Introduction
Primary liver cancer is the second leading cause of
cancer-related mortality in men globally and the sixth leading
cause of cancer-related mortality in women (1). According to the statistics from the
International Agency for Research on Cancer of the World Health
Organization, there were >782,000 cases of primary liver cancer
in 2012, making liver cancer the fifth and ninth most common cancer
among men and women, respectively (2). Most risk factors for liver cancer,
such as alcohol, are commoner in men than in women, so the
prevalence of hepatocellular carcinoma (HCC) in men is 2–3 times
higher than in women (1).
Cell characteristics are under control of and
maintained by both genetic and epigenetic mechanisms (3). Epigenetics is defined as inheritable
changes in gene expression with no alterations in DNA sequence
(4). Methylation, the most
important and common epigenetic modification, is an important means
of regulating genome function (5).
Decreases in the genome-wide methylation level is an important
indicator of early cancer and may be significantly correlated with
the severity and metastasis of cancer (6). The main reason underlying this
phenomenon is the demethylation of repeat sequences, which leads to
genome instability (7,8). Aberrant hypermethylation of CpG
islands in specific promoter regions is another important
phenomenon in the late stage of cancer, leading to changes in
chromosomal structure, silencing of tumor suppressor genes and
other cancer-related genes, thereby promoting cancer cells to adapt
to the microenvironment and metastasis (9,10).
Previous studies have been conducted to examine the implication of
methylation in the diagnosis, treatment response and prognosis of
various malignancies, such as lung (11), breast (12), and ovarian cancer (13). It has also been reported that
coactivator-associated arginine methyltransferase 1-mediated GAPDH
methylation is a key regulatory mechanism of glucose metabolism in
liver cancer (14). In addition,
it has also been found that the mRNA expression of tumor suppressor
P14ARF is regulated by DNA methylation in primary liver cancer; DNA
methylation of the P14ARF gene may be related to the occurrence and
tumor-node-metastasis stage of primary liver cancer (15).
The Cancer Genome Atlas (TCGA) (16), a public database used in
bioinformatics analysis, has complete patient data and is conducive
to the analysis of cancer progression and prognosis. In the present
study, HCC-related methylation-driven genes were identified from
TCGA database, and the correlation between the identified genes and
HCC progression and prognosis was analyzed. A series of
bioinformatic and survival analyses were conducted to identify the
biomarkers associated with methylation and constructed a prognostic
risk model for predicting the prognosis of patients with HCC.
Materials and methods
Study population and data
processing
The HCC-related level 3 RNA-sequencing (Seq) data
and methylation data were downloaded from TCGA (https://gdc.cancer.gov/). Specifically, the Perl
programming language (http://www.perl.org/) was used to extract matrix files
from the RNA-seq and methylation data files. There were 122 women
and 255 men; 204 patients were aged ≥60 and 172 patients aged
<60. The clinical data of the patients with HCC are summarized
in Table I. The RNA-Seq and
methylation matrix files were merged using the Limma package
(17) and the MethylMix algorithm.
Following the merge, the genes meeting all of the following three
conditions were defined as methylation-driven genes: i) Gene
expression levels differ between normal and cancer tissues; ii)
methylation levels differ between normal and cancer tissues; and
iii) the degree of DNA methylation is correlated with gene
expression. The Pheatmap package (1.0.12, http://CRAN.R-project.org/package=pheatmap) was
used to cluster the methylation-driven genes.
 | Table I.Summary of the clinical data for
hepatocellular carcinoma from The Cancer Genome Atlas. |
Table I.
Summary of the clinical data for
hepatocellular carcinoma from The Cancer Genome Atlas.
|
Characteristics | No. of samples, n
(%) |
|---|
| Age, years |
|
≤50 | 32 (8.5) |
|
>50 | 342 (91.5) |
| Sex |
|
Female | 122 (32.4) |
|
Male | 255 (67.6) |
| Stage |
| I | 175 (49.6) |
| II | 87 (24.6) |
|
III | 86 (24.4) |
| IV | 5 (1.4) |
| Survival state |
|
Alive | 245 (65.0) |
|
Deceased | 132 (35.0) |
| Race |
|
American | 19 (5.2) |
|
Asian | 161 (43.9) |
|
Caucasian | 187 (50.9) |
Gene Ontology (GO) and pathway
enrichment analysis of aberrantly expressed methylation-driven
genes
The Database for Annotation, Visualization and
Integrated Discovery (DAVID 6.7; http://david.ncifcrf.gov/) was used to perform GO
enrichment analysis of the aberrant methylation-driven genes. A
list of differentially expressed methylation-driven genes was
submitted to the functional annotations tool in DAVID (screening
criteria, P<0.01).
Differentially expressed methylation-driven genes
were analyzed using the overexpression analysis function of
ConsensusPathDB 3.4.1 (http://cpdb.molgen.mpg.de/). The Kyoto Encyclopedia of
Genes and Genomes database (conducted in ConsensusPathDB 3.4.1) was
used to perform pathway enrichment analysis for the differentially
expressed methylation-driven genes taking P<0.01 as a cutoff
value.
