., Ltd., Chengdu, China) was
administered at a dose of 5 mg/kg 30 sec prior to reperfusion. To
establish a MI/R model, during the experiment, left anterior
descending arteries were obstructed for 40 min in the MI/R group
rabbits, but not in the sham group rabbits. Left anterior
descending arteries were untied 40 min later and reperfused.
TCC and Evans blue staining
As previously described (29), TTC/Evans blue (Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany) staining was used to measure the
cardiomyocyte risk area. At the end of the myocardial I/R
protocols, evans blue (3%; 0.5 ml) was injected into the vena cava
at room temperature in order to detect the area-at-risk (AAR). When
the remaining blood had been washed out, the right ventricle was
trimmed away and the left ventricle was cut into 2-mm-thick slices.
The slices were subsequently stained with 2% TTC at 37°C for 15 min
in order to measure the area of necrosis (AN). The AN area was
identified by the non-staining region, whereas the live area was
stained red. The cardiac injury was presented as AN/AAR.
Establishment of cardiomyocyte MI/R
model via OGD/R injury
The MI/R cell model was established on the basis of
previous investigations (30–33).
SD rats (8–12 weeks; male:female, 1:4; n=15) were obtained from
Guangdong Medical Laboratory Animal Center. The animals had free
access to food and water and were housed at 25°C, with 45–65%
humidity and a 12 h light/dark cycle. The animals were mated to
produce the neonatal rats, as described previously. Six 1–3 day-old
neonatal SD rats were used to isolate cardiomyocytes, as previously
described (32). In brief, the
collected hearts were minced into pieces of ~1 mm3.
Minced tissue was resuspended in dissociation buffer (60 mg trypsin
and 40 mg collagenase type II in 100 ml H2O2)
and incubated in preheated tissue processing unit (InGeneron Inc.,
Houston, TX, USA) for 30 min at 37°C. The dissociation enzyme
activity was inhibited by incubation with cold horse serum
(Sigma-Aldrich; Merck KGaA) at 37°C for 5 min. Fresh dissociation
buffer was added to the remaining tissue samples. The above steps
were repeated until tissue fragments were completely dissolved.
Cell suspensions were collected and centrifuged for 10 min at 350 ×
g at 4°C. Finally, cell pellets were resuspended in cold 1× ADS
solution (6.8 g NaCl, 4.76 g HEPES, 0.138 g Na2HPO4, 0.6
g glucose, 0.4 g KCl and 0.051 g MgSO4-7H2O
in 1000 ml ultrapure water, pH: 7.35–7.45). To establish an in
vitro model of MI/R, the neonatal rat cardiomyocytes were
treated with oxygen-glucose deprivation for 6 h followed by
recovery for 1 h. Neonatal rat cardiomyocytes were randomly
assigned to 4 groups. In the control group, cells were cultured in
Dulbecco's modified Eagle's medium (DMEM; Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) containing 10% fetal bovine
serum (FBS; Gibco; Thermo Fisher Scientific, Inc.) under 5%
CO2 and 37°C. In the OGD/R group, the cells were
incubated in glucose-free Earle's balanced salt solution (Thermo
Fisher Scientific, Inc.) and maintained under 95% N2 and
5% CO2 at 37°C for 4 h. Cells were subsequently removed
and incubated in fresh DMEM containing 10% FBS under 5%
CO2 at 37°C for 4 h. In the RS + OGD/R group, cells were
incubated with RS (1 µM) in high glucose DMEM for 3 h at 37°C and
washed with PBS, then OGD/R was induced as described for the OGD/R
group. In the ATR + RS + OGD/R group, cells were incubated with ATR
(1 µM) in high glucose DMEM for 1 h at 37°C and washed with PBS.
Subsequently, the medium was replaced with DMEM containing RS (1
µM) and cells were incubated for 3 h at 37°C. Cells were
subsequently washed with PBS and OGD/R was induced as described
above. Cells in the four groups were subsequently transfected with
50 nM scramble small interfering (si)RNA negative control (NC) or
50 nM PPAR-γ-siRNA (sequences unavailable; MyBioSource, Inc., San
Diego, CA, USA) using Lipofectamine 3000 (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) for 6 h at 37°C, according to
the manufacturer's protocol. Thus, the grouping was as follows:
Control (cells transfected with NC siRNA), control + siPPAR-γ
(cells transfected with siPPAR-γ only), OGD/R (NC siRNA), OGD/R +
siPPAR-γ, RS + OGD/R (NC siRNA), RS + OGD/R + siPPAR-γ, ATR + RS +
OGD/R (NC siRNA), ATR + RS + OGD/R + siPPAR-γ.
Superoxide dismutase (SOD), lactate
dehydrogenase (LDH), creatine kinase-muscle/brain (CK-MB) and
malondialdehyde (MDA) activity detection
Blood (10 ml) was collected from the central artery
via the rabbit ear using a syringe. The needle was parallel to the
artery. A cotton ball was subsequently pressed to the ear to stop
the bleeding. Anticoagulants were added and blood was centrifuged
at 1,000 × g at 4°C for 10 min. The supernatant from blood samples
was collected for subsequent experimentation. The cells from in
vitro experiments were also collected and centrifuged for 10
min at 3,000 × g and 4°C, and the supernatant was stored at −80°C.
The activities of SOD, MDA and LDH were detected using SOD activity
detection kit, MDA activity detection kit and LDH-cytotoxicity
assay kit, respectively, according to the manufacturer's protocol.
The three kits were purchased from Beyotime Institute of
Biotechnology (Jiangsu, China). CK-MB activity was measured using
Creatine Kinase Activity Assay kit (Sigma-Aldrich; Merck KGaA)
according to the manufacturer's protocol.
Cell viability assay
Cell viability in each group was detected by MTT
assay. Cells (2×103 cells/well) were seeded into 96-well
plates (100 µl/well) in serum-free DMEM and incubated at 37°C in an
incubator with 5% CO2 for 48 h. Following incubation,
cells were treated with 20 µl MTT (5 mg/ml; cat. no. M-2128;
Sigma-Aldrich; Merck KGaA) solution for 4 h at 37°C. Cells were
subsequently treated with 10 µl dimethylsulfoxide (DMSO) for 15 min
at room temperature. Optical density (OD) value was measured at a
wavelength of 490 nm using a spectrophotometer (Sigma-Aldrich;
Merck KGaA).
Enzyme linked immunosorbent assay
(ELISA)
The activity of troponin I (cat. no. MBS765393) and
troponin T (cat. no. MBS056907; both MyBioSource, Inc.) was
detected by ELISA according to the manufacturer's protocol.
Cultured cells were added into the corresponding wells and the
wells were sealed using adhesive tape and maintained at 37°C for 90
min. A total of 100 µl biotinylated antibody fluids were added into
wells. The wells were sealed using adhesive tape and incubated for
60 min at 4°C. Chromogenic substrate was added into all wells with
the exception of blank wells. Plates were maintained for 10–15 min
in the dark at 37°C. Subsequently, stop solution was added into
each well and mixed immediately for 10 min. Finally, the OD450
value was detected using a microplate reader (Bio-Rad Laboratories,
Inc., Hercules, CA, USA).
Evaluation of reactive oxygen species
(ROS)
Cells were seeded at a density of
1×104/well into a 6-well plate in a 37°C incubator.
After 24 h, 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA; 10
µM; Sigma-Aldrich; Merck KGaA) was added into the wells and was
incubated at 37°C for 30 min. The cells were washed in PBS three
times (2–3 min each time) to remove the DCFH-DA dye. The evaluation
of mitochondrial reactive oxygen species (ROS) was performed using
mitoSOX dye (Molecular Probes, USA), which selectively targets the
mitochondrial matrix and emits red fluorescence when oxidized by
ROS. The cells were stained with 2 µM mitoSOX dye and incubated for
15 min at 37°C. The cells were subsequently suspended in PBS and
analyzed by flow cytometry. ROS levels were measured by FACSCalibur
with Cell Quest software version 3.1 (BD Biosciences, San Jose, CA,
USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) assay
The mRNA expression levels of caspase-9, cytochrome
c (cyt c), UCP2 and PPAR-γ were detected by RT-qPCR. Total RNA from
cells and tissues was extracted with TRIzol reagent (Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol.
