Introduction
NOP2/Sun domain family member 2 (NSUN2), also known
as Misu, is a type of RNA methyltransferase that is targeted by Myc
and mediates Myc-induced cell proliferation and growth (1). Numerous studies have indicated that
NSUN2 may be implicated in multiple biological processes, including
cell proliferation, migration, testis differentiation, stem cell
differentiation, and diseases such as intellectual disability and
human cancer (1–4).
Previous studies have demonstrated that the NSUN2
gene is upregulated in several types of cancer, including
esophageal, stomach, liver, pancreas, uterine cervix, prostate,
kidney, bladder, thyroid, and breast cancers (5). High expression of NSUN2 leads to
increased proliferation and metastasis of tumor cells (1,2),
indicating that NSUN2 may be a valuable target for cancer therapy
and a potential cancer diagnostic marker. Therefore, understanding
NSUN2 function is extremely important in basic and clinical
studies.
NSUN2 was recently reported to methylate various
types of RNAs and modulate their functions. NSUN2 is able to
stabilize p16 mRNA by methylating its 3′ untranslated region at
A988 (6). Cyclin-dependent kinase
1 and p21 mRNAs are also methylated by NSUN2, which impacts the
efficiency of their translation (7,8).
NSUN2 may also methylate transfer RNAs (tRNAs) and microRNAs
(miRNA) and influence their processing (9,10).
However, it remains unknown whether lncRNAs are modulated by
NSUN2.
lncRNAs, which are longer than 200 nucleotides (nt),
have been identified as the most abundant non-protein-coding RNAs.
Functional studies have revealed that lncRNAs are implicated in
diverse biological processes and disease-associated pathways,
including cell proliferation and differentiation, stem cell
pluripotency, and tumorigenesis and metastasis (11,12).
Multiple lines of evidence have suggested that the dysregulation of
lncRNA is associated with cancer, indicating an important role of
lncRNAs in tumorigenesis (12,13).
Thus, it was speculated that NSUN2 may be implicated in
tumorigenesis through its modulation of lncRNAs.
To investigate the effect of NSUN2 on lncRNA
expression in cancer, a NSUN2-deficient HepG2 cell line was
established with the clustered regularly interspaced short
palindromic repeats/caspase 9 (CRISPR/Cas9) system, and expression
profiles of lncRNAs were obtained by RNA-sequencing (RNA-seq)
analysis.
Materials and methods
Cell line
The HepG2 cell line (a liver cancer cell line) was
purchased from the American Type Culture Collection (cat. no.
HB-8065; Manassas, VA, USA). Cells were maintained in
Dulbecco's-modified Eagle's medium (DMEM, HyClone; GE Healthcare
Life Sciences, Logan, UT, USA) supplemented with 10% fetal bovine
serum (HyClone; GE Healthcare Life Sciences), 100 U/ml penicillin,
and 100 mg/ml streptomycin at 37°C with 5% CO2.
The NSUN2-deficient HepG2 cell line was established
as previously described (14). In
brief, two selection markers and a poly(A) signal were inserted
into the first exon of the NSUN2 gene via CRISPR/Cas9-mediated
homology-directed repair. Double marker selection was employed for
screening the clonal cell lines for successful biallelic
integration of the poly(A) signal. Finally, the efficiency of gene
silencing was evaluated by reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) using primers specific for
NSUN2 (Table I).
 | Table I.Primers for reverse-transcription
quantitative polymerase chain reaction. |
Table I.
Primers for reverse-transcription
quantitative polymerase chain reaction.
| Gene symbol | Forward primer
(5′-3′) | Reverse primer
(5′-3′) |
|---|
| ACTB |
AAGACCTGTACACCAACACAG |
AGGGCAGTGATCTCCTTCT |
| NSUN2 |
ATCTTGAGAAAATCGCCACAC |
ATCATTCGCAATAACAAATCCCT |
|
ENST00000623282 |
TTTAACTGGACTCTTGGCACT |
TGTTAAGCATACCCCTACCTG |
|
NONHSAT194852.1 |
GGAGTGTGTCAGTGTCCACC |
GCACCAAATCTGCTTTCGCA |
|
NONHSAT016752.2 |
AGCACACAGGCATCTAGTGG |
CAGGCTGCTCTTCCATTCCA |
|
NONHSAT053044.2 |
AACCTTAGGCAAGTTACGTT |
GATAACTATGTGCTAGGCTCT |
|
NONHSAT087855.2 |
GGGACGGCTTCTCGGCAAT |
TTCTGGGTGTATCCAGTTGTGC |
|
ENST00000585065 |
GAAGTTAGTCCCTGGGGTGTC |
TGGGGCACAAATCCACATCT |
|
NONHSAT180118.1 |
CCAGTTCAGCCAGTACGTGT |
CCTTTCCCTTTTAGATCCCTGC |
|
ENST00000366365 |
GTTGAGCCTGCCAAGTTGTG |
ACGTAGGTCCTGTTTGCAGT |
| H19 |
TTTGGTTACAGGACGTGGCA |
CCTCGATCCCCTAAACCTCC |
| HIST1H4H |
GAGGAGCTAAGCGTCATCGC |
AGAAATTCGCTTGACACCGC |
| LOXL2 |
CTCCACTGTACTGGCAACGA |
GCGGTAGGTTGAGAGGATGG |
| PPARG |
TCTCCGTAATGGAAGACCACT |
AGGCTCCACTTTGATTGCACT |
| CGA |
TTCGGATCCACAGTCAACCG |
CACATCAGGAGCGGAATGGA |
| TGFA |
GCGAGTGCCAGCAGAGAG |
GCACGCAGCCAACACAATAC |
| FN1 |
TTGCTCCTGCACATGCTTTG |
TCGGGAATCTTCTCTGTCAGC |
| TNC |
AAAGCGGGGAATGTTGGGAT |
GCCTGTAAGCTTTTCCCAAGTG |
| TGFB1 |
CGACTCGCCAGAGTGGTTAT |
AGTGAACCCGTTGATGTCCA |
RNA library construction and
sequencing
Total RNA of HepG2 cells and NSUN2-deficient HepG2
cells was extracted using TRIzol® Reagent (Invitrogen;
Thermo Fisher Scientific, Inc., Waltham, MA, USA), and quantified
and qualified with an Agilent 2100 Bioanalyzer (Agilent
Technologies, Inc., Santa Clara, CA, USA). For next generation
sequencing, cDNA libraries were constructed using the
NEBNext® Ultra™ RNA Library Prep kit for
Illumina® (Illumina, Inc., San Diego, CA, USA),
according to the manufacturer's protocol.