Screening for prognosis-related
signatures and risk score calculations
The univariate Cox regression model was used to
calculate the association between the methylation level of each
aberrant methylation-driven gene and the overall survival (OS) of
the patient. Genes were considered statistically significant in
univariate Cox analysis when P<0.05. The amount of Z-value
represents the distance between the original score and the parent
mean, measured in standard deviation. When the original score is
lower than the average, Z is negative and vice versa. Multivariate
Cox regression was used in the R language (3.5.1, http://www.r-project.org/) Survival Package (2.44–1.1,
http://CRAN.R-project.org/package=survival) to screen
the independent prognostic ability of the aberrant
methylation-driven genes (18). A
prognostic risk assessment model was constructed by integrating the
information of the aberrant methylation-driven genes.
The log-rank test was used to calculate P-values and
the prognostic correlation coefficient β. The risk score was
defined as follows: Risk score=βgene1 ×
methylgene1 + βgene2 × methylgene2
+ ····· + βgene (n) × methylgene (n), where β
is the prognostic correlation coefficient and methyl is the
methylation level of the corresponding gene. The risk score of each
sample was calculated and the median risk score was set as the
cutoff value for assigning the samples into the low- and high-risk
groups (11,19). The Kaplan-Meier method was used to
plot the survival curves for the low- and high-risk groups.
Differences between groups were evaluated using the log-rank test.
Receiver operating characteristic (ROC) curves were drawn to
predict the survival time of patients in terms of the degree of
gene methylation.
Survival and correlation analysis
Survival analysis was performed by combining the
identified prognostic aberrantly expressed methylation-driven genes
with the corresponding gene expression data. The survival curves
were rendered using the Survival Package in R. The resulting
prognostic genes were considered as key genes in HCC because their
methylation degree and gene expression level were significantly
correlated with prognosis. The abnormal methylation was thought to
be associated with gene expression. In order to examine
correlation, methylation related loci of the key genes were
extracted from the downloaded HCC methylation data to assess the
correlation between the key gene methylation loci and gene
expression (the screening criteria were |Cor|. P>|0.3|; Cor is
correlation).
Results
TCGA data processing and screening of
differentially expressed methylation-driven genes
HCC-related level 3 RNA-Seq data (374 HCC samples
and 50 control samples) and methylation data (380 HCC samples and
50 control samples) were downloaded from TCGA. A total of 238
methylation-driven genes were identified according to the
predefined conditions using Perl script and the R package. The
Pheatmap package was used to cluster the methylation-driven genes
(Fig. 1). The distribution map of
the degree of methylation of some of the differentially methylated
genes is shown in Fig. 2.
GO enrichment and pathway analysis of
aberrantly expressed methylation-driven genes
Functional enrichment analysis was conducted on the
identified genes to understand the functional role of the aberrant
methylation-driven genes in HCC. The results showed that the
aberrantly expressed methylation-driven genes were associated with
molecular functions (MFs), biological processes (BPs) and cellular
components (CCs). In the BP cluster, the genes were mainly
associated with ‘epithelial cell morphogenesis in placental
branching’, ‘positive regulation of lipid catabolism’, ‘cholesterol
homeostasis’, ‘cholesterol metabolic process’ and ‘lipoprotein
metabolic process’. The MF included ‘endopeptidase inhibitor
activity’, ‘cholesterol transporter activity’ and ‘transcription
factor activity’, ‘sequence-specific DNA binding’. The CC cluster
included ‘extracellular space’, ‘blood microparticles’ and the
‘apical plasma membrane’ (Table
II).
| Table II.GO functional enrichment of
aberrantly expressed methylation-driven genes. |
Table II.
GO functional enrichment of
aberrantly expressed methylation-driven genes.
| Function | Term | Enrichment | Count | P-value | FDR |
|---|
| BP | GO:0060672 | Epithelial cell
morphogenesis in placental branching | 3 | 0.00036 | 0.581 |
|
| GO:0050996 | Positive regulation
of lipid catabolic process | 3 | 0.00118 | 1.897 |
|
| GO:0002819 | Regulation of
adaptive immune response | 3 | 0.00176 | 2.813 |
|
| GO:0042632 | Cholesterol
homeostasis | 5 | 0.00542 | 8.427 |
|
| GO:0008203 | Cholesterol
metabolic process | 5 | 0.00671 | 10.339 |
|
| GO:0042157 | Lipoprotein
metabolic process | 4 | 0.00836 | 12.724 |
| CC | GO:0072562 | Blood
microparticle | 8 | 0.00146 | 1.822 |
|
| GO:0005615 | Extracellular
space | 26 | 0.00682 | 8.244 |
|
| GO:0016324 | Apical plasma
membrane | 9 | 0.0152 | 17.546 |
|
| GO:0005576 | Extracellular
region | 28 | 0.0176 | 19.967 |
| MF | GO:0004866 | Endopeptidase
inhibitor activity | 5 | 0.00106 | 1.438 |
|
| GO:0017127 | Cholesterol
transporter activity | 3 | 0.0121 | 15.423 |
|
| GO:0003700 | Transcription
factor activity, Sequence-specific DNA binding | 20 | 0.0128 | 16.189 |
Pathway enrichment analysis was performed on the 238
differentially expressed methylation-driven genes using
ConsensusPathDB. A total of 14 pathways with significance
(P<0.01) were identified, the most relevant of which was the
pathway responsible for the regulation of insulin-like growth
factor (IGF) transport and uptake by insulin-like growth factor
binding proteins (IGFBPs), plasma lipoprotein remodeling and
statins (Fig. 3).