Concentration of extracted RNA was determined using a UV
spectrophotometer (Thermo Fisher Scientific Inc.). First-strand
cDNA was synthesized using Revert Aid First Strand cDNA Synthesis
kit (Thermo Fisher Scientific Inc.). The temperature protocol was
set as 25°C for 10 min, 42°C for 50 min and 70°C for 8 min. The
mRNA expression levels were evaluated by qPCR using SYBR-Green PCR
Master Mix (Applied Biosystems; Thermo Fisher Scientific Inc.) in
ABI 7500 Real-time PCR system (Applied Biosystems; Thermo Fisher
Scientific Inc.). The PCR thermocycling conditions were as follows:
95°C for 5 min, 35 cycles of 95°C for 30 sec and 60°C for 60 sec,
followed by a final extension at 72°C for 7 min. The results were
quantified using the 2−∆∆Cq calculation (34). The following specific primers were
used: Caspase-9 (product size, 196 bp), 5′-CAGGACCTTGGACAGTGACT-3′
(forward), 5′-AATGCCATCCAAGGTCTCGA-3′ (reverse); cyt c (product
size, 249 bp), 5′-GTTCAGAAGTGTGCCCAGTG-3′ (forward),
5′-GTCTGCCCTTTCTCCCTTCT-3′ (reverse); UCP2 (product size, 170 bp),
5′-AGACCATTGCACGAGAGGAA-3′ (forward), 5′-AGAAGTGAAGTGGCAAGGGA-3′
(reverse); PPAR-γ (product size, 164 bp),
5′-AGGGCGATCTTGACAGGAAA-3′ (forward), 5′-CGAAACTGGCACCCTTGAAA-3′
(reverse); GAPDH (product size, 155 bp), 5′-AACGACCCCTTCATTGACCT-3′
(forward), 5′-ATGTTAGTGGGGTCTCGCTC-3′ (reverse).
Western blot analysis
The proteins from tissue were extracted using tissue
protein extraction kit (Beijing ComWin Biotech Co., Ltd., Beijing,
China). The proteins from cells were extracted using a total
protein extraction kit (Beijing Solarbio Science & Technology
Co., Ltd.). The concentrations of proteins were detected using
Pierce BCA Protein Assay kit (Pierce; Thermo Fisher Scientific,
Inc.). Equivalent proteins (30 µg/lane) were separated by 10%
SDS-PAGE gel and transferred onto polyvinylidene fluoride
membranes. Proteins were blocked with 5% skimmed milk at room
temperature for 2 h. Primary antibodies against caspase-9 (1:5,000;
cat. no. ab2324), cyt c (1:5,000; cat. no. ab28146), UCP2 (1:1,000;
cat. no. ab97931), PPAR-γ (1:1,000; cat. no. ab223137) and GAPDH
(1:2,500; cat. no. ab9485) were from Abcam (Cambridge, MA) and were
incubated with the membranes at 4°C overnight. Horseradish
peroxidase-conjugated mouse anti-rabbit IgG secondary antibody
(cat. no. sc-2357; 1:2,000) were from Santa Cruz Biotechnology,
Inc. (Dallas, TX, USA). The secondary antibodies were incubated
with the membranes at room temperature for 1 h. The images were
obtained using enhanced chemiluminescence western blotting
detection system (GE Healthcare, Chicago, IL, USA). The data were
analyzed using Image Lab Software (version 4.1; Bio-Rad
Laboratories, Inc.).
Statistical analysis
All experimental data were analyzed by one-way
analysis of variance followed by Turkey's multiple comparisons
test. All values are presented as the mean ± standard deviation of
three experiments. P<0.05 was considered to indicate a
statistically significant difference.
Results
RS increases SOD activity and
decreases LDH, CK-MB, MDA and troponin I/T activities
To determine whether pretreatment with RS can induce
myocardial protection, a rabbit MI/R model was established. The
plasma concentrations of SOD, LDH, CK-MB, MDA and troponin I/T at
the end of the MI/R period were measured to evaluate the extent of
myocardial injury. The results indicated that the activity of SOD
significantly decreased in the MI/R group compared with the sham
group. Pretreatment with RS significantly increased SOD activity
(Fig. 1A). LDH, CK-MB, MDA and