Then, the libraries with different indices were
multiplexed and loaded on an Illumina HiSeq instrument according to
the manufacturer's instructions (Illumina, Inc.). Sequencing was
performed using a 2×150 bp paired-end configuration, and image
analysis and base calling were conducted using HiSeq Control
Software (HCS) + OLB + GAPipeline-1.6 (Illumina, Inc.) on the HiSeq
instrument. The sequences were processed by Genewiz, Inc. (South
Plainfield, NJ, USA).
Differential expression analysis
To remove technical sequences, including adapters,
PCR primers, or fragments thereof, and quality of bases <20,
pass filter data of fastq format were processed by Trimmomatic
(v0.30) (15) to obtain high
quality clean data.
Reference genome sequences (hg38) and gene model
annotation files of humans were downloaded from ENSEMBL (http://asia.ensembl.org/info/data/ftp/index.html).
Hisat2 (v2.0.1) was used to index the reference genome sequence
(16). Finally, clean data were
aligned to the reference genome using Hisat2 software (v2.0.1).
lncRNAs were identified based on the NONCODE database (version:
NONCODE 2016; http://www.bioinfo.org/NONCODE2016/).
To further analyze the expression levels of genes,
Stringtie (version 1.3.0) (17)
was used to count the number of fragments of each gene following
Hisat2 comparison, which were normalized with the trimmed mean of M
values method (18). The fragments
per kilobase of exon model per million mapped reads (FPKM) value of
each gene was calculated by the script.
Differential expression analysis was performed using
the DESeq Bioconductor package (http://www-huber.embl.de/users/anders/DESeq), a model
based on the negative binomial distribution. Fold-change was
calculated based on the FPKM value. Following adjustment with the
Benjamini and Hochberg method (19) for controlling the false discovery
rate, genes with P<0.01 and log2 (fold-change) >2 were
considered differentially expressed genes. For analysis of the
differentially expressed lncRNAs, a more stringent standard
(fold-change >4; P<0.01) was set to narrow the scope of
candidate lncRNAs.
Prediction of target genes by
lncRNAs
Target gene prediction was performed by evaluating
trans- and cis-regulation. Trans-regulation predicts whether the
database is the mRNA database of the species. First, Basic Local
Alignment Search Tool (Magic-BLAST 1.3.0) was used to select
complementary or similarity sequences (20). RNAplex (v0.2) was subsequently used
to calculate the complementary energy between the two sequences,
and sequences above the threshold were selected. RNAplex was
designed to quickly identify possible hybridization sites for a
query RNA in large RNA databases and is also useful for searching
RNA-RNA interactions (21).
Cis-regulation refers to an lncRNA acting on neighboring target
genes. Genes with distances <10 kb upstream and downstream of
lncRNAs were selected as targets for cis-regulation.
Gene Ontology (GO) and Kyoto
Encyclopedia of Genes and Genomes (KEGG) enrichment analysis
GO::TermFinder comprises a set of object-oriented
Perl modules for accessing GO information and evaluating and
visualizing the collective annotation of a list of genes to GO
terms (22). It was used to
identify GO terms that annotate a list of enriched genes with
P<0.05. KEGG is a collection of databases dealing with genomes,
biological pathways, diseases, drugs and chemical substances
(http://en.wikipedia.org/wiki/KEGG)
(23). KOBAS (v3.0) software
(http://kobas.cbi.pku.edu.cn/index.php) was used to
test the statistical enrichment of the differentially expressed
genes in KEGG pathways. The background gene set comprised all genes
expressed in HepG2 cells and NSUN2-deficient HepG2 cells, according
to the FPKM value.
Construction of coexpression
network
Genes enriched in KEGG pathways and their
coexpressed lncRNAs were used to construct the coexpression network
using Cytoscape software (v3.3.0) (24).
RT-qPCR analysis
Total RNA of HepG2 cells and NSUN2-deficient HepG2
cells was extracted using TRIzol® according to the
manufacturer's instructions. Complementary DNAs were synthesized
using the PrimeScript™ RT reagent kit plus gDNA Eraser
(Takara, Kusatsu, Japan) and random primers. qPCR was performed
using a CFX Connect™ real-time system (Bio-Rad
Laboratories, Inc., Hercules, CA, USA), and the RNA level was
quantified using a SYBR Green Master Mix (Vazyme, Piscataway, NJ,
USA). The thermocycling conditions were: 95°C for 5 min, followed
by 40 cycles of 94°C for 15 sec and 60°C for 30 sec. Comparative
quantification of each RNA was normalized to the β-actin gene using
the 2−∆∆Cq method (25). The specific primers used for
amplification are presented in Table
I.
Statistical analysis
All data were analyzed using SPSS software (version
20.0; IBM Corp., Armonk, NY, USA). Spearman's correlation test was
used to estimate the coexpression relationship between lncRNAs and
mRNAs (26,27). The RT-qPCR data are expressed as
the mean ± standard error of the mean. Multigroup comparisons of
the means were analyzed using one-way analysis of variance and
post-hoc contrasts were performed using Tukey's test. P<0.05 was
considered to indicate a statistically significant difference. Each
group contained three samples, and all experiments were repeated at
least three times.