Prognostic implications of aberrantly
expressed methylation-driven genes
Univariate Cox regression analysis of the aberrantly
expressed methylation-driven genes in patients with HCC showed that
28 genes were significantly associated with OS (P<0.05; Table III). Multivariate Cox regression
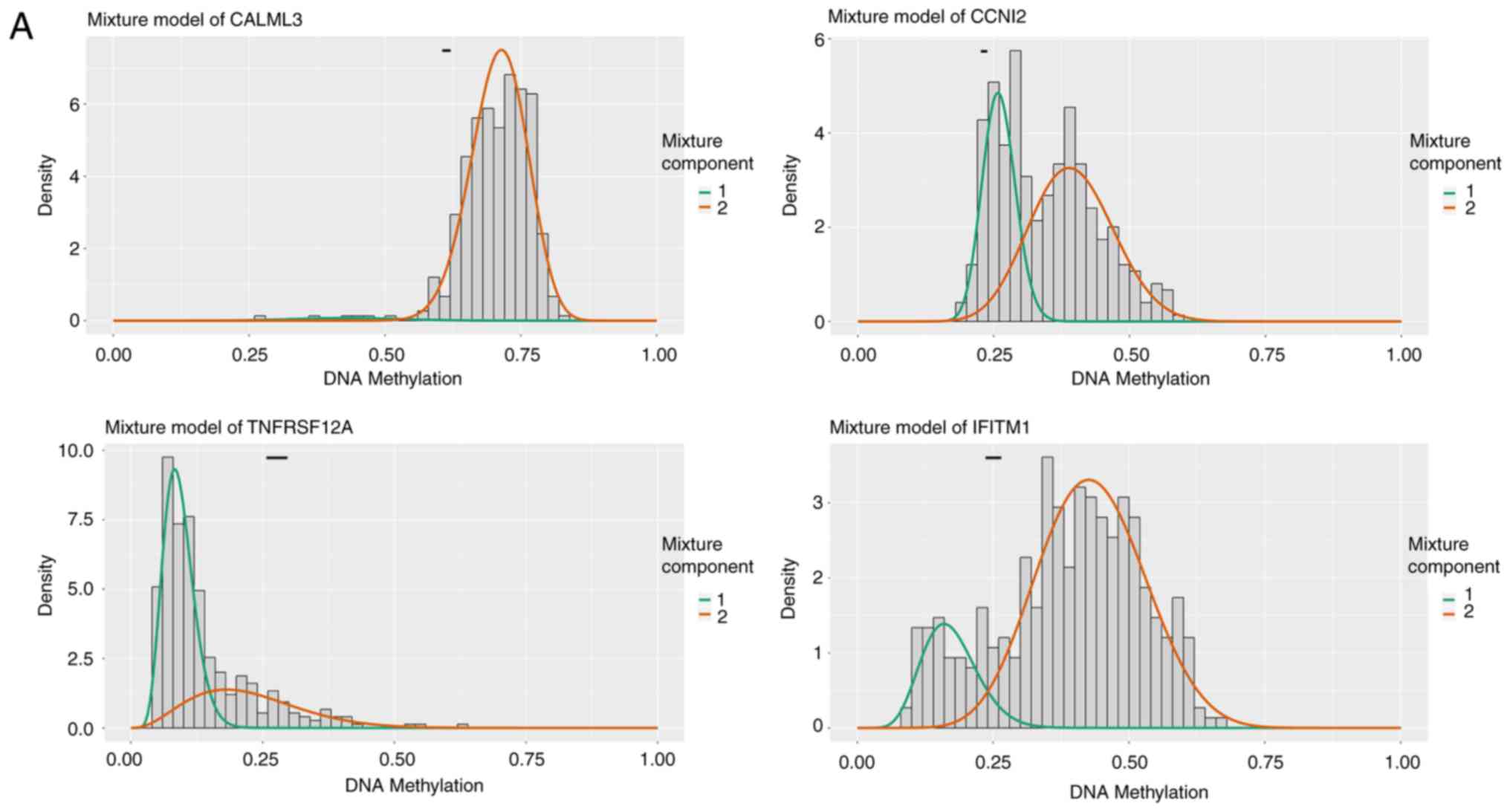
analysis was used to construct a prognostic risk model. As shown in
Table IV, eight genes were found
with prognostic significance: Calmodulin-like protein 3 (CALML3),
cyclin I family member 2 (CCNI2), tumor necrosis factor receptor
superfamily member 12A (TNFRSF12A), interferon induced
transmembrane protein 1 (IFITM1), ectonucleotide
pyrophosphatase/phosphodiesterase 7 pseudogene 13 (ENPP7P13),
D-dopachrome decarboxylase (DDT), RASAL2-antisense RNA1
(RASAL2-AS1) and ankyrin repeat domain 22 (ANKRD22). The following
risk model was constructed: Risk score=CALML3 (degree of
methylation) × (−4.860) + CCNI2 × (2.071) + TNFRSF12A × (−3.369) +
IFITM1 × (1.203) + ENPP7P13 × (−1.366) + DDT × (2.139) + RASAL2-AS1
× (−1.384) + ANKRD22 × (−3.215). The median risk score derived from
this model was set as the cutoff value for stratifying the 376
patients into the high-risk (n=188) or low-risk groups (n=188). The
relationship between HCC risk and the degree of methylation was
illustrated in Fig. 4. The
Kaplan-Meier method was used to plot the survival curves for the
low- and high-risk HCC groups (Fig.