troponin I/T activities were significantly increased in the MI/R
group compared with the sham group, and RS significantly inhibited
LDH, CK-MB, MDA and troponin I/T activities caused by MI/R.
Furthermore, ATR significantly reversed whereas ATR markedly
reversed the effect of RS (Fig.
1B-E).
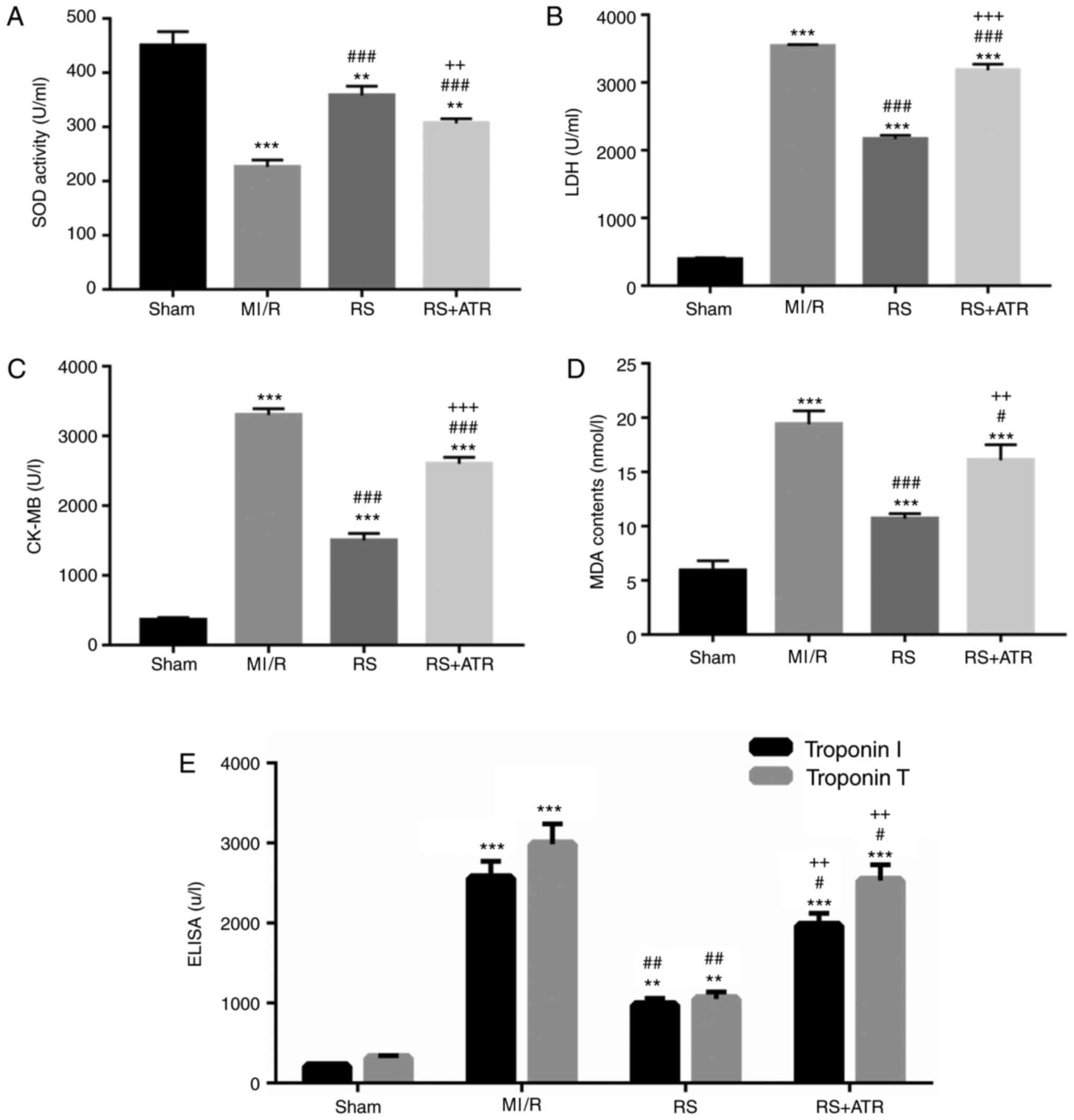 | Figure 1.RS increases SOD activity, and
decreases LDH, CK-MB, MDA and troponin I/T activities. A total of
48 rabbits were randomly divided into four groups, including the
sham group, MI/R group, RS group and RS + ATR group. (A) SOD
activity was detected using a commercial kit. (B) LDH activity was
measured using a cytotoxicity assay. (C) CK-MB activity was
analyzed using a CK-MB assay. (D) MDA activity was analyzed using a
MDA activity assay kit. (E) Troponin I/T activities were measured
by ELISA. **P<0.01 and ***P<0.001 vs. the sham group;
#P<0.05, ##P<0.01 and
###P<0.001 vs. the MI/R group; ++P<0.01
and +++P<0.001 vs. the RS group. RS, rosuvastatin;
ATR, atractyloside; SOD, superoxide dismutase; LDH, lactate
dehydrogenase; CK-MB, creatine kinase-muscle/brain, MDA,
malondialdehyde; MI/R, myocardial ischemia/reperfusion. |
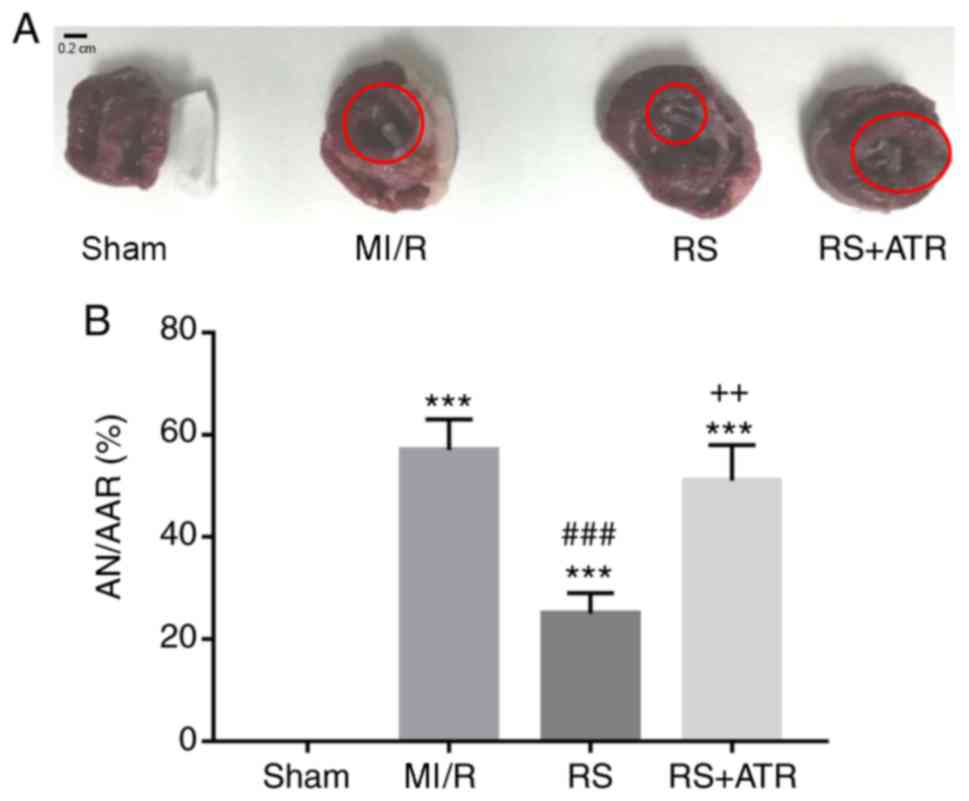
RS inhibits myocardial infarct
size
To further confirm the direct effects of RS on
myocardial MI/R injury, ischemic area and infarct size were
measured in rabbits using the Evans blue/TTC method (Fig. 2A). The infarct size was analyzed
and expressed as the percentage of AN/AAR. The results indicated
that the infarct size was significantly increased in the MI/R group
compared with the sham group. Treatment with RS significantly
ameliorated the injury, whereas ATR markedly reversed the
protective effect of RS (Fig.
2B).
RS downregulates the expression of
caspase-9 and cyt c, and upregulates the expression of UCP2 and
PPAR-γ
The effects of pretreatment with RS on expression of
cell apoptosis-associated proteins caspase-9 and cyt c. UCP2 and
PPAR-γ expression levels were determined to elucidate the effects
of RS on mitochondrial protection. The results revealed that
compared with the sham group, caspase-9, cyt c, UCP2 and PPAR-γ
mRNA and protein expression levels significantly increased in the
MI/R group (Fig. 3). Compared with
the MI/R group, mRNA and protein expression of UCP2 and PPAR-γ
further increased, while caspase-9 and cyt-c expression decreased
in response to RS treatment. Furthermore, ATR reversed the effects
of RS (Fig. 3).
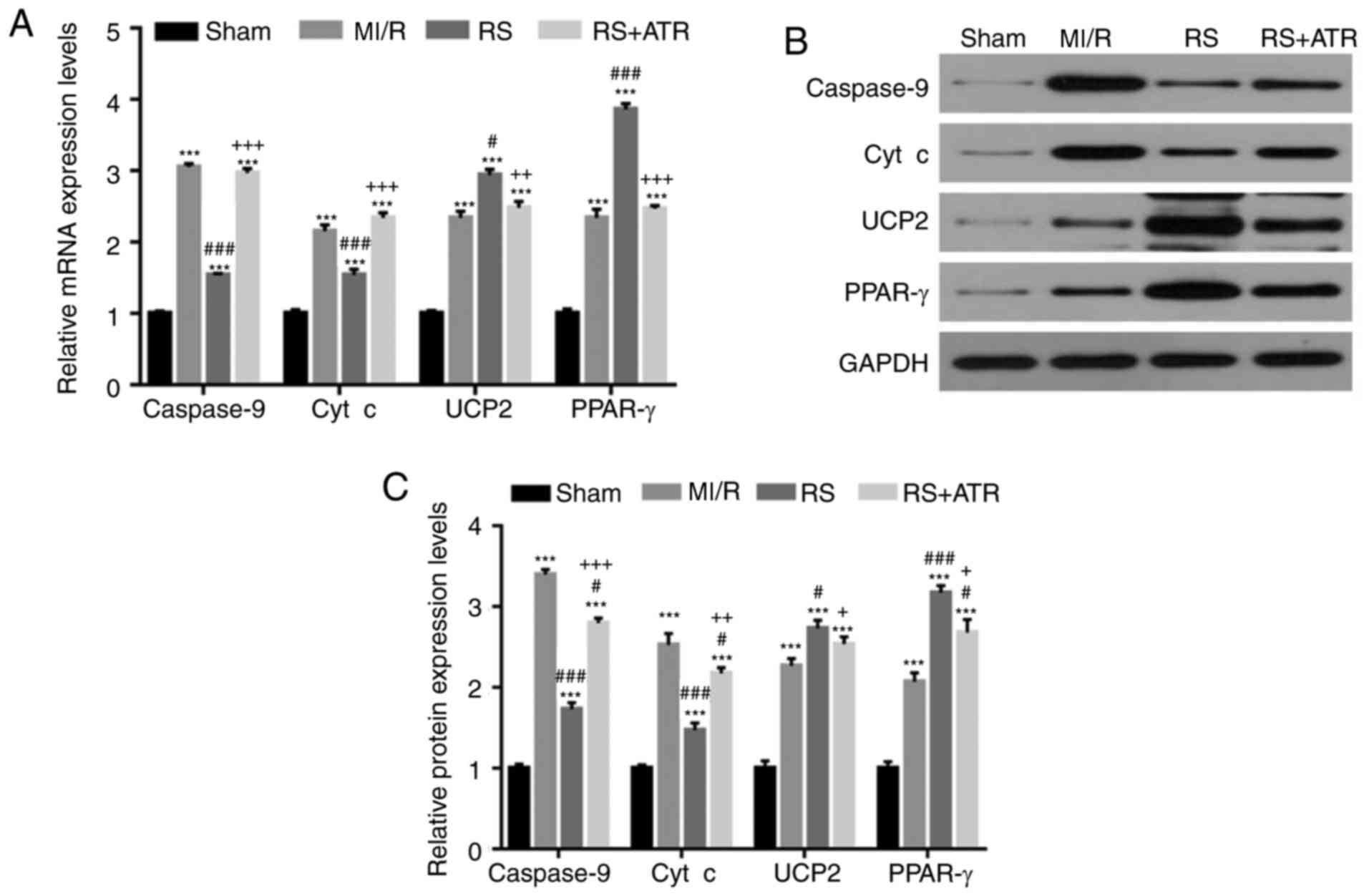 | Figure 3.RS downregulates caspase-9 and cyt c
expression, and upregulates UCP2 and PPAR-γ expression. A total of
48 rabbits were randomly divided into four groups, including the
sham group, MI/R group, RS group and RS + ATR group. mRNA and
protein expression levels of caspase-9, cyt c, UCP2 and PPAR-γ were
detected by (A) reverse transcription-quantitative polymerase chain
reaction and (B) western blotting. (C) The results of western
blotting were quantitatively analyzed. ***P<0.001 vs. the sham
group; #P<0.05 and ###P<0.001 vs. the
MI/R group; +P<0.05, ++P<0.01 and
+++P<0.001 vs. the RS group. Cyt c, cytochrome c;
UCP2, mitochondrial uncoupling protein 2; PPAR-γ, peroxisome
proliferator-activated receptor-γ; MI/R, myocardial
ischemia/reperfusion; RS, rosuvastatin; ATR, atractyloside. |
Pretreatment with RS promotes
cardiomyocyte viability, inhibits LDH release and reduces ROS
production following OGD/R damage
The protective effect of preconditioning with RS was
further studied on rat cardiomyocytes with OGD/R injury.