Results
Differential expression of lncRNAs and
mRNAs associated with NSUN2
From the RNA-seq data, an average of 13.6 and 11.9
million clean reads were generated in NSUN2-deficient HepG2 cells
and HepG2 cells. In NSUN2-deficient HepG2 cells and normal HepG2
cells, 94.8 and 95.5% of clean reads were uniquely mapped,
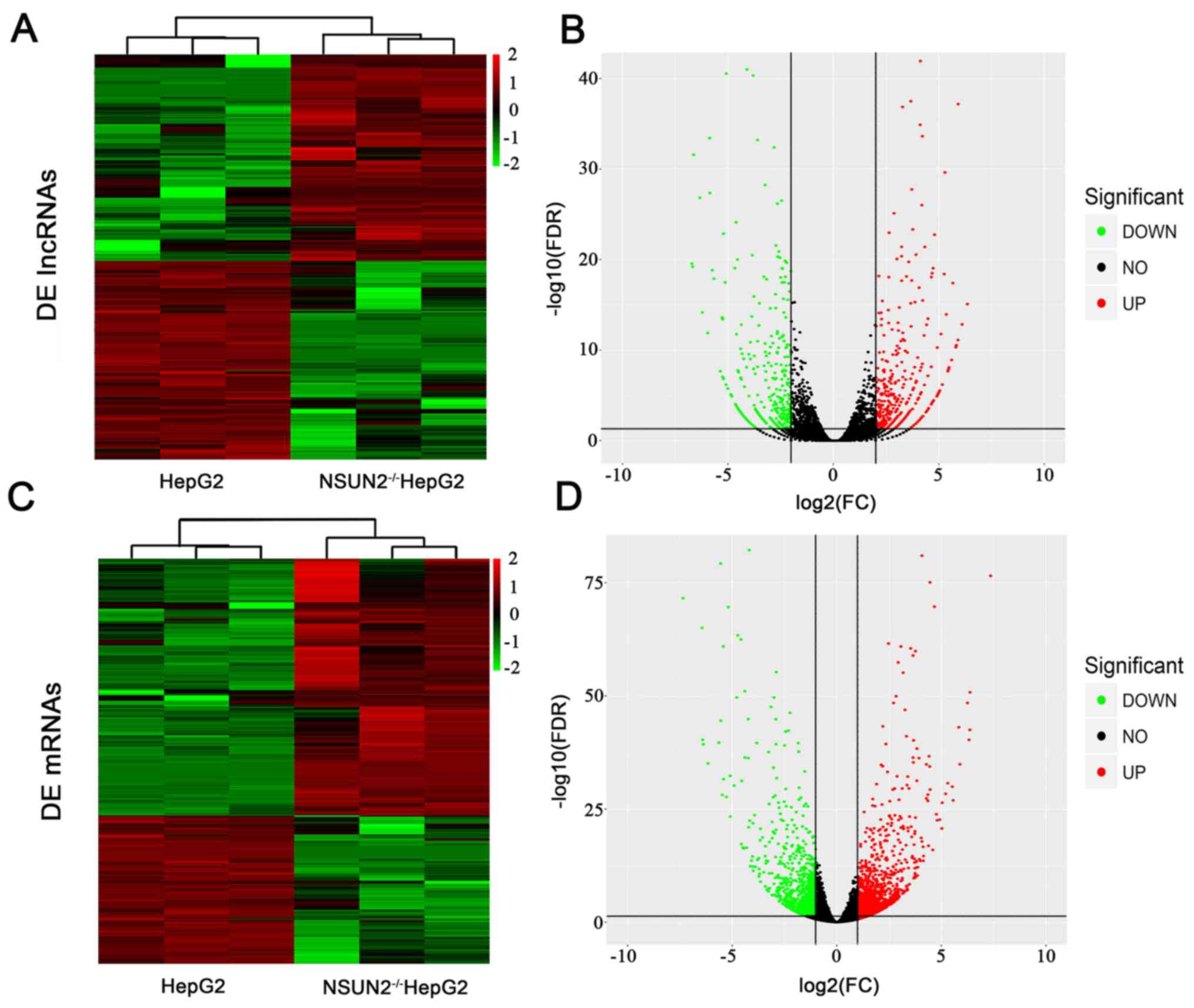
respectively. A total of 757 lncRNAs were differentially expressed
(fold-change >4; P<0.01) between NSUN2-deficient HepG2 cells
and HepG2 cells, of which 392 lncRNAs were upregulated and 365
lncRNAs were downregulated (Fig. 1A
and B; Table II).
| Table II.Top 10 downregulated and upregulated
differentially expressed lncRNAs in NOP2/Sun domain family member
2-deficient HepG2 cells compared with normal controls. |
Table II.
Top 10 downregulated and upregulated
differentially expressed lncRNAs in NOP2/Sun domain family member
2-deficient HepG2 cells compared with normal controls.
| A,
Downregulated |
|---|
|
|---|
| lncRNA | log2 fold
change | P-value |
|---|
|
NONHSAT061515.2 | −9.1672 |
8×10−210 |
|
NONHSAT208991.1 | −7.89821 |
2.73×10−87 |
|
NONHSAT053947.2 | −7.52574 |
1.94×10−49 |
|
NONHSAT120697.2 | −7.35533 |
8.05×10−67 |
|
NONHSAT175932.1 | −6.83085 |
7.51×10−46 |
|
NONHSAT124573.2 | −6.70262 |
2.79×10−20 |
|
NONHSAT186327.1 | −6.6746 |
6.06×10−20 |
|
NONHSAT141130.2 | −6.62423 |
2.57×10−32 |
|
ENST00000397381 | −6.48342 |
5.29×10−76 |
|
NONHSAT053044.2 | −6.3331 |
1.46×10−27 |
|
| B,
Upregulated |
|
| lncRNA | log2 fold
change | P-value |
|
|
NONHSAT179381.1 | 5.670156 |
3.91×10−18 |
|
ENST00000443205 | 5.805754 |
4.68×10−11 |
|
NONHSAT199040.1 | 5.831291 |
2.94×10−11 |
|
ENST00000521369 | 5.905295 |
7.55×10−12 |
|
NONHSAT202740.1 | 5.914346 |
6.53×10−38 |
|
NONHSAT185459.1 | 6.106938 |
1.43×10−13 |
|
NONHSAT060224.2 | 6.355958 |
8.03×10−16 |
|
NONHSAT112221.2 | 6.63073 |
8.95×10−47 |
|
NONHSAT103133.2 | 6.949018 |
1.39×10−53 |
|
NONHSAT180405.1 | 7.830624 |
1.41×10−74 |
mRNA expression was also compared between
NSUN2-deficient and wild-type cells. A total of 1,834 mRNAs were
differentially expressed (fold-change >2; P<0.05) in
NSUN2-deficient HepG2 cells compared with wild-type HepG2 cells
(Fig. 1C and D; Table III). Of these genes, 1,163 mRNAs
were upregulated and 671 mRNAs were downregulated in
NSUN2-deficient HepG2 cells.
| Table III.Top 10 downregulated and upregulated
differentially expressed mRNAs in NOP2/Sun domain family member
2-deficient HepG2 cells compared with normal controls. |
Table III.