5). The 5-year survival rate of patients was 35.8% [95%
confidence interval (CI)=27.1–47.4%] in the high-risk group and
61.7% (95% CI=51.4–74.2%) in the low-risk group. The difference
between the groups was significant (P<0.0001). The ROC curve had
an area under the curve of 0.742, indicating that this model is
able to predict the survival rate of patients with HCC.
| Table III.Methylation-driven genes
significantly associated with overall survival in hepatocellular
carcinoma using univariate Cox regression analysis. |
Table III.
Methylation-driven genes
significantly associated with overall survival in hepatocellular
carcinoma using univariate Cox regression analysis.
| Gene | HR | Z | P-value |
|---|
| ANKRD22 | 0.033 | −3.352 | 0.001 |
| LIME1 | 7.012 | 3.181 | 0.001 |
| RASAL2-AS1 | 0.187 | −3.086 | 0.002 |
| LINC00346 | 0.064 | −3.054 | 0.002 |
| ENPP7P13 | 0.164 | −2.971 | 0.003 |
| TNFRSF12A | 0.050 | −2.944 | 0.003 |
| GPR75 | 6.951 | 2.830 | 0.005 |
| DDT | 24.444 | 2.442 | 0.015 |
| PABPC1P4 | 6.664 | 2.441 | 0.015 |
| EPHX3 | 5.015 | 2.379 | 0.017 |
| SFN | 0.248 | −2.352 | 0.019 |
| TMPRSS5 | 0.059 | −2.343 | 0.019 |
| RABGGTB | 0.235 | −2.249 | 0.024 |
| FITM1 | 10.908 | 2.242 | 0.025 |
| CAPS | 0.260 | −2.199 | 0.028 |
| IFITM1 | 4.161 | 2.179 | 0.029 |
| CCNI2 | 7.200 | 2.152 | 0.031 |
| RPL39L | 6.896 | 2.148 | 0.032 |
| USP44 | 4.258 | 2.138 | 0.033 |
| LCAT | 9.746 | 2.130 | 0.033 |
| RARRES2P8 | 0.311 | −2.069 | 0.039 |
| HPX | 6.227 | 2.057 | 0.040 |
| NQO1 | 0.398 | −2.055 | 0.040 |
| GSTM1 | 3.443 | 2.047 | 0.041 |
| RNF135 | 3.878 | 2.030 | 0.042 |
| PPIEL | 6.030 | 2.030 | 0.042 |
| LDHB | 3.933 | 1.998 | 0.046 |
| CALML3 | 0.067 | −1.971 | 0.049 |
| Table IV.Methylation-driven genes
significantly associated with overall survival in hepatocellular
carcinoma using multivariate Cox regression analysis. |
Table IV.
Methylation-driven genes
significantly associated with overall survival in hepatocellular
carcinoma using multivariate Cox regression analysis.
| Gene | coef | exp(coef) | se(coef) | Z-value | P-value |
|---|
| CALML3 | −4.860 | 0.008 | 1.344 | −3.617 | 0.0003 |
| CCNI2 | 2.071 | 7.931 | 1.095 | 1.891 | 0.059 |
| TNFRSF12A | −3.369 | 0.034 | 1.008 | −3.342 | 0.0008 |
| IFITM1 | 1.203 | 3.329 | 0.716 | 1.680 | 0.093 |
| ENPP7P13 | −1.366 | 0.255 | 0.711 | −1.920 | 0.055 |
| DDT | 2.139 | 8.493 | 1.351 | 1.584 | 0.113 |
| RASAL2-AS1 | −1.384 | 0.251 | 0.570 | −2.426 | 0.015 |
| ANKRD22 | −3.215 | 0.040 | 1.060 | −3.033 | 0.002 |
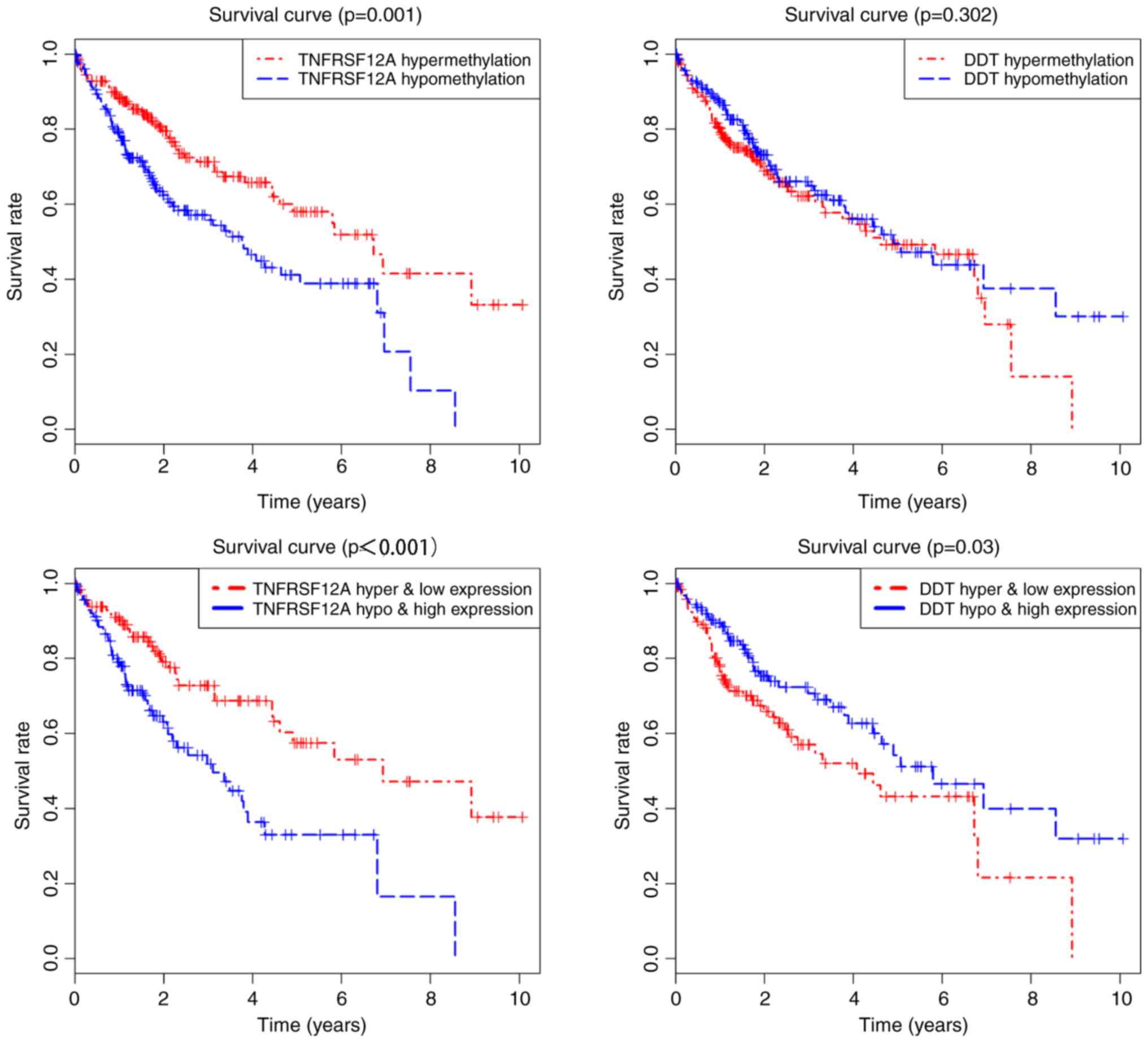
Association between different
methylation loci and gene expression
P<0.05 was used as a screening standard for
combinatorial survival. The methylation degree and gene expression
level of TNFRSF12A and DDT were significantly associated with the
prognosis of HCC (Fig. 6). In
addition, the methylation loci of the prognostic genes were
identified using TCGA. The correlation between the different
methylation loci and gene expression was analyzed using |Cor|.