Cardiomyocytes were divided into the following groups: Control,
control + siPPAR-γ, OGD/R, OGD/R + siPPAR-γ, RS + OGD/R, RS + OGD/R
+ siPPAR-γ, ATR + RS + OGD/R, ATR + RS + OGD/R + siPPAR-γ. The cell
viability was evaluated using the MTT and LDH release assays. OGD/R
significantly inhibited myocardial viability compared with the
control group and PPAR-γ silencing in OGD/R cells further inhibited
myocardial viability compared with the control and control +
siPPAR-γ groups. Myocardial cell viability in the RS + OGD/R group
was significantly increased compared with the OGD/R group. The
results also indicated that treatment with ATR reversed the effect
of RS. Furthermore, OGD/R and OGD/R + siPPAR-γ significantly
increased LDH activity compared with the control group. While LDH
activity was significantly decreased in the RS + OGD/R group
compared with the OGD/R group. Treatment with ATR significantly
reversed the effect mediated by RS (Fig. 4 A, B). Furthermore, it was revealed
that the mitochondrial and cellular ROS levels were significantly
elevated in OGD/R and OGD/R + siPPAR-γ groups, compared with the
control group. ROS content in RS + OGD/R group was significantly
decreased compared with the OGD/R group. Treatment with ATR
significantly reversed the effect mediated by RS (Fig. 5A, B).
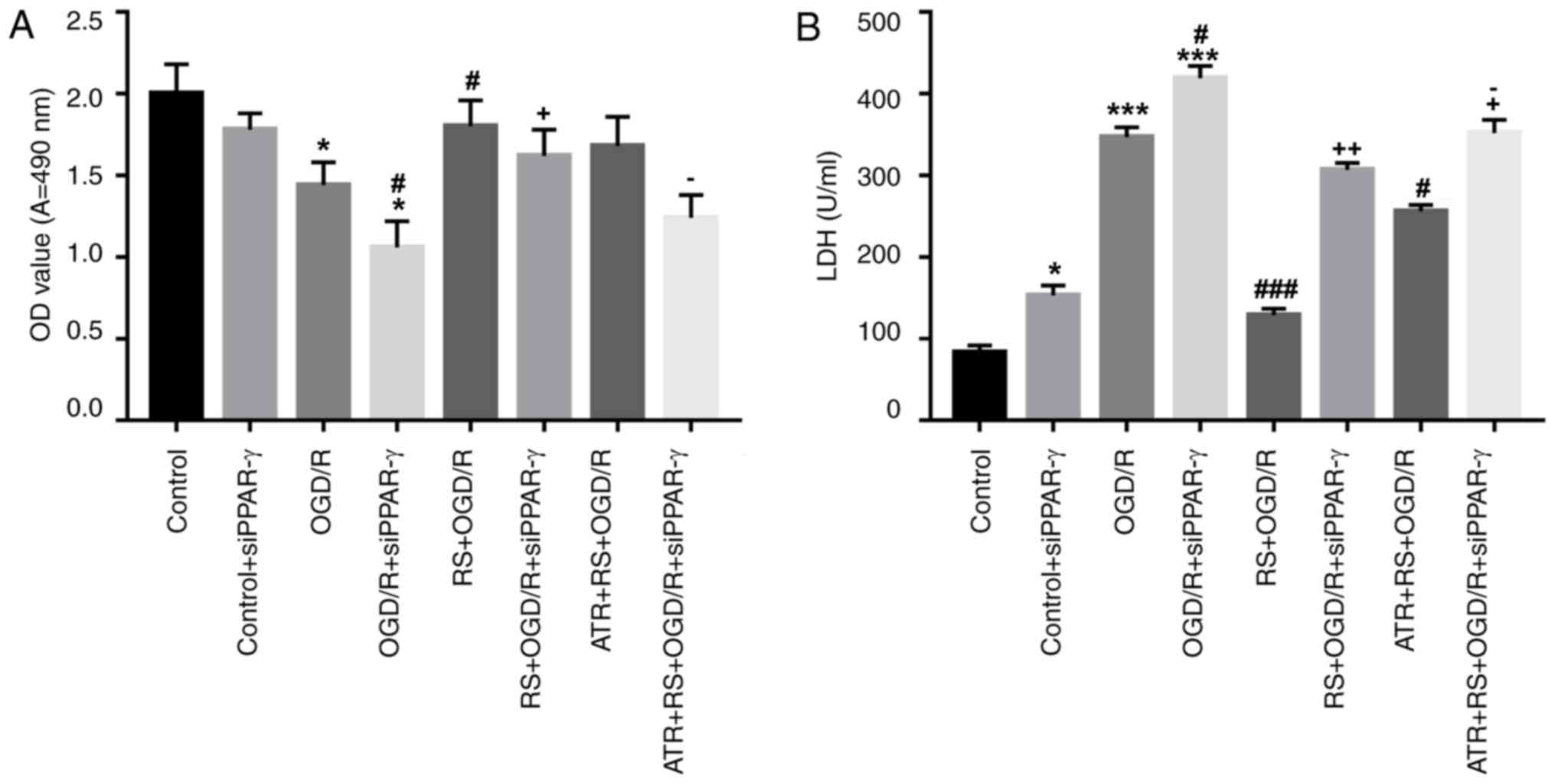 | Figure 4.Pretreatment with RS promotes
cardiomyocyte viability and inhibits LDH release following OGD/R
damage. Cardiomyocytes were divided into the following groups:
Control, control + siPPAR-γ, OGD/R, OGD/R + siPPAR-γ, RS + OGD/R,
RS + OGD/R + siPPAR-γ, ATR + RS + OGD/R, ATR + RS + OGD/R +
siPPAR-γ. (A) Following reperfusion, cell viability was evaluated
by MTT assay. (B) LDH activity was measured using LDH cytotoxicity
assay. *P<0.05 and ***P<0.001 vs. the control group;
#P<0.05 and ###P<0.001 vs. the OGD/R
group; +P<0.05 and ++P<0.01 vs. the
OGD/R + siPPAR-γ group; OGD/R, oxygen-glucose
deprivation/reperfusion; LDH, lactate dehydrogenase; OD, optical
density; si, small interfering RNA; PPAR-γ, peroxisome
proliferator-activated receptor-γ; RS, rosuvastatin; ATR,
atractyloside. |
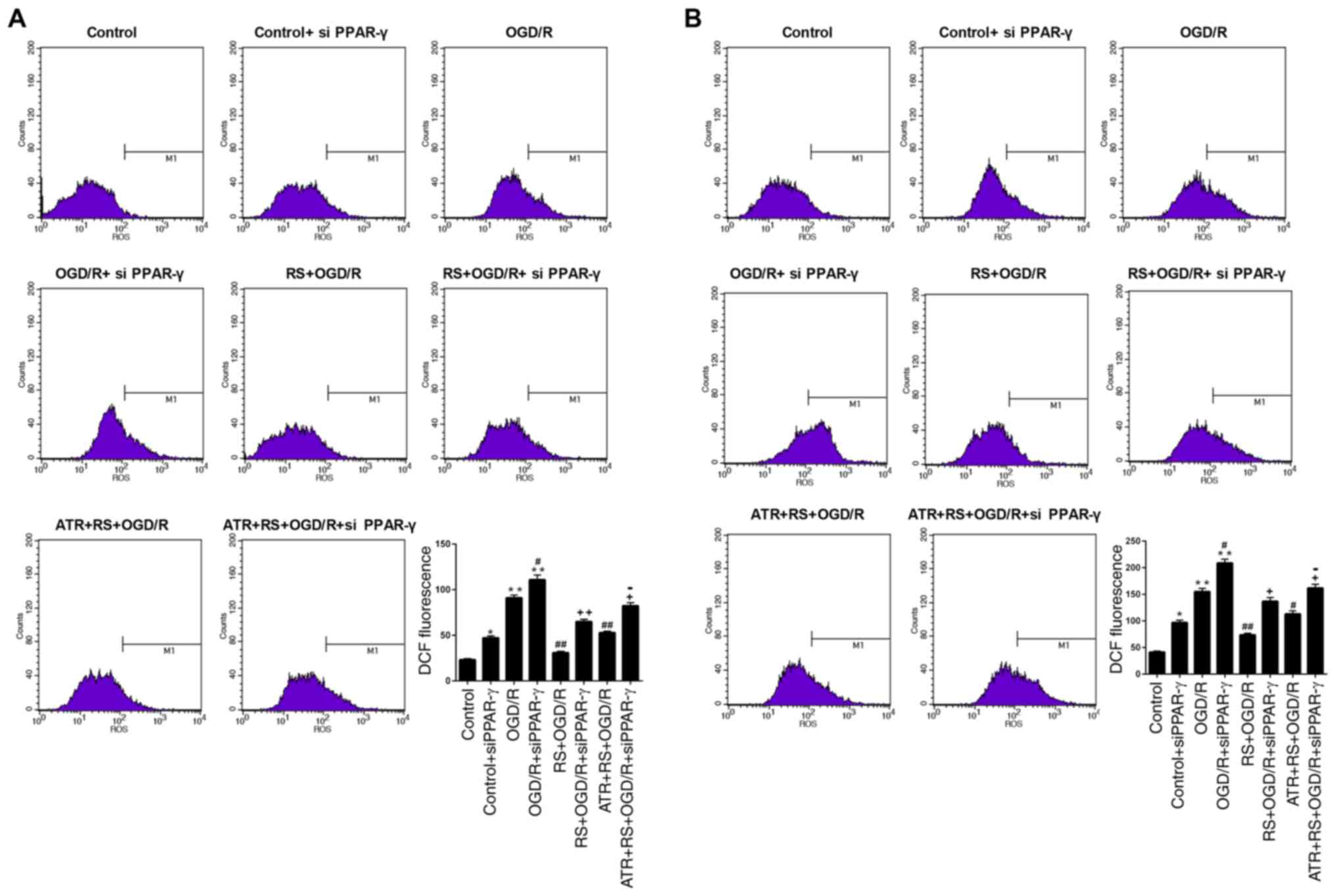 | Figure 5.Pretreatment with RS reduces ROS
production following OGD/R damage. Cardiomyocytes were divided into
the following groups: Control, control + siPPAR-γ, OGD/R, OGD/R +
siPPAR-γ, RS + OGD/R, RS + OGD/R + siPPAR-γ, ATR + RS + OGD/R, ATR