Top 10 downregulated and upregulated
differentially expressed mRNAs in NOP2/Sun domain family member
2-deficient HepG2 cells compared with normal controls.
| A,
Downregulated |
|---|
|
|---|
| Gene | Gene symbol | log2 fold
change | P-value |
|---|
|
ENSG00000135373 | EHF | −7.352147492 |
2.47×10−72 |
|
ENSG00000175874 | CREG2 | −7.111981449 |
5.31×10−88 |
|
ENSG00000079308 | TNS1 | −6.554792085 |
4.72×10−153 |
|
ENSG00000121797 | CCRL2 | −6.440975865 |
9.08×10−66 |
|
ENSG00000161249 | DMKN | −6.403903015 |
4.81×10−41 |
|
ENSG00000154997 | SEPT14 | −6.158418718 |
1.14×10−89 |
|
ENSG00000127324 | TSPAN8 | −6.069707964 |
4.77×10−40 |
|
ENSG00000178172 | SPINK6 | −5.636422774 |
8.06×10−36 |
|
ENSG00000078098 | FAP | −5.556570307 |
6.05×10−189 |
|
ENSG00000099998 | GGT5 | −5.544869611 |
2.19×10−40 |
|
| B,
Upregulated |
|
| Gene | Gene
symbol | log2 fold
change | P-value |
|
|
ENSG00000147246 | HTR2C | 5.306442373 |
1.95×10−31 |
|
ENSG00000124253 | PCK1 | 5.524179098 |
1.08×10−30 |
|
ENSG00000121904 | CSMD2 | 5.563322974 |
1.17×10−27 |
|
ENSG00000164128 | NPY1R | 5.880433118 |
8.29×10−44 |
|
ENSG00000105894 | PTN | 6.248807612 |
1.27×10−35 |
|
ENSG00000163530 | DPPA2 | 6.323234879 |
3.53×10−49 |
|
ENSG00000125740 | FOSB | 7.352207927 |
4.93×10−41 |
|
ENSG00000170345 | FOS | 7.442578093 |
3.12×10−43 |
|
ENSG00000006611 | USH1C | 7.895447434 |
1.58×10−51 |
|
ENSG00000135346 | CGA | 10.37870244 |
2.89×10−77 |
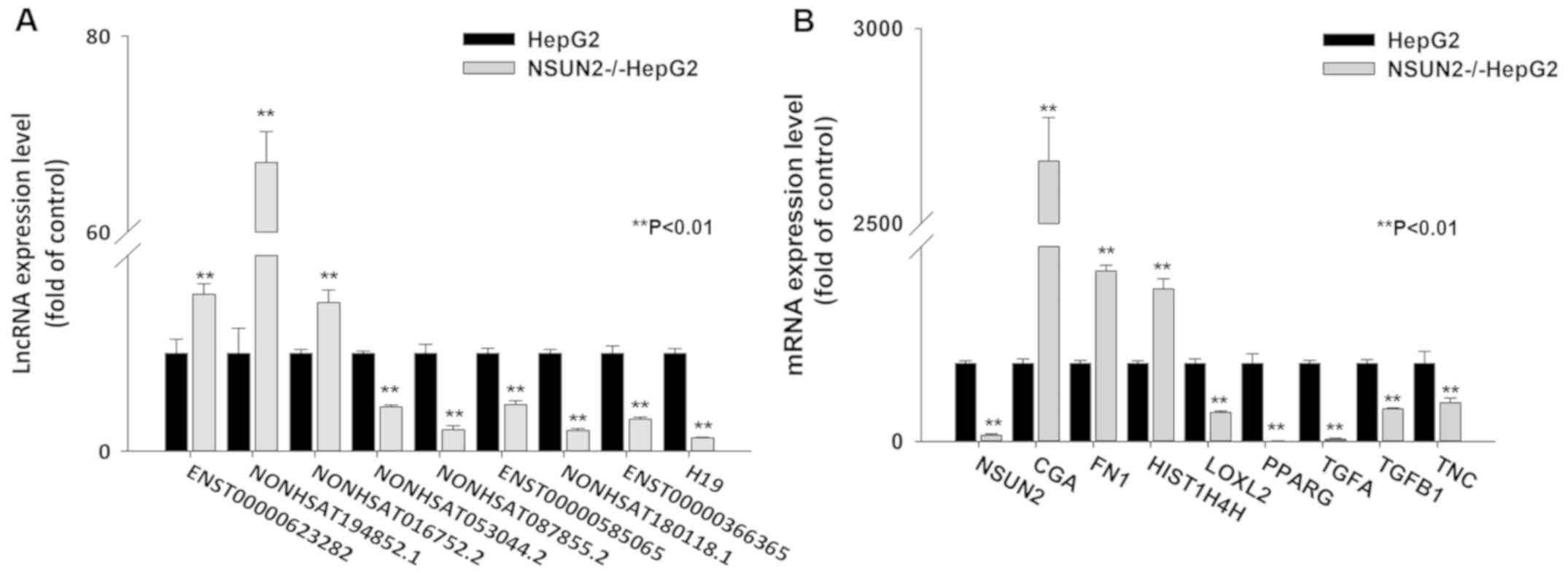
To further evaluate the reliability of the RNA-seq
results, a total of 16 differentially expressed (DE) lncRNA and
mRNA transcripts were randomly selected to validate the relative
expression levels in the NSUN2-deficient HepG2 and wild-type HepG2
cells using RT-qPCR. The results demonstrated that all of the data
for differential expression of lncRNAs and mRNAs were consistent
with the RNA-seq results (Fig. 2A and
B), indicating that the RNA-seq data were reliable.
Correlation analysis of DE lncRNAs and
target mRNAs
To better understand the functions of differentially
expressed lncRNAs, target genes of 757 lncRNAs were predicted
through cis and trans action. As a result, a total of 212 lncRNAs
with 368 target mRNAs (rs>0.9; P<0.05) exhibited
significant correlations (Table
IV), among which the correlation of 253 mRNAs was negative and
the correlation of 290 mRNAs was positive.
| Table IV.Top 20 significantly co-expressed
lncRNAs and targeted mRNAs. |
Table IV.