P>|0.3| was used as the screening cutoff value. It was found
that the expression of TNFRSF12A and DDT was related to the
methylation level of multiple loci (Fig. 7). TNFRSF12A was generally in a
hypomethylated high expression state in patients with HCC, while
DDT was in a hypermethylated low expression state, suggesting that
TNFRSF12A may be a proto-oncogene and DDT may be a tumor suppressor
gene.
Discussion
The incidence of hepatitis B (HBV) related HCC is
expected to decrease with the increasing rate of vaccinations
against HBV and the cumulative impact of new generation antiviral
drugs (20). However, alcoholism,
diabetes, obesity and metabolic syndrome play an important role in
hepatocellular carcinoma in regions with a low prevalence of HBV,
especially in western populations. As a result, the prevalence of
HCC will continue to increase (21). Diet is also a primary factors
affecting DNA methylation (22).
DNA methylation is dependent on the availability of
s-adenosylmethionine (SAM), while methyl donors in food (including
folate, betaine and choline) are linked to the synthesis of SAM
(23,24). A methyl-deficient diet has been
reported to reduce the concentration of SAM in the liver, leading
to the methylation of CpG islands in 164 genes in the livers of
mice; these genes are involved in DNA damage and repair, lipid and
glucose metabolism, and the progression of fibrosis, which can
ultimately lead to HCC (25,26).
At present, the pathogenesis of HCC has not been
clearly elucidated. HCC is related with multiple gene mutations and
epigenetic aberrations (27).
Mechanistically, DNA methylation leads to transcriptional silencing
in one of two ways: i) Methylation at the CpG site hinders the
spatial accessibility of transcription factors to homologous
binding sites in various gene promoters (28); and ii) the direct binding of
methyl-CpG-binding domain proteins to methylated DNA, causing
transcriptional inhibition (29).
It was hypothesized that the methylation-driven genes aberrantly
expressed between patient with HCC and normal samples may be useful
for predicting the prognosis of HCC. To test this, R script was
used to combine methylation and transcriptome data, identifying 238
methylation driven-genes. The subsequent functional enrichment of
these genes revealed the potential functional targets, such as
‘blood microparticles’, ‘extracellular space’ and ‘apical plasma
membranes’, interfering with lipid metabolism in hepatocytes and
the ‘regulation of adaptive immune responses’ by affecting
‘endopeptidase inhibitor activity’, ‘cholesterol transporter
activity’, ‘transcription factor activity’, ‘sequence-specific DNA
binding’. The resulting pathways predominantly regulated IGF
transport and IGFBP uptake, plasma lipoprotein remodeling and
statins. These results suggested that high-fat, hypomethylated
diets can affect DNA methylation, further affecting lipid
metabolism in the body. This cycle can lead to liver steatosis,
inflammation, fibrosis and cancer.
Multivariate Cox regression was used to generate a
prognostic risk model, which was used to evaluate prognosis. The
5-year survival rate was 35.8% (95% CI=27.1–47.4%) in the high-risk
group and 61.7% (95% CI=51.4–74.2%) in the low-risk group
(P<0.0001). The prognostic model constructed showed a level of
accuracy and sensitivity in evaluating the prognosis of patients
with HCC.
The expression of TNFRSF12A is increased in various
tumors, especially in HCC and breast cancer (30). As the sole signal receptor of the
proinflammatory cytokine TNF superfamily member 12, TNFRSF12A is
involved in stimulating many signal transduction pathways,
including the nuclear factor-κB pathway. It has been reported that
the knockout of the differentially expressed TNFRSF12A gene
inhibited the proliferation and migration of HCC cells in
vitro (31). High levels of
TNFRSF12A expression are associated with the upregulation of matrix
metallopeptidase-9 and may be important for the progression of
breast cancer; furthermore, TNFRSF12A-targeted therapy can improve
survival (32). In the present
study, TNFRSF12A was found to be hypomethylated in patients with
HCC and that this was significantly correlated with prognosis. The
methylation status of TNFRSF12A was negatively correlated with gene
expression. The hypomethylated status of TNFRSF12A and its high
expression corresponded to a significantly lower survival rate.
Therefore, TNFRSF12A can be used as an independent prognostic
predictor of HCC. Further analysis of the methylation loci showed
that cg00510447 and cg26808293 were also negatively correlated with
gene expression. It is, therefore, speculated that the
hypomethylation of cg00510447 and cg26808293 resulted in the high
expression of the TNFRSF12A gene, which may affect metabolism in
HCC, and the proliferation and migration of tumor cells.
DDT (MIF2), the second member of the
multi-functional proinflammatory protein macrophage mobility
inhibitor superfamily, is an important mediator of the
inflammation-cancer axis and plays a role in angiogenesis by
inducing angiogenic factors, such as vascular endothelial growth
factor and interleukin-8 (33). In
lung adenocarcinoma cells, MIF and DDT have a cumulative effect on
the induction of these carcinogenic factors, indicating that MIF
and DDT may play a synergistic role in malignant diseases (34), however, little is known about the
methylation of DDT in HCC. In the present study, it was found that
DDT was hypermethylated in HCC, and that the prognosis of HCC was
poorer in the hypermethylated low expression group. Further
analysis indicated that cg11060661 and cg16132339 were also
negatively correlated with gene expression. The results suggested
that DDT plays a protective role in HCC and may act as a tumor
suppressor gene. Further studies are required to elucidate the
specific mechanisms underlying the preliminary findings of the
present study.
Unlike mutations, epigenetic changes are reversible,
especially DNA methylation and histone modifications (35). The expression of genes silenced by
DNA methylation can be promoted in cancer cell lines using
demethylating agents, such as 5-azacytidine and
5-azacytidine-2′-deoxycytidine (36), which are used in myelodysplastic
syndromes and acute myeloid leukemia (37). As a novel DNA methyltransferase
inhibitor, Zebularine can inhibit DNA methylation by forming a
covalent complex with DNA methyltransferase 1. The stability and
half-life of Zebularine in neutral aqueous solutions is
significantly better than azatadine and histamine (38). In animal experiments, Andersen
et al (39) demonstrated
that Zebularine response genes and demethylation signatures
predictive of clinical outcome in patients with HCC, could be used
as a new diagnostic and prognostic tool for selecting patients with
HCC who may benefit from epigenetic therapy. In the target group
with the highest degree of DNA CpG methylation, epigenetic therapy
with Zebularine successfully inhibited tumor cell proliferation and
increased apoptosis.
There have been a number of previous studies on DNA
methylation in HCC. For example, the methylation of P14ARF, growth
arrest and DNA damage inducible GADD45 β and protocadherin 8 is
associated with HCC (15,40,41).
In addition, Tao et al (42) found that seven genes (WNK2,
EMILIN2, TLX3, TM6SF1, TRIM58, HIST1H4F and GRASP) were
hypermethylated in HBV-related HCC and hypomethylated in paired
adjacent liver tissues. Fan et al (43), using |logFC|≥2 and P≤0.05,
identified six hub genes that were altered in HCC from The Gene
Expression Omnibus; patients with high expression of mitotic arrest
deficient 2-like 1, cell division cycle 20 and cyclin B1 and low
expression cyclin D1, androgen receptor and estrogen receptor 1 had
a shorter OS. However, these previous studies, including (15,40–43),
only assessed the value of DNA methylation of single or multiple
genes in HCC. Although DNA methylation determines when, where and
how genes are expressed, DNA methylation at a single site does not
necessarily affect the prognosis of a patient as there are multiple
methylation sites on each gene (44). Only when DNA methylation affects
gene expression can it affect the prognosis of a patient.
Therefore, the present study combined gene expression and DNA
methylation (Cor≤-0.3) from TCGA to conduct a comprehensive
analysis, using this a prognostic risk model was constructed for
HCC using univariate and multivariate COX regression. This model
can also be used to quantify the survival time of patients with
HCC, and has strong clinical applicability. However, the present
study is comprised only of bioinformatics analysis with no
experimental verification. Therefore, future studies should collect
cancer samples and the clinical data of patients with HCC in order
to verify the accuracy of the model by detecting the DNA
methylation status of the eight prognostic genes identified in the
present study.
It can be concluded from the analysis conducted in
the present study that the occurrence and development of HCC are
closely related to the eight methylation-driven genes identified,
including CALML3, CCNI2, TNFRSF12A, IFITM1, ENPP7P13, DDT,
RASAL2-AS1 and ANKRD22. The degree of methylation and gene
expression level of TNFRSF12A and DDT are significantly correlated
with the prognosis of HCC and were negatively correlated with the
methylation status of the following loci: cg00510447, cg26808293,
cg11060661 and cg16132339. Experimental and clinical trials are
required to further investigate the findings of the present study,
which may be important for the diagnosis, treatment and prognosis
of patients with HCC.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81671946) and the
Medical Scientific Research Foundation of Guangdong Province (grant
no. A2015479).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
JL conceived and designed the study. Data analysis
was conducted by XG, JL and XG, and NC contributed to study
methodology, software use, study administration and data
validation. JL and XG wrote, reviewed and edited the manuscript.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
BP
|
biology process
|
|
CC
|
cellular component
|
|
DAVID
|
The Database for Annotation,
Visualization and Integrated Discovery
|
|
GO
|
Gene Ontology
|
|
HCC
|
hepatocellular carcinoma
|
|
IGF
|
insulin-like growth factor
|
|
IGFBPs
|
insulin-like growth factor binding
proteins
|
|
MF
|
molecular function
|
References
|
1
|
McGlynn KA, Petrick JL and London WT:
Global epidemiology of hepatocellular carcinoma: An emphasis on
demographic and regional variability. Clin Liver Dis. 19:223–238.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shen H and Laird PW: Interplay between the
cancer genome and epigenome. Cell. 153:38–55. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kanwal R, Gupta K and Gupta S: Cancer
epigenetics: An introduction. Methods Mol Biol. 1238:3–25. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yang IV and Schwartz DA: Epigenetic
control of gene expression in the lung. Am J Respir Crit Care Med.
183:1295–1301. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Widschwendter M, Jiang G, Woods C, Müller
HM, Fiegl H, Goebel G, Marth C, Müller-Holzner E, Zeimet AG, Laird
PW and Ehrlich M: DNA hypomethylation and ovarian cancer biology.
Cancer Res. 64:4472–4480. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yoder JA, Walsh CP and Bestor TH: Cytosine
methylation and the ecology of intragenomic parasites. Trends
Genet. 13:335–340. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jones PA: Functions of DNA methylation:
Islands, start sites, gene bodies and beyond. Nat Rev Genet.
13:484–492. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Baylin SB and Chen WY: Aberrant gene
silencing in tumor progression: Implications for control of cancer.
Cold Spring Harb Symp Quant Biol. 70:427–433. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jones PA and Baylin SB: The epigenomics of
cancer. Cell. 128:683–692. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gao C, Zhuang J, Zhou C, Ma K, Zhao M, Liu
C, Liu L, Li H, Feng F and Sun C: Prognostic value of aberrantly
expressed methylation gene profiles in lung squamous cell
carcinoma: A study based on The Cancer Genome Atlas. J Cell
Physiol. 234:6519–6528. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Feng L and Jin F: Screening of
differentially methylated genes in breast cancer and risk model
construction based on TCGA database. Oncol Lett. 16:6407–6416.
2018.PubMed/NCBI
|
|
13
|
Udali S, Guarini P, Ruzzenente A,
Ferrarini A, Guglielmi A, Lotto V, Tononi P, Pattini P, Moruzzi S,
Campagnaro T, et al: DNA methylation and gene expression profiles
show novel regulatory pathways in hepatocellular carcinoma. Clin
Epigenetics. 7:432015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhong XY, Yuan XM, Xu YY, Yin M, Yan WW,
Zou SW, Wei LM, Lu HJ, Wang YP and Lei QY: CARM1 methylates GAPDH
to regulate glucose metabolism and is suppressed in liver cancer.
Cell Rep. 24:3207–3223. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang H, Nie W and Huang F: The
correlation relationship between P14ARF gene DNA methylation and
primary liver cancer. Med Sci Monit. 21:3077–3082. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tomczak K, Czerwińska P and Wiznerowicz M:
The Cancer Genome Atlas (TCGA): An immeasurable source of
knowledge. Contemp Oncol (Pozn). 19:A68–A77. 2015.PubMed/NCBI
|
|
17
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: Limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Singh R and Mukhopadhyay K: Survival
analysis in clinical trials: Basics and must know areas. Perspect
Clin Res. 2:145–148. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang CB, Zhu P, Yang P, Cai JQ, Wang ZL,
Li QB, Bao ZS, Zhang W and Jiang T: Identification of high risk
anaplastic gliomas by a diagnostic and prognostic signature derived
from mRNA expression profiling. Oncotarget. 6:36643–36651.
2015.PubMed/NCBI
|
|
20
|
Chan SL, Wong VW, Qin S and Chan HL:
Infection and cancer: The case of Hepatitis B. J Clin Oncol.
34:83–90. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wallace MC, Preen D, Jeffrey GP and Adams
LA: The evolving epidemiology of hepatocellular carcinoma: A global
perspective. Expert Rev Gastroenterol Hepatol. 9:765–779. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tian Y, Wong WS, Chan LY and Cheng AS:
Epigenetic regulation of hepatocellular carcinoma in non-alcoholic
fatty liver disease. Semin Cancer Biol. 23:471–482. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kalhan SC, Edmison J, Marczewski S,
Dasarathy S, Gruca LL, Bennett C, Duenas C and Lopez R: Methionine
and protein metabolism in non-alcoholic steatohepatitis: Evidence
for lower rate of transmethylation of methionine. Clin Sci (Lond).
121:179–189. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Niculescu MD and Zeisel SH: Diet, methyl
donors and DNA methylation: Interactions between dietary folate,
methionine and choline. J Nutr. 132 (8 Suppl):2333S–2335S. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tryndyak VP, Han T, Muskhelishvili L,
Fuscoe JC, Ross SA, Beland FA and Pogribny IP: Coupling global
methylation and gene expression profiles reveal key
pathophysiological events in liver injury induced by a
methyl-deficient diet. Mol Nutr Food Res. 55:411–418. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pogribny IP, James SJ and Beland FA:
Molecular alterations in hepatocarcinogenesis induced by dietary
methyl deficiency. Mol Nutr Food Res. 56:116–125. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bruix J, Gores GJ and Mazzaferro V:
Hepatocellular carcinoma: Clinical frontiers and perspectives. Gut.
63:844–855. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jones PA and Takai D: The role of DNA
methylation in mammalian epigenetics. Science. 293:1068–1070. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Karpf AR and Jones DA: Reactivating the
expression of methylation silenced genes in human cancer. Oncogene.
21:5496–5503. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wiley SR, Cassiano L, Lofton T,
Davis-Smith T, Winkles JA, Lindner V, Liu H, Daniel TO, Smith CA
and Fanslow WC: A novel TNF receptor family member binds TWEAK and
is implicated in angiogenesis. Immunity. 15:837–846. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang T, Ma S, Qi X, Tang X, Cui D, Wang Z,
Chi J, Li P and Zhai B: Knockdown of the differentially expressed
gene TNFRSF12A inhibits hepatocellular carcinoma cell proliferation
and migration in vitro. Mol Med Rep. 15:1172–1178. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yang J, Min KW, Kim DH, Son BK, Moon KM,
Wi YC, Bang SS, Oh YH, Do SI, Chae SW, et al: High TNFRSF12A level
associated with MMP-9 overexpression is linked to poor prognosis in
breast cancer: Gene set enrichment analysis and validation in
large-scale cohorts. PLoS One. 13:e02021132018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
O'Reilly C, Doroudian M, Mawhinney L and
Donnelly SC: Targeting MIF in cancer: Therapeutic strategies,
current developments, and future opportunities. Med Res Rev.
36:440–460. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Coleman AM, Rendon BE, Zhao M, Qian MW,
Bucala R, Xin D and Mitchell RA: Cooperative regulation of
non-small cell lung carcinoma angiogenic potential by macrophage
migration inhibitory factor and its homolog, D-dopachrome
tautomerase. J Immunol. 181:2330–2337. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
O'Brien CA, Pollett A, Gallinger S and
Dick JE: A human colon cancer cell capable of initiating tumour
growth in immunodeficient mice. Nature. 445:106–110. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang M, Xiao J, Shen M, Yahong Y, Tian R,
Zhu F, Jiang J, Du Z, Hu J, Liu W and Qin R: Isolation and
characterization of tumorigenic extrahepatic cholangiocarcinoma
cells with stem cell-like properties. Int J Cancer. 128:72–81.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Klepin HD: Myelodysplastic syndromes and
acute myeloid leukemia in the elderly. Clin Geriat Med. 32:155–173.
2016. View Article : Google Scholar
|
|
38
|
Zhou L, Cheng X, Connolly BA, Dickman MJ,
Hurd PJ and Hornby DP: Zebularine: A novel DNA methylation
inhibitor that forms a covalent complex with DNA
methyltransferases. J Mol Biol. 321:591–599. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Andersen JB, Factor VM, Marquardt JU,
Raggi C, Lee YH, Seo D, Conner EA and Thorgeirsson SS: An
integrated genomic and epigenomic approach predicts therapeutic
response to zebularine in human liver cancer. Sci Transl Med.
2:54ra772010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hou XJ, Zhao QD, Jing YY, Han ZP, Yang X,
Wei LX, Zheng YT, Xie F and Zhang BH: Methylation mediated Gadd45β
enhanced the chemosensitivity of hepatocellular carcinoma by
inhibiting the stemness of liver cancer cells. Cell Biosci.
7:632017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cheng Z, Yunfei P, Fan Y, Qin R, Liu W and
Zhang C: PCDH8 is frequently inactivated by promoter
hypermethylation in liver cancer: Diagnostic and clinical
significance. J Cancer. 7:446–452. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tao R, Li J, Xin J, Wu J, Guo J, Zhang L,
Jiang L, Zhang W, Yang Z and Li L: Methylation profile of single
hepatocytes derived from hepatitis B virus-related hepatocellular
carcinoma. PLoS One. 6:e198622011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Fan G, Tu Y, Chen C, Sun H, Wan C and Cai
X: DNA methylation biomarkers for hepatocellular carcinoma. Cancer
Cell Int. 18:1402018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zeidler R, de Freitas Soares BL, Bader A
and Giri S: Molecular epigenetic targets for liver diseases:
Current challenges and future prospects. Drug Discov Today.
22:1620–1636. 2017. View Article : Google Scholar : PubMed/NCBI
|