+ RS + OGD/R + siPPAR-γ. Flow cytometry was used to determine the
(A) mitochondrial and (B) cellular ROS levels. M1 indicates the
cells that emitted a DCF signal and therefore the level of ROS
production. *P<0.05 and **P<0.01 vs. the control group;
#P<0.05 and ##P<0.01 vs. the OGD/R
group; +P<0.05 and ++P<0.01 vs. the
OGD/R + siPPAR-γ group; −P<0.05 vs. the RS + OGD/R +
siPPAR-γ group. OGD/R, oxygen-glucose deprivation/reperfusion; LDH,
lactate dehydrogenase; DCF, 2′,7′-dichlorofluorescein; ROS,
reactive oxygen species; RS, rosuvastatin; ATR, atractyloside; si,
small interfering RNA; PPAR-γ, peroxisome proliferator-activated
receptor-γ. |
Pretreatment with RS decreases
caspase-9 and cyt c expression, and increases UCP2 and PPAR-γ
expression following OGD/R damage
To further elucidate the mechanism of RS
preconditioning on myocardial OGD/R injury in rats, RT-qPCR and
western blotting were performed. As presented in Fig. 6, compared with the control group,
caspase-9, cyt c, UCP2 and PPAR-γ expression levels were
significantly increased in the OGD/R group. The expression levels
of caspase-9 and cyt c were lower in the RS + OGD/R and RS + OGD/R
+ si-PPAR-γ groups compared with the OGD/R and OGD/R + si-PPAR-γ
groups, respectively. The expression of UCP2 and PPAR-γ increased
following RS preconditioning in the RS + OGD/R and RS + OGD/R +
si-PPAR-γ groups, compared with OGD/R and OGD/R+si-PPAR-γ groups,
respectively. The effect of RS on the expression of caspase-9, cyt
c and UCP2 was reversed by ATR (Fig.
6). The WB results in the expression of caspase-9, cyt c and
PPAR-γ had a similar trend to the mRNA data. However, although ATR
reversed the effect of RS on UCP2 mRNA expression, this effect was
not observed at the protein level (Fig. 7).
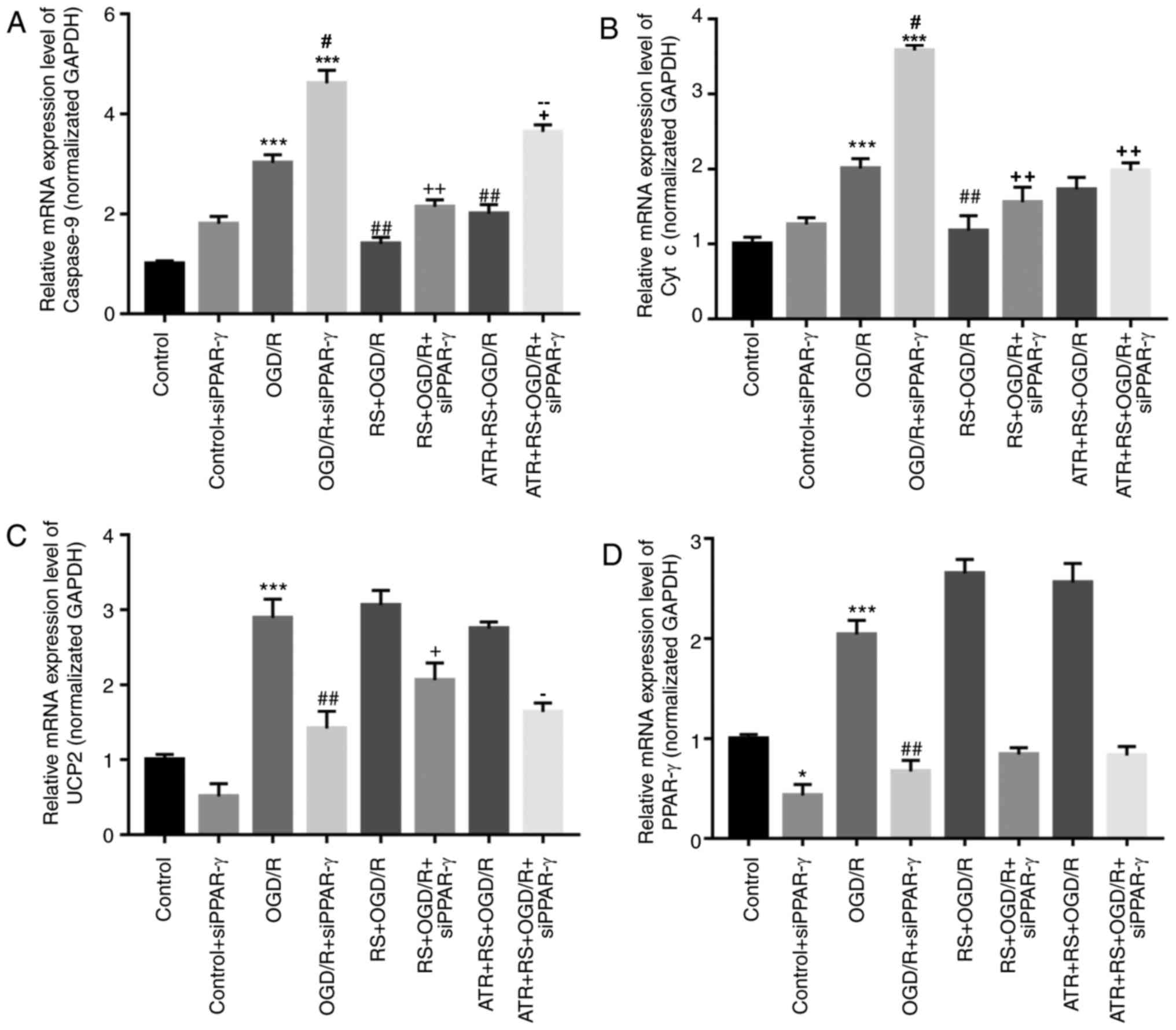 | Figure 6.Pretreatment with RS decreases
caspase-9 and cyt c mRNA expression, and increases UCP2 and PPAR-γ
mRNA expression following OGD/R damage. Cardiomyocytes were divided
into the following groups: Control, control + siPPAR-γ, OGD/R,
OGD/R + siPPAR-γ, RS + OGD/R, RS + OGD/R + siPPAR-γ, ATR + RS +
OGD/R, ATR + RS + OGD/R + siPPAR-γ. Reverse
transcription-quantitative polymerase chain reaction was performed
to analyze the mRNA expression levels of (A) caspase-9, (B) cyt c,
(C) UCP2 and (D) PPAR-γ in cardiomyocytes. *P<0.05 and
***P<0.001 vs. the control group; #P<0.05 and
##P<0.01 vs. the OGD/R group; +P<0.05