Top 20 significantly co-expressed
lncRNAs and targeted mRNAs.
| lncRNA | log2 FC | Coef | Target gene | Gene name | log2 FC |
|---|
|
ENST00000366365 | −2.23253 | −0.9868 |
ENSG00000188732 | FAM221A | 2.2387726 |
|
NONHSAT086754.2 | −2.73917 | −0.978 |
ENSG00000152689 | RASGRP3 | 2.7018123 |
|
NONHSAT017591.2 | 2.622214 | −0.978 |
ENSG00000103381 | CPPED1 | −2.591183 |
|
NONHSAT105304.2 | 3.80573 | −0.978 |
ENSG00000125730 | C3 | −3.818577 |
|
NONHSAT161933.1 | 3.905272 | −0.978 |
ENSG00000134830 | C5AR2 | −3.92987 |
|
NONHSAT031026.2 | 3.936562 | −0.978 |
ENSG00000134830 | C5AR2 | −3.92987 |
|
ENST00000564650 | 2.39005 | −0.9779 |
ENSG00000188505 | NCCRP1 | −2.352221 |
|
NONHSAT165291.1 | −2.28239 | −0.9765 |
ENSG00000188732 | FAM221A | 2.2387726 |
|
NONHSAT103857.2 | −2.07256 | −0.9765 |
ENSG00000105650 | PDE4C | 2.1183298 |
|
NONHSAT042028.2 | 3.698807 | −0.9747 |
ENSG00000173698 | ADGRG2 | −3.652078 |
|
NONHSAT000158.2 | −2.30185 | 0.978 |
ENSG00000188112 | C6orf132 | −2.196761 |
|
NONHSAT185304.1 | 2.199372 | 0.978 |
ENSG00000008311 | AASS | 2.279951 |
|
NONHSAT202599.1 | 2.73477 | 0.978 |
ENSG00000121236 | TRIM6 | 2.8359967 |
|
NONHSAT179528.1 | 2.426634 | 0.9824 |
ENSG00000009950 | MLXIPL | 2.4755839 |
|
NONHSAT018086.2 | 2.637192 | 0.9824 |
ENSG00000139174 | PRICKLE1 | 2.6800691 |
|
NONHSAT165633.1 | 2.144573 | 0.9845 |
ENSG00000105650 | PDE4C | 2.1183298 |
|
NONHSAT057058.2 | 2.729542 | 0.9845 |
ENSG00000152689 | RASGRP3 | 2.7018123 |
|
NONHSAT024073.2 | −3.80956 | 0.9868 |
ENSG00000125730 | C3 | −3.818577 |
|
NONHSAT058560.2 | 4.390725 | 0.9868 |
ENSG00000241644 | INMT | 4.3854325 |
|
NONHSAT159228.1 | 2.027489 | 0.9912 |
ENSG00000066468 | FGFR2 | 2.0244554 |
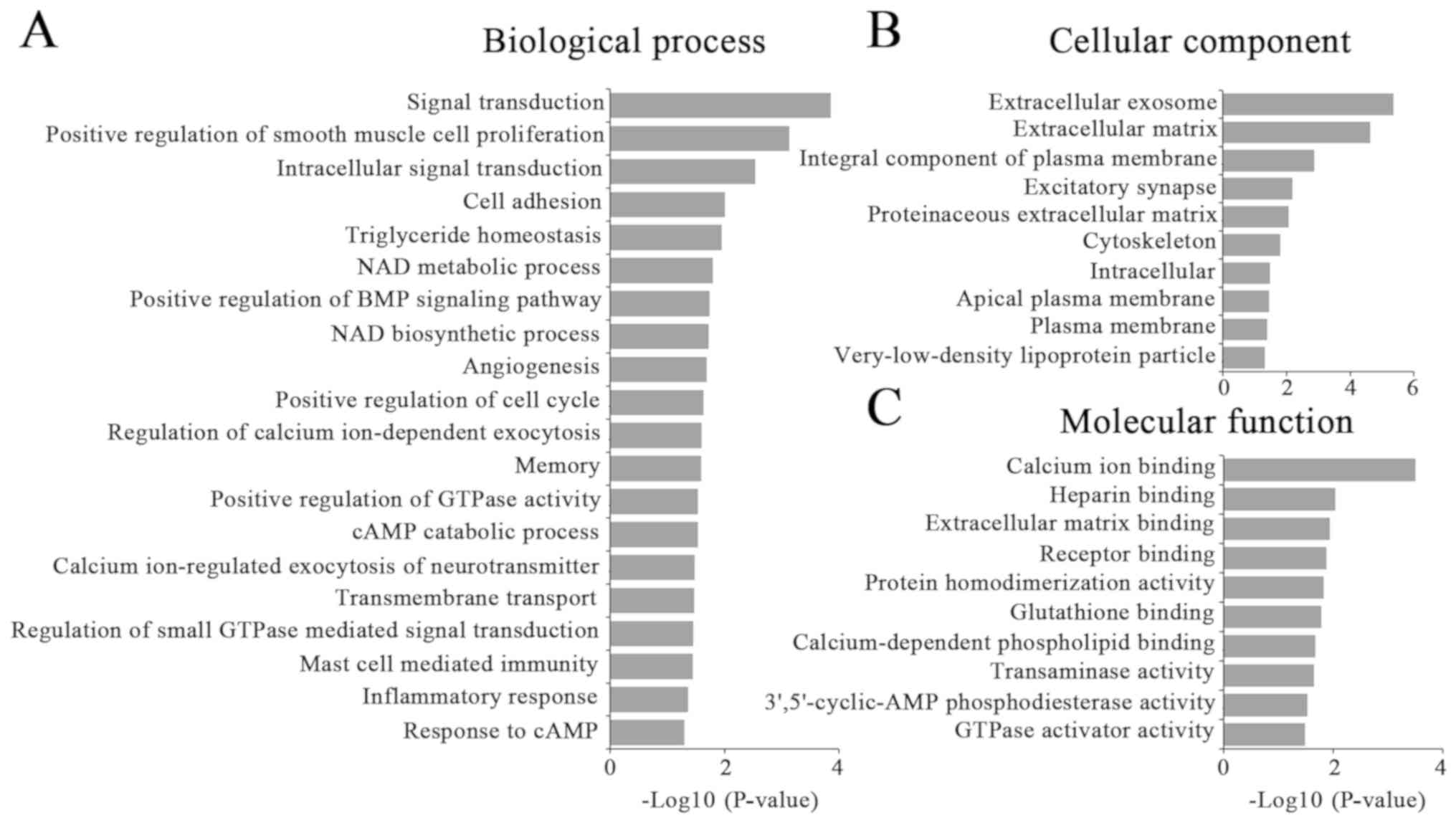
A total of 368 coexpressed mRNAs were selected for
GO enrichment and KEGG pathway analysis. The results demonstrated
that the most enriched GOs were involved in 42 biological
processes, 18 cellular components and 23 molecular functions
(Fig. 3). These dysregulated
lncRNAs by NSUN2 were associated with ‘signal transduction’
(ontology: biological process), ‘extracellular exosome’ (ontology:
cellular component) and ‘calcium ion binding’ (ontology: molecular
function). KEGG pathway analysis demonstrated that the correlated
mRNAs were primarily enriched in ‘pathways in cancer’ (hsa05200),
‘focal adhesion’ (hsa05032), the ‘PI3K-Akt signaling pathway’
(hsa04151) and ‘hematopoietic cell lineage’ (hsa04640) (Table V).
| Table V.Top 15 significantly enriched
pathways for the significantly correlated mRNAs targeted by
lncRNAs. |
Table V.
Top 15 significantly enriched
pathways for the significantly correlated mRNAs targeted by
lncRNAs.
| Term | Count | P-value | Input |
|---|
| hsa05200: Pathways
in cancer | 15 | 0.0009457 | FN1, WNT2B, LAMA4,
GNB4, PGF, ARHGEF12, EGLN3, ARNT2, WNT7B, FGFR2, FLT3LG, PLCB1,
ITGA2B, RASGRP3, JUN |
| hsa04921: Oxytocin
signaling pathway | 8 | 0.0028716 | MYLK3, KCNJ12,
PLCB1, CACNB2, CAMK4, CAMK1D, PPP1R12A, JUN |
| hsa04510: Focal
adhesion | 9 | 0.0038045 | FN1, LAMA4, PGF,
MYLK3, ITGA2B, THBS1, ARHGAP5, PPP1R12A, JUN |
| hsa04672:
Intestinal immune network for IgA production | 4 | 0.0055313 | MAP3K14, TNFRSF13C,
ICOSLG, CCL28 |
| hsa04810:
Regulation of actin cytoskeleton | 9 | 0.005559 | FN1, ARHGEF4,
ARHGEF12, LIMK1, MYLK3, ITGA2B, FGFR2, PIKFYVE, PPP1R12A |
| hsa04610:
Complement and coagulation cascades | 5 | 0.0069775 | C1S, PROCR, C1R,
THBD, C3 |
| hsa04512:
ECM-receptor interaction | 5 | 0.0084585 | SV2B, FN1, LAMA4,
THBS1, ITGA2B |
| hsa04640:
Hematopoietic cell lineage | 5 | 0.0097028 | FLT3LG, CD37, IL6R,
IL11, ITGA2B |
| hsa04611: Platelet
activation | 6 | 0.011229 | MYLK3, ARHGEF12,
ITGA2B, COL3A1, PLCB1, PPP1R12A |
| hsa05032: Morphine
addiction | 5 | 0.0120466 | GNB4, GABRG3,
PDE10A, PDE11A, PDE4C |
| hsa00561:
Glycerolipid metabolism | 4 | 0.0123949 | DGKH, LPL, MGLL,
AKR1B1 |
| hsa04151: PI3K-Akt
signaling pathway | 11 | 0.0130241 | FN1, LAMA4, GNB4,
PGF, IL6R, ITGA2B, THBS1, JAK3, FGFR2, IFNAR2, NR4A1. |
| hsa01100: Metabolic
pathways | 28 | 0.0145589 | PTGES, KYNU, GGT5,
QPRT, ETNPPL, GCNT3, SPTLC3, HKDC1, GALNT16, NMNAT1, UGDH, RDH10,
AKR1B1, NDUFA9, AASS |
| hsa05146:
Amoebiasis | 5 | 0.0171801 | FN1, SERPINB9,
LAMA4, COL3A1, PLCB1 |
| hsa04530: Tight
junction | 6 | 0.017745 | INADL, PARD6B,
RAB3B, OCLN, MYH3, PRKCH |
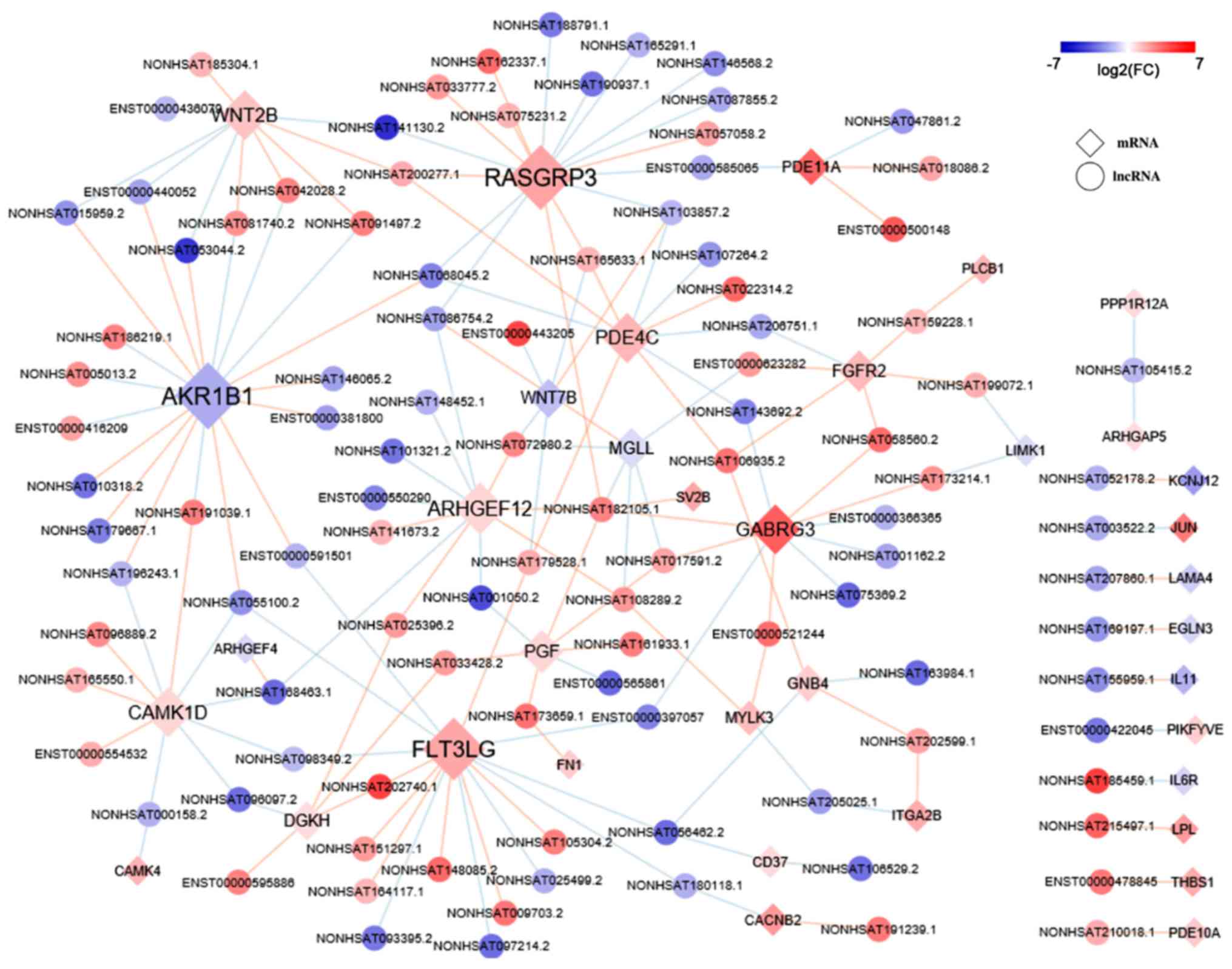
Coexpression network of KEGG enriched
mRNAs
The coexpression network comprised 128 nodes and 137
edges between 93 lncRNAs and 239 mRNAs. Among them, 70 pairs were
positively correlated, and 324 pairs were negatively correlated
(Fig. 4).
Discussion
NSUN2 is upregulated in numerous types of tumors and
modulates the biological functions of multiple RNA species,
including tRNA, mRNA, vault RNA, and miRNA (9,10,28,29).
However, little is known about the association between NSUN2 and
lncRNAs, and investigating this relationship may help further the
understanding of the molecular role of NSUN2 in tumorigenesis. In
the present study, the lncRNA expression profiles in
NSUN2-deficient HepG2 cells and wild-type HepG2 cells were examined
by genome-wide RNA-seq, and lncRNAs with statistically significant
differential expression were identified. Integrated analyses of
these lncRNAs, concentrating on target gene prediction, gene
coexpression, GO and pathway analyses, were performed to elucidate
their potential functions. As a result, a total of 757 lncRNAs were
differentially expressed in response to NSUN2 knockout in HepG2
cells.
The biological role of lncRNAs is complicated due to
their diverse and complex functions. Emerging evidence has
indicated that lncRNAs may serve as signal, decoy, guide or
scaffold molecules in the regulation of gene expression (30). Therefore, the function of lncRNAs
may be assessed by analyzing the mRNAs that they regulate. The
present study used cis and trans action to predict mRNAs targeted
by DE lncRNAs. Furthermore, Spearman's correlation test was used to
estimate the coexpression between lncRNAs and mRNAs. Ultimately,
212 DE lncRNAs were identified to be coexpressed with 368 target
mRNAs, with 290 pairs positively correlated and 253 pairs
negatively correlated. For example, the lncRNAs NONHSAT103857.2,
NONHSAT001050.2, NONHSAT146065.2, ENSG00000130600 and NONHSAT
159228.1 were coexpressed with the Wnt7B, placental growth factor,
epiregulin, insulin-like growth factor 2 and fibroblast growth
factor receptor 2 (FGFR2) genes, which have been reported to be
important regulators in tumors.
GO enrichment was employed to further examine the
biological role of these DE lncRNAs. The coexpressed mRNAs were
classified into hierarchical categories to determine gene
regulatory networks based on the biological processes, cellular
components and molecular functions. GO analysis revealed that these
genes were significantly enriched in many cancer-associated
biological processes, including cell proliferation, cell adhesion
and angiogenesis. Furthermore, pathway analysis highlighted a
number of pathways closely associated with carcinogenesis,
including ‘pathways in cancer’, ‘focal adhesion’, ‘extracellular
matrix (ECM)-receptor interaction’ and ‘PI3K-Akt signaling’.
Focal adhesions are large, dynamic protein complexes
through which the cytoskeleton of a cell connects to the ECM. The
focal adhesion and ECM-receptor pathways serve important roles in
various biological processes such as cell proliferation, migration
and angiogenesis, which are crucial for tumorigenesis (31–33).
The phosphatidylinositol-3-kinase-protein kinase B
(PI3K-Akt) pathway is a signal transduction pathway that promotes
survival and growth in response to extracellular signals. The
PI3K-Akt pathway may be activated by a range of signals, including
hormones, growth factors and components of the ECM, and further
mediate various downstream responses, including survival, growth,
cell proliferation and migration (34–37).
The PI3K-Akt pathway also serves an important role in the
regulation of angiogenesis (38,39),
which is essential for embryonic development, tumor growth and
metastasis. Recently, numerous studies have demonstrated
dysfunction of the PI3K-Akt pathway in carcinogenesis. Various
components of the PI3K-Akt pathway are frequently altered in
different types of human cancer types, including breast, gastric,
colon and hepatocellular carcinoma (HCC) (40–42).
The PI3K pathway may transmit oncogenic signals to Akt, thus
promoting tumorigenesis through a number of signaling pathways
including the nuclear factor κ-light-chain-enhancer of activated B
cells (NF-κB) pathway (43). Taken
together, these results suggest that these dysregulated lncRNAs by
NSUN2 deletion may be involved in tumor cell proliferation and
angiogenesis by modulating focal adhesion, ECM-receptor
interactions and PI3K-Akt signaling pathways.
A total of two well-studied lncRNAs, human
urothelial carcinoma associated 1 (UCA1) and H19, were also
demonstrated to be differentially expressed in the present study.
Previous studies indicated that UCA1 and H19 may affect
tumorigenesis by modulating the PI3K-Akt pathway. UCA1 is a known
oncogene and is highly expressed in various types of cancer,
including bladder cancer, gastric cancer, colorectal cancer and HCC
(44–47). UCA1 is able to enhance the
proliferation and metastasis of bladder cancer cells through the
PI3K-Akt or Wnt signaling pathways (48). In HCC, UCA1 may serve an oncogenic
role in promoting the proliferation and metastasis of HCC cells
through the inhibition of miRNA-216b and activation of the
FGFR1/extracellular signal-regulated kinases signaling pathway
(47). Numerous studies have
reported that NSUN2 is involved in regulating cell proliferation
(1,7,49).
In the present study, UCA1 was the most significantly downregulated
lncRNA in NSUN2-deficient HepG2 cells, indicating that NSUN2 may
affect cell proliferation by regulating the expression of UCA1.
As an important imprinting gene exclusively
expressed from the maternal allele (50), H19 was also significantly
downregulated in NSUN2-deficient HepG2 cells. The function of H19
is controversial. One study reported that H19 serves a role in the
negative regulation of body weight and cell proliferation (51). Mutations in the H19 gene were also
demonstrated to be associated with Beckwith-Wiedemann syndrome
(52) and Wilms' tumorigenesis
(53). However, increased H19
expression has been observed in various types of cancer, including
colorectal cancer, esophageal cancer (54), lung cancer (55) and HCC (56). Downregulation of H19 lncRNA
inhibits the migration and invasiveness of melanoma cells by
inactivating the NF-κB and PI3K-Akt signaling pathways. On the
other hand, overexpression of H19 lncRNA promotes invasion and
autophagy via the PI3K/AKT/mammalian target of rapamycin pathways
in trophoblast cells (57). In
HCC, H19 overexpression might be a risk factor for tumor
aggressiveness and poorer outcomes (56). Moreover, c-Myc, an oncogene that
functions as a regulator of gene transcription, was also reported
to directly induce H19 expression (55). A previous study demonstrated that
NSUN2 was directly targeted by Myc and mediated Myc-induced cell
proliferation and growth (1). The
present study revealed that the H19 expression level was
significantly decreased following NSUN2 deletion in HepG2 cells,
indicating that NSUN2 may mediate the interaction between Myc and
H19 lncRNA, further implicating NSUN2 in the regulation of tumor
proliferation and metastasis.
It is noteworthy that the question of whether these
lncRNAs are directly modulated by NSUN2 requires further
experimental support. For example, an NSUN2-overexpression
experiment may be performed to narrow the scope of candidate
lncRNAs targeted by NSUN2. The application of technologies to
investigate RNA-protein interaction, including RNA electrophoretic
mobility shift assays and RNA binding protein immunoprecipitation
assays, may help elucidate the molecular mechanisms behind NSUN2
modulation of these lncRNAs. Moreover, further research may address
the functional significance of NSUN2 expression in other types of
cancer cells in order to elucidate whether there is a common
tumorigenic mechanism.
In conclusion, the present study revealed the
expression patterns of lncRNAs in response to NSUN2 deletion in
HepG2 cells. NSUN2 is an RNA m5C methyltransferase with important
roles in tumor proliferation and metastasis, but its molecular
function remains largely unknown. The results presented provide
novel insight into the roles of lncRNAs associated with NSUN2 in
carcinogenesis. Furthermore, a highly correlated expression pattern
of lncRNAs induced by NSUN2 deficiency was observed, with coding
genes that are involved in cancer development. These findings may
be important for understanding the molecular function of NSUN2.
Further studies are required to define the precise role of lncRNAs
regulated by NSUN2 in tumorigenesis, in addition to the molecular
mechanisms behind the regulation of lncRNAs and/or mRNAs by NSUN2.
These will further our understanding of how the NSUN2 gene is
involved in human diseases and cancer.
Acknowledgements
Not applicable.
Funding
This research was supported by the National Natural
Science Foundation of China (grant nos. 81773013, 91540117), the
National Key Research and Development Program in China (grant no.
2016YFC1303604), the Postgraduate Research and Practice Innovation
Program of Jiangsu Province (grant no. KYLX15_1376) and the
Priority Academic Program Development of Jiangsu Higher Education
Institutions (Animal Science and Veterinary Medicine).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZS performed the major experiments, and wrote and
revised the manuscript. HC contributed to the experimental design,
and revised the manuscript. SX, HX, YY and JO performed the
experiments. XH, SC and ZY conducted the data analysis. All authors
read and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Frye M and Watt FM: The RNA
methyltransferase Misu (NSun2) mediates Myc-induced proliferation
and is upregulated in tumors. Curr Biol. 16:971–981. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yi J, Gao R, Chen Y, Yang Z, Han P, Zhang
H, Dou Y, Liu W, Wang W, Du G, et al: Overexpression of NSUN2 by
DNA hypomethylation is associated with metastatic progression in
human breast cancer. Oncotarget. 8:20751–20765. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hussain S, Tuorto F, Menon S, Blanco S,
Cox C, Flores JV, Watt S, Kudo NR, Lyko F, Frye M, et al: The mouse
cytosine-5 RNA Methyltransferase NSun2 Is a component of the
chromatoid body and required for testis differentiation. Mol Cell
Biol. 33:15612013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Khan MA, Rafiq MA, Noor A, Hussain S,
Flores JV, Rupp V, Vincent AK, Malli R, Ali G, Khan FS, et al:
Mutation in NSUN2, which encodes an RNA methyltransferase, causes
autosomal-recessive intellectual disability. Am J Hum Genet.
90:856–863. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Okamoto M, Hirata S, Sato S, Koga S, Fujii
M, Qi G, Ogawa I, Takata T, Shimamoto F and Tatsuka M: Frequent
increased gene copy number and high protein expression of tRNA
(cytosine-5-)-methyltransferase (NSUN2) in human cancers. DNA Cell
Biol. 31:660–671. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang X, Liu Z, Yi J, Tang H, Xing J, Yu
M, Tong T, Shang Y, Gorospe M and Wang W: The tRNA
methyltransferase NSun2 stabilizes p16INK(4) mRNA by methylating
the 3′-untranslated region of p16. Nat Commun. 3:7122012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Xing J, Yi J, Cai X, Tang H, Liu Z, Zhang
X, Martindale JL, Yang X, Jiang B, Gorospe M and Wang W: NSun2
promotes cell growth via elevating cyclin-dependent kinase 1
translation. Mol Cell Biol. 35:4043–4052. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li Q, Li X, Tang H, Jiang B, Dou Y,
Gorospe M and Wang W: NSUN2-mediated m5C methylation and
METTL3/METTL14-mediated m6A methylation cooperatively enhance p21
Translation. J Cell Biochem. 118:2587–2598. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hussain S, Sajini AA, Blanco S, Dietmann
S, Lombard P, Sugimoto Y, Paramor M, Gleeson JG, Odom DT, Ule J and
Frye M: NSun2-mediated cytosine-5 methylation of vault noncoding
RNA determines its processing into regulatory small RNAs. Cell Rep.
4:255–261. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yuan S, Tang H, Xing J, Fan X, Cai X, Li
Q, Han P, Luo Y, Zhang Z, Jiang B, et al: Methylation by NSun2
represses the levels and function of microRNA 125b. Mol Cell Biol.
34:3630–3641. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li L and Chang HY: Physiological roles of
long noncoding RNAs: Insight from knockout mice. Trends Cell Biol.
24:594–602. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Deng K, Guo X, Wang H and Xia J: The
lncRNA-MYC regulatory network in cancer. Tumour Biol. 35:9497–9503.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ma C, Nong K, Zhu H, Wang W, Huang X, Yuan
Z and Ai K: H19 promotes pancreatic cancer metastasis by
derepressing let-7's suppression on its target HMGA2-mediated EMT.
Tumour Biol. 35:9163–9169. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu Y, Han X, Yuan J, Geng T, Chen S, Hu
X