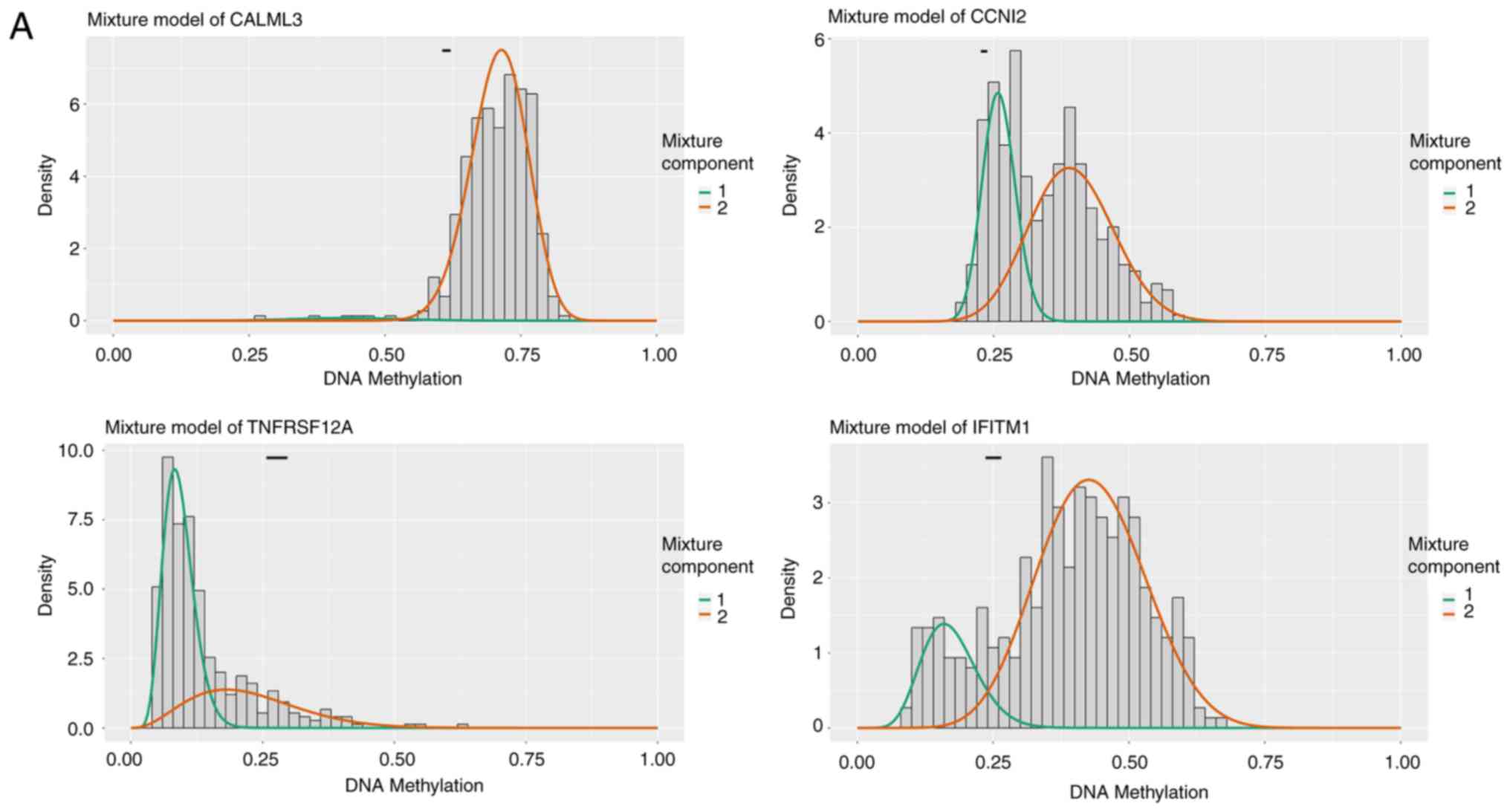
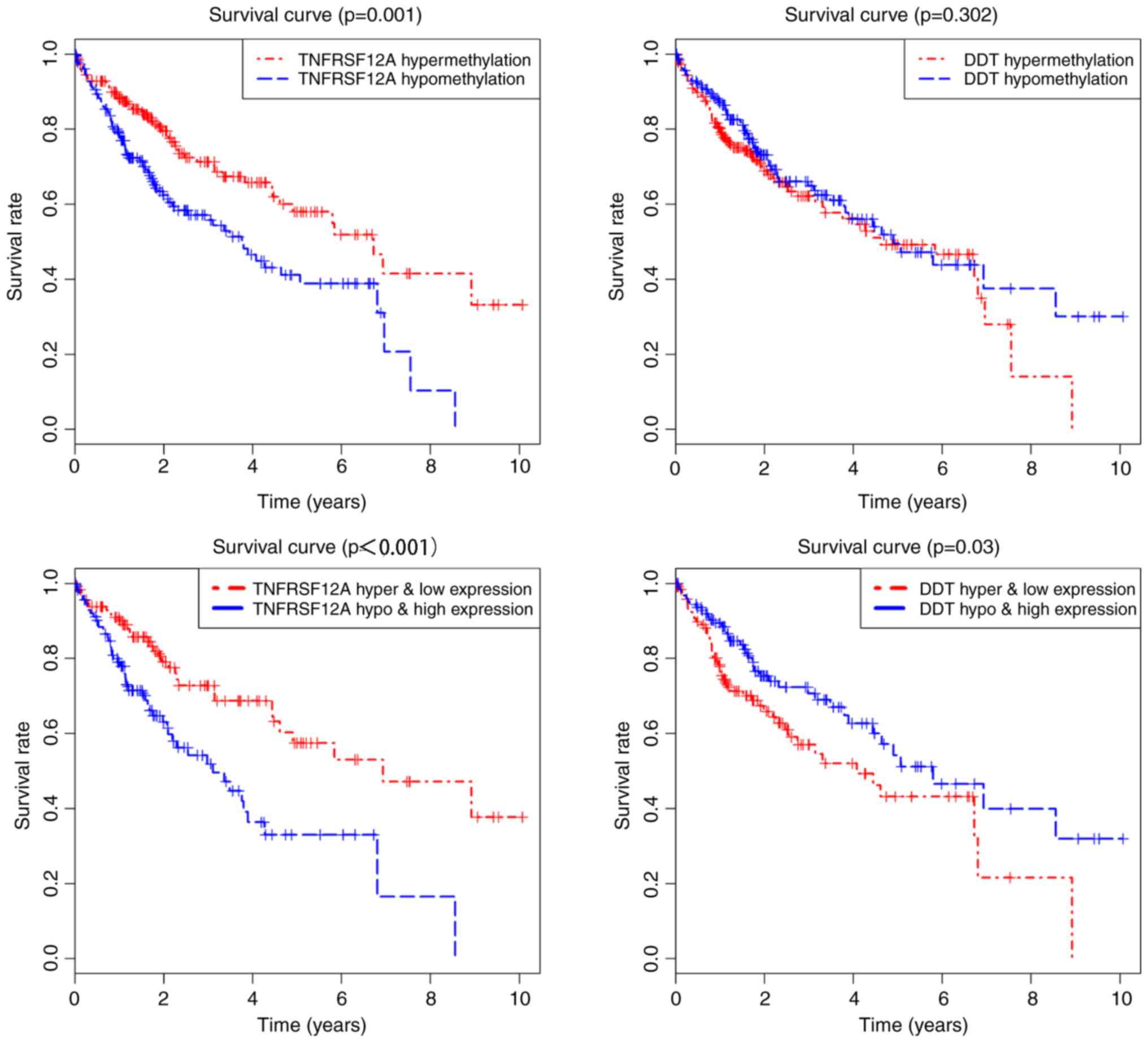
|
1
|
McGlynn KA, Petrick JL and London WT:
Global epidemiology of hepatocellular carcinoma: An emphasis on
demographic and regional variability. Clin Liver Dis. 19:223–238.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shen H and Laird PW: Interplay between the
cancer genome and epigenome. Cell. 153:38–55. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kanwal R, Gupta K and Gupta S: Cancer
epigenetics: An introduction. Methods Mol Biol. 1238:3–25. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yang IV and Schwartz DA: Epigenetic
control of gene expression in the lung. Am J Respir Crit Care Med.
183:1295–1301. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Widschwendter M, Jiang G, Woods C, Müller
HM, Fiegl H, Goebel G, Marth C, Müller-Holzner E, Zeimet AG, Laird
PW and Ehrlich M: DNA hypomethylation and ovarian cancer biology.
Cancer Res. 64:4472–4480. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yoder JA, Walsh CP and Bestor TH: Cytosine
methylation and the ecology of intragenomic parasites. Trends
Genet. 13:335–340. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jones PA: Functions of DNA methylation:
Islands, start sites, gene bodies and beyond. Nat Rev Genet.
13:484–492. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Baylin SB and Chen WY: Aberrant gene
silencing in tumor progression: Implications for control of cancer.
Cold Spring Harb Symp Quant Biol. 70:427–433. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jones PA and Baylin SB: The epigenomics of
cancer. Cell. 128:683–692. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gao C, Zhuang J, Zhou C, Ma K, Zhao M, Liu
C, Liu L, Li H, Feng F and Sun C: Prognostic value of aberrantly
expressed methylation gene profiles in lung squamous cell
carcinoma: A study based on The Cancer Genome Atlas. J Cell
Physiol. 234:6519–6528. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Feng L and Jin F: Screening of
differentially methylated genes in breast cancer and risk model
construction based on TCGA database. Oncol Lett. 16:6407–6416.
2018.PubMed/NCBI
|
|
13
|
Udali S, Guarini P, Ruzzenente A,
Ferrarini A, Guglielmi A, Lotto V, Tononi P, Pattini P, Moruzzi S,
Campagnaro T, et al: DNA methylation and gene expression profiles
show novel regulatory pathways in hepatocellular carcinoma. Clin
Epigenetics. 7:432015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhong XY, Yuan XM, Xu YY, Yin M, Yan WW,
Zou SW, Wei LM, Lu HJ, Wang YP and Lei QY: CARM1 methylates GAPDH
to regulate glucose metabolism and is suppressed in liver cancer.
Cell Rep. 24:3207–3223. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang H, Nie W and Huang F: The
correlation relationship between P14ARF gene DNA methylation and
primary liver cancer. Med Sci Monit. 21:3077–3082. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tomczak K, Czerwińska P and Wiznerowicz M:
The Cancer Genome Atlas (TCGA): An immeasurable source of
knowledge. Contemp Oncol (Pozn). 19:A68–A77. 2015.PubMed/NCBI
|
|
17
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: Limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Singh R and Mukhopadhyay K: Survival
analysis in clinical trials: Basics and must know areas. Perspect
Clin Res. 2:145–148. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang CB, Zhu P, Yang P, Cai JQ, Wang ZL,
Li QB, Bao ZS, Zhang W and Jiang T: Identification of high risk
anaplastic gliomas by a diagnostic and prognostic signature derived
from mRNA expression profiling. Oncotarget. 6:36643–36651.
2015.PubMed/NCBI
|
|
20
|
Chan SL, Wong VW, Qin S and Chan HL:
Infection and cancer: The case of Hepatitis B. J Clin Oncol.
34:83–90. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wallace MC, Preen D, Jeffrey GP and Adams
LA: The evolving epidemiology of hepatocellular carcinoma: A global
perspective. Expert Rev Gastroenterol Hepatol. 9:765–779. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tian Y, Wong WS, Chan LY and Cheng AS:
Epigenetic regulation of hepatocellular carcinoma in non-alcoholic
fatty liver disease. Semin Cancer Biol. 23:471–482. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kalhan SC, Edmison J, Marczewski S,
Dasarathy S, Gruca LL, Bennett C, Duenas C and Lopez R: Methionine
and protein metabolism in non-alcoholic steatohepatitis: Evidence
for lower rate of transmethylation of methionine. Clin Sci (Lond).
121:179–189. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Niculescu MD and Zeisel SH: Diet, methyl
donors and DNA methylation: Interactions between dietary folate,
methionine and choline. J Nutr. 132 (8 Suppl):2333S–2335S. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tryndyak VP, Han T, Muskhelishvili L,
Fuscoe JC, Ross SA, Beland FA and Pogribny IP: Coupling global
methylation and gene expression profiles reveal key
pathophysiological events in liver injury induced by a
methyl-deficient diet. Mol Nutr Food Res. 55:411–418. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pogribny IP, James SJ and Beland FA:
Molecular alterations in hepatocarcinogenesis induced by dietary
methyl deficiency. Mol Nutr Food Res. 56:116–125. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bruix J, Gores GJ and Mazzaferro V:
Hepatocellular carcinoma: Clinical frontiers and perspectives. Gut.
63:844–855. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jones PA and Takai D: The role of DNA
methylation in mammalian epigenetics. Science. 293:1068–1070. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Karpf AR and Jones DA: Reactivating the
expression of methylation silenced genes in human cancer. Oncogene.
21:5496–5503. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wiley SR, Cassiano L, Lofton T,
Davis-Smith T, Winkles JA, Lindner V, Liu H, Daniel TO, Smith CA
and Fanslow WC: A novel TNF receptor family member binds TWEAK and
is implicated in angiogenesis. Immunity. 15:837–846. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang T, Ma S, Qi X, Tang X, Cui D, Wang Z,
Chi J, Li P and Zhai B: Knockdown of the differentially expressed
gene TNFRSF12A inhibits hepatocellular carcinoma cell proliferation
and migration in vitro. Mol Med Rep. 15:1172–1178. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yang J, Min KW, Kim DH, Son BK, Moon KM,
Wi YC, Bang SS, Oh YH, Do SI, Chae SW, et al: High TNFRSF12A level
associated with MMP-9 overexpression is linked to poor prognosis in
breast cancer: Gene set enrichment analysis and validation in
large-scale cohorts. PLoS One. 13:e02021132018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
O'Reilly C, Doroudian M, Mawhinney L and
Donnelly SC: Targeting MIF in cancer: Therapeutic strategies,
current developments, and future opportunities. Med Res Rev.
36:440–460. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Coleman AM, Rendon BE, Zhao M, Qian MW,
Bucala R, Xin D and Mitchell RA: Cooperative regulation of
non-small cell lung carcinoma angiogenic potential by macrophage
migration inhibitory factor and its homolog, D-dopachrome
tautomerase. J Immunol. 181:2330–2337. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
O'Brien CA, Pollett A, Gallinger S and
Dick JE: A human colon cancer cell capable of initiating tumour
growth in immunodeficient mice. Nature. 445:106–110. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang M, Xiao J, Shen M, Yahong Y, Tian R,
Zhu F, Jiang J, Du Z, Hu J, Liu W and Qin R: Isolation and
characterization of tumorigenic extrahepatic cholangiocarcinoma
cells with stem cell-like properties. Int J Cancer. 128:72–81.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Klepin HD: Myelodysplastic syndromes and
acute myeloid leukemia in the elderly. Clin Geriat Med. 32:155–173.
2016. View Article : Google Scholar
|
|
38
|
Zhou L, Cheng X, Connolly BA, Dickman MJ,
Hurd PJ and Hornby DP: Zebularine: A novel DNA methylation
inhibitor that forms a covalent complex with DNA
methyltransferases. J Mol Biol. 321:591–599. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Andersen JB, Factor VM, Marquardt JU,
Raggi C, Lee YH, Seo D, Conner EA and Thorgeirsson SS: An
integrated genomic and epigenomic approach predicts therapeutic
response to zebularine in human liver cancer. Sci Transl Med.
2:54ra772010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hou XJ, Zhao QD, Jing YY, Han ZP, Yang X,
Wei LX, Zheng YT, Xie F and Zhang BH: Methylation mediated Gadd45β
enhanced the chemosensitivity of hepatocellular carcinoma by
inhibiting the stemness of liver cancer cells. Cell Biosci.
7:632017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cheng Z, Yunfei P, Fan Y, Qin R, Liu W and
Zhang C: PCDH8 is frequently inactivated by promoter
hypermethylation in liver cancer: Diagnostic and clinical
significance. J Cancer. 7:446–452. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tao R, Li J, Xin J, Wu J, Guo J, Zhang L,
Jiang L, Zhang W, Yang Z and Li L: Methylation profile of single
hepatocytes derived from hepatitis B virus-related hepatocellular
carcinoma. PLoS One. 6:e198622011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Fan G, Tu Y, Chen C, Sun H, Wan C and Cai
X: DNA methylation biomarkers for hepatocellular carcinoma. Cancer
Cell Int. 18:1402018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zeidler R, de Freitas Soares BL, Bader A
and Giri S: Molecular epigenetic targets for liver diseases:
Current challenges and future prospects. Drug Discov Today.
22:1620–1636. 2017. View Article : Google Scholar : PubMed/NCBI
|