and ++P<0.01 vs. the OGD/R + siPPAR-γ group;
−P<0.05 and −−P<0.01 vs. the RS + OGD/R
+ siPPAR-γ group. OGD/R, oxygen-glucose deprivation/reperfusion;
cyt c, cytochrome c; RS, rosuvastatin; ATR, atractyloside; si,
small interfering RNA; PPAR-γ, peroxisome proliferator-activated
receptor-γ; UCP2, mitochondrial uncoupling protein 2. |
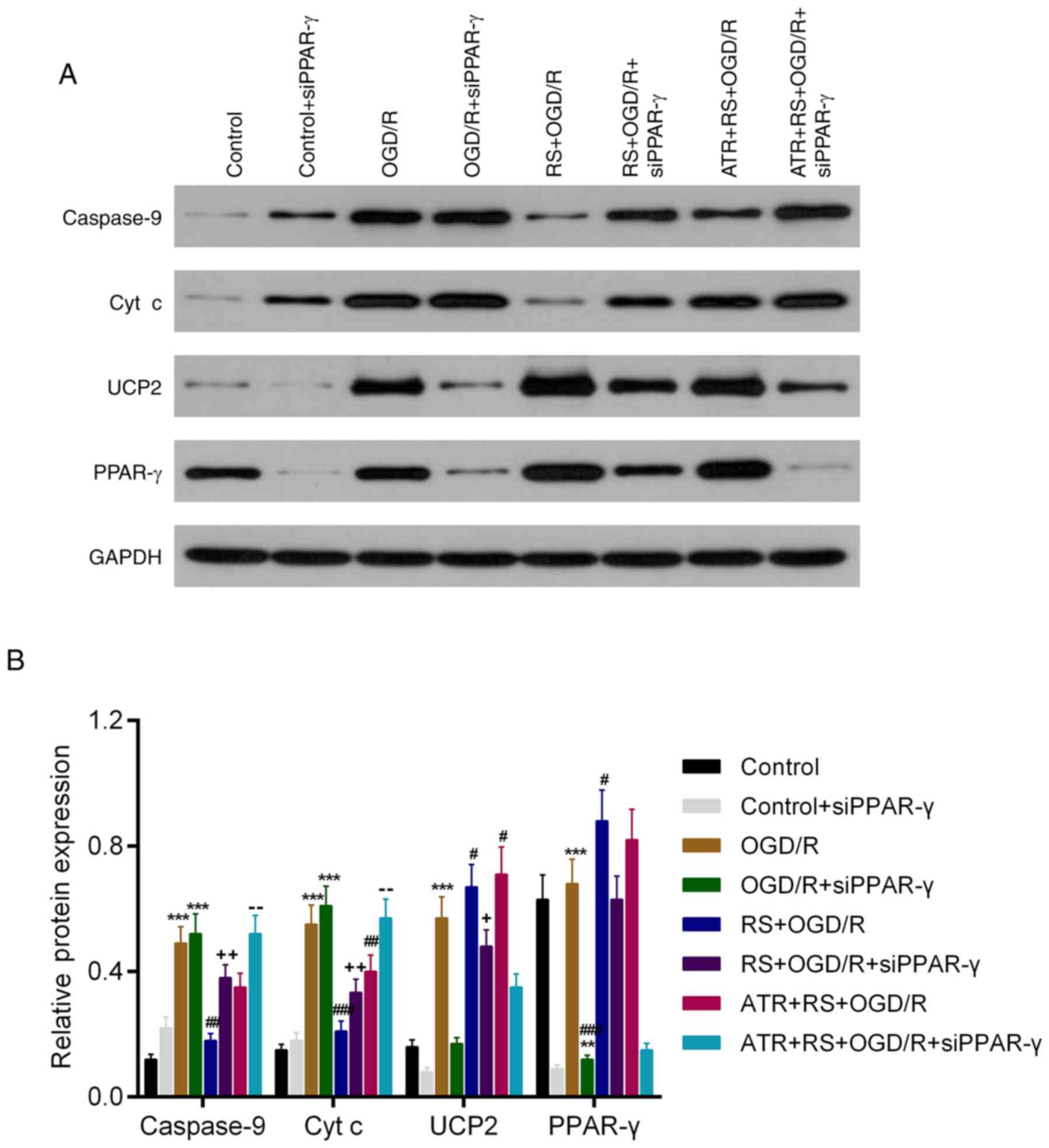 | Figure 7.RS downregulates protein expression
levels of caspase-9 and cyt c, and upregulates protein expression
levels of UCP2 and PPAR-γ following OGD/R damage. Cardiomyocytes
were divided into the following groups: Control, control +
siPPAR-γ, OGD/R, OGD/R + siPPAR-γ, RS + OGD/R, RS + OGD/R +
siPPAR-γ, ATR + RS + OGD/R, ATR + RS + OGD/R + siPPAR-γ. (A)
Western blot analysis was used to detect the protein expression
levels of caspase-9, cyt c, UCP2 and PPAR-γ in cardiomyocytes. (B)
The protein expression levels were quantitatively analyzed
according to the protein gray values. **P<0.01 and ***P<0.001
vs. the control group; #P<0.05 and
##P<0.01 vs. the OGD/R group; +P<0.05
and ++P<0.01 vs. the OGD/R + siPPAR-γ group;
−−P<0.01 vs. the RS + OGD/R + siPPAR-γ group. OGD/R,
oxygen-glucose deprivation/reperfusion; cyt c, cytochrome c;
PPAR-γ, peroxisome proliferator-activated receptor-γ; UCP2,
mitochondrial uncoupling protein 2; RS, rosuvastatin; ATR,
atractyloside; si, small interfering RNA. |
Discussion
Coronary heart disease (CHD) and atherosclerosis
(AS) affect human health worldwide (35). Interventional cardiovascular
therapy has greatly improved the clinical outcomes of patients with
CHD (36). Restoring blood flow is
indispensable for rescuing the ischemic myocardium, however,
myocardial revascularization also causes damage, known as MI/R
injury (37). MI/R could aggravate
the hemodynamic dysfunction and cause ischemia/reperfusion injury
in the patient, which may eventually lead to mortality (38). Statins, commonly used drugs for
treatment of AS and acute coronary syndrome (ACS), regulate the
lipid metabolism and serve multiple pharmacological roles
associated with the stability of atherosclerotic plaques,
endothelial function and immune regulation (39). These pharmacological effects
indicate that further investigation of statins in the context of
cardiovascular disease may be beneficial for development of novel
treatment methods. Therefore, the present study analyzed the direct
effect and potential mechanism of RS in MI/R injury.
In the current study, an in vivo rabbit model
of MI/R was established using protocols described in previous
studies (40,41). Subsequently, the activities of SOD,
LDH, CK-MB, MDA and troponin I/T in serum samples from each
treatment group were evaluated. The results indicated that RS
significantly enhanced the SOD activity, and reduced the LDH,
CK-MB, MDA and troponin I/T activities compared with the MI/R
group. ATR reversed the effects of RS. Furthermore, following Evans
blue/TTC staining, it was revealed that RS markedly inhibited the
myocardial infarct size compared to MI/R group. Expression levels
of UCP2 and PPAR-γ in serum samples from each treatment group were
also determined. RS increased the UCP2 and PPAR-γ expression levels
compared to MI/R group; while the effect of RS was reversed by ATR.
Based on these results, it can be hypothesized that treatment with
RS prior to MI/R can reduce MI/R injury via upregulation of UCP2
and PPAR-γ in vivo. ATR partially reversed the protective
effects of RS.
The results obtained using RS in vivo were
further tested using an in vitro model of MI/R to further
elucidate the underlying mechanisms of action. In vitro, the
OGD/R injury was used to mimic the I/R damage in cardiomyocytes.
The viability of cardiomyocytes from each treatment group was
measured. The results revealed that silencing of PPAR-γ inhibited
the viability of cardiomyocytes (OGD/R group vs. OGD/R + siPPAR-γ
group). RS enhanced the viability of myocardial cells suppressed by
OGD/R. Mitochondria have been hypothesized to be the primary source
of ROS following I/R injury (42).
Excessive ROS production can cause damage of the antioxidative
system, increase membrane permeability and cause calcium overdose
during reperfusion (43). The
increase in Ca2+ levels in the mitochondria may lead to
the opening of the mitochondrial permeability transition pore, loss
of MMP and increased ROS production (44). The interaction between ROS and
Ca2+ may aggravate apoptotic cell injury during I/R
(45). Therefore, the present
study measured the mitochondrial and cellular ROS content in
cardiomyocytes from each treatment group. According to the flow
cytometry data, RS markedly suppressed ROS production enhanced by
OGD/R both in mitochondria and intracellular space.
Caspase-9 and cyt c are known apoptosis-associated
proteins (46,47) and a recent study has indicated that
the release of cyt c could induce the activation of caspase-9 in
gastric carcinoma cells (48). In
the present study, the effects of RS on myocardial cells were
further examined by detecting alterations in the mRNA and protein
expression levels of caspase-9, cyt c, UCP2 and PPAR-γ. The results
indicated that RS upregulated the expression levels of UCP2 and
PPAR-γ following OGD/R damage, while this pretreatment
significantly reduced the expression of apoptosis-associated
proteins, caspase-9 and cyt c in myocardial cells. The expression
levels of UCP2 and PPAR-γ, were up-regulated following OGD/R damage
and further increased in the RS group. It may be hypothesized that
OGD/R promoted cell apoptosis and enhanced the expression of
apoptosis-associated proteins, which may have activated the
expression of UCP2 and PPAR-γ to protect the cells against
apoptosis. It may be concluded that RS protected the myocardial
cells against OGD/R injury by up-regulating the expression of
PPAR-γ and UCP2.
In the present study, RS mitigated MI/R injury,
increased SOD activity and decreased LDH, CK-MB, MDA and troponin
I/T activities. RS also downregulated the expression of
apoptosis-associated genes (caspase-9 and cyt c). RS suppressed the
production of ROS, and ATR reversed the effect of RS. Therefore, it
may be hypothesized that RS induces cardioprotective effects by
suppressing ROS production and inhibiting mitochondria-mediated
apoptosis.
In conclusion, in the present study RS inhibited
myocardial infarct size and ROS in vivo and protected
primary myocardial cells against OGD/R injury in vitro.
Furthermore, RS decreased the expression levels of
apoptosis-associated genes (caspase-9 and cyt c), and increased the
expression of UCP2 and PPAR-γ. Furthermore, the effect of RS was
reversed by ATR. The present study provided evidence for the use of
RS as a potential agent for the treatment of cardiac injury.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Quanzhou
Science and Technology Planning Project (grant no. 2015Z80).
Availability of data and materials
All data generated and/or analyzed during this study
are included in this published article.
Authors' contributions
LW wrote the main manuscript and analyzed the data.
RL and LG performed the experiments. MH designed the study. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
The project protocol was approved by the
Institutional Review Board of Fujian Province Medical
Association.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Cheng TO: Coronary arteriosclerotic
disease existed in China over 2,200 years ago. Methodist Debakey
Cardiovasc J. 8:47–48. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Libby P, Tabas I, Fredman G and Fisher EA:
Inflammation and its resolution as determinants of acute coronary
syndromes. Circ Res. 114:1867–1879. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ma P, Han L, Lv Z, Chen W, Hu H, Tu J,
Zhou X and Liu SM: In-hospital free fatty acids levels predict the
severity of myocardial ischemia of acute coronary syndrome. BMC
Cardiovasc Disord. 16:292016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Arnold JR, Karamitsos TD, van Gaal WJ,
Testa L, Francis JM, Bhamra-Ariza P, Ali A, Selvanayagam JB,
Westaby S, Sayeed R, et al: Residual ischemia after
revascularization in multivessel coronary artery disease: Insights
from measurement of absolute myocardial blood flow using magnetic
resonance imaging compared with angiographic assessment. Circ
Cardiovasc Interv. 6:237–245. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Torosoff MT, Sidhu MS and Boden WE: Impact
of myocardial ischemia on myocardial revascularization in stable
ischemic heart disease. Lessons from the COURAGE and FAME 2 trials.
Herz. 38:382–386. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Neto Frias CA, Koike MK, Saad KR, Saad PF
and Montero EF: Effects of ischemic preconditioning and cilostazol
on muscle ischemia-reperfusion injury in rats. Acta Cir Bras. 29
Suppl 3:S17–S21. 2014. View Article : Google Scholar
|
|
7
|
Halladin NL: Oxidative and inflammatory
biomarkers of ischemia and reperfusion injuries. Dan Med J.
62:B50542015.PubMed/NCBI
|
|
8
|
Hermida N and Balligand JL: Low-density
lipoprotein-cholesterol-induced endothelial dysfunction and
oxidative stress: The role of statins. Antioxid Redox Signal.
20:1216–1237. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kosmas CE, Alkhawam H, El-Hunjul M, Wagman
G, Kahn MR, Grady KM and Vittorio TJ: Statin-mediated low-density
lipoprotein lowering in chronic congestive heart failure. Am J Med
Sci. 347:14–22. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Barone E, Di Domenico F and Butterfield
DA: Statins more than cholesterol lowering agents in Alzheimer
disease: Their pleiotropic functions as potential therapeutic
targets. Biochem Pharmacol. 88:605–616. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gazzerro P, Proto MC, Gangemi G, Malfitano
AM, Ciaglia E, Pisanti S, Santoro A, Laezza C and Bifulco M:
Pharmacological actions of statins: A critical appraisal in the
management of cancer. Pharmacol Rev. 64:102–146. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kamada A, Yoshikawa Y, Domae E, Goda S,
Okazaki J, Kawamoto A, Komasa Y and Ikeo T: Effect of statin on
synthesis of bone morphogenetic protein-2 and extracellular matrix
in human osteosarcoma cells. J Osaka Odontolo Soc. 67:2004.
|
|
13
|
Adams SP, Sekhon SS and Wright JM:
Lipid-lowering efficacy of rosuvastatin. Cochrane Database Syst
Rev: CD010254. 2014. View Article : Google Scholar
|
|
14
|
Robertsen I, Asberg A, Granseth T, Vethe
NT, Akhlaghi F, Ghareeb M, Molden E, Reier-Nilsen M, Holdaas H and
Midtvedt K: More potent lipid-lowering effect by rosuvastatin
compared with fluvastatin in everolimus-treated renal transplant
recipients. Transplantation. 97:1266–1271. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen K, Li D, Zhang X, Hermonat PL and
Mehta JL: Anoxia-reoxygenation stimulates collagen type-I and MMP-1
expression in cardiac fibroblasts: Modulation by the PPAR-gamma
ligand pioglitazone. J Cardiovasc Pharmacol. 44:682–687. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shao X, Wang M, Wei X, Deng S, Fu N, Peng
Q, Jiang Y, Ye L, Xie J and Lin Y: Peroxisome
proliferator-activated receptor-γ: Master regulator of adipogenesis
and obesity. Curr Stem Cell Res Ther. 11:282–289. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ma T, Ma ZQ, Du XH, Yu QS, Wang R and Liu
L: Effect of valsartan on ACAT-1 and PPAR-γ expression in intima
with carotid artery endothelial balloon injury in rabbit. Int J
Clin Exp Med. 8:5527–5533. 2015.PubMed/NCBI
|
|
18
|
Balaji Prathab S, Chand Vijay C, Justin A
and Ramanathan M: Telmisartan mediates anti-inflammatory and not
cognitive function through PPAR-γ agonism via SARM and MyD88
signaling. Pharmacol Biochem Behav. 137:60–68. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jin H, Gebska MA, Blokhin IO, Wilson KM,
Ketsawatsomkron P, Chauhan AK, Keen HL, Sigmund CD and Lentz SR:
Endothelial PPAR-γ protects against vascular thrombosis by
downregulating P-selectin expression. Arterioscler Thromb Vasc
Biol. 35:838–844. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Boss O, Muzzin P and Giacobino JP: The
uncoupling proteins, a review. Eur J Endocrinol. 139:1–9. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
De Marchi U, Castelbou C and Demaurex N:
Uncoupling protein 3 (UCP3) modulates the activity of
Sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) by decreasing
mitochondrial ATP production. J Biol Chem. 286:32533–32541. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ozcan C, Palmeri M, Horvath TL, Russell KS
and Russell RR III: Role of uncoupling protein 3 in
ischemia-reperfusion injury, arrhythmias and preconditioning. Am J
Physiol Heart Circ Physiol. 304:H1192–H1200. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang X, Gong J, Liu X, Zhan R, Kong R,
Zhao Y, Wan D, Leng X, Chen M and Qian L: Expression of uncoupling
protein 3 in mitochondria protects against stress-induced
myocardial injury: A proteomic study. Cell Stress Chaperones.
15:771–779. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hoerter J, Gonzalez-Barroso MD, Couplan E,
Mateo P, Gelly C, Cassard-Doulcier AM, Diolez P and Bouillaud F:
Mitochondrial uncoupling protein 1 expressed in the heart of
transgenic mice protects against ischemic-reperfusion damage.
Circulation. 110:528–533. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen GG, Yan JB, Wang XM, Zheng MZ, Jiang
JP, Zhou XM, Cai B and Shen YL: Mechanism of uncoupling protein 2
mediated myocardial injury in hypothermic preserved rat hearts. Mol
Med Rep. 14:1857–1864. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ji XB, Li XR, Hao-Ding, Sun Q, Zhou Y, Wen
P, Dai CS and Yang JW: Inhibition of uncoupling protein 2
attenuates cardiac hypertrophy induced by transverse aortic
constriction in mice. Cell Physiol Biochem. 36:1688–1698. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Villarroya F, Iglesias R and Giralt M:
PPARs in the control of uncoupling proteins gene expression. PPAR
Res. 2007:743642007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Oberkofler H, Klein K, Felder TK, Krempler
F and Patsch W: Role of peroxisome proliferator-activated
receptor-gamma coactivator-1alpha in the transcriptional regulation
of the human uncoupling protein 2 gene in INS-1E cells.
Endocrinology. 147:966–976. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sachdeva J, Dai W, Gerczuk PZ and Kloner
RA: Combined remote perconditioning and postconditioning failed to
attenuate infarct size and contractile dysfunction in a rat model
of coronary artery occlusion. J Cardiovasc Pharmacol Ther.
19:567–573. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang F, Yin J, Lu Z, Zhang G, Li J, Xing
T, Zhuang S and Wang N: Limb ischemic preconditioning protects
against contrast-induced nephropathy via renalase. EBioMedicine.
9:356–365. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang F, Zhang G, Lu Z, Geurts AM, Usa K,
Jacob HJ, Cowley AW, Wang N and Liang M: Antithrombin III/SerpinC1
insufficiency exacerbates renal ischemia/reperfusion injury. Kidney
Int. 88:796–803. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rutering J, Ilmer M, Recio A, Coleman M,
Vykoukal J and Alt E: Improved method for isolation of neonatal rat
cardiomyocytes with increased Yield of C-Kit+ cardiac progenitor
cells. J Stem Cell Res Ther. 5:1–8. 2015.PubMed/NCBI
|
|
33
|
Tao L, Bei Y, Li Y and Xiao J: Neonatal
Rat Cardiomyocytes isolation, culture and determination of
MicroRNAs' effects in proliferation. Methods Mol Biol.
1733:203–213. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Dalen JE, Alpert JS, Goldberg RJ and
Weinstein RS: The epidemic of the 20 (th) century: Coronary heart
disease. Am J Med. 127:807–812. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Faxon DP and Williams DO: Interventional
cardiology: Current status and future directions in coronary
disease and valvular heart disease. Circulation. 133:2697–2711.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Karu I, Tähepõld P, Ruusalepp A and
Starkopf J: Pretreatment by hyperoxia-a tool to reduce
ischaemia-reperfusion injury in the myocardium. Curr Clin
Pharmacol. 5:125–132. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ferdinandy P, Schulz R and Baxter GF:
Interaction of cardiovascular risk factors with myocardial
ischemia/reperfusion injury, preconditioning and postconditioning.
Pharmacol Rev. 59:418–458. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Nissen SE, Tuzcu EM, Brewer HB, Sipahi I,
Nicholls SJ, Ganz P, Schoenhagen P, Waters DD, Pepine CJ, Crowe TD,
et al: Effect of ACAT inhibition on the progression of coronary
atherosclerosis. N Engl J Med. 354:1253–1263. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhao XJ, Liu XL, He GX and Xu HP: Effects
of single-dose atorvastatin on interleukin-6, interferon gamma and
myocardial no-reflow in a rabbit model of acute myocardial
infarction and reperfusion. Braz J Med Biol Res. 47:245–251. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhao ZG, Tang ZZ, Zhang WK and Li JG:
Protective effects of embelin on myocardial ischemia-reperfusion
injury following cardiac arrest in a rabbit model. Inflammation.
38:527–533. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhao ZQ: Oxidative stress-elicited
myocardial apoptosis during reperfusion. Curr Opin Pharmacol.
4:159–165. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhao K, Zhao GM, Wu D, Soong Y, Birk AV,
Schiller PW and Szeto HH: Cell-permeable peptide antioxidants
targeted to inner mitochondrial membrane inhibit mitochondrial
swelling, oxidative cell death and reperfusion injury. J Biol Chem.
279:34682–34690. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ding WX, Shen HM and Ong CN: Pivotal role
of mitochondrial Ca (2+) in microcystin-induced mitochondrial
permeability transition in rat hepatocytes. Biochem Biophys Res
Commun. 285:1155–1161. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Simon HU, Haj-Yehia A and Levi-Schaffer F:
Role of reactive oxygen species (ROS) in apoptosis induction.
Apoptosis. 5:415–418. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Brentnall M, Rodriguez-Menocal L, De
Guevara RL, Cepero E and Boise LH: Caspase-9, caspase-3 and
caspase-7 have distinct roles during intrinsic apoptosis. BMC Cell
Biol. 14:322013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Skemiene K, Rakauskaite G, Trumbeckaite S,
Liobikas J, Brown GC and Borutaite V: Anthocyanins block
ischemia-induced apoptosis in the perfused heart and support
mitochondrial respiration potentially by reducing cytosolic
cytochrome c. Int J Biochem Cell Biol. 45:23–29. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhu X, Zhang K, Wang Q, Chen S, Gou Y, Cui
Y and Li Q: Cisplatin-mediated c-myc overexpression and cytochrome
c (cyt release result in the up-regulation of the death receptors
DR4 and DR5 and the activation of caspase 3 and caspase 9, likely
responsible for the TRAIL-sensitizing effect of cisplatin. Med
Oncol. 32:1332015. View Article : Google Scholar : PubMed/NCBI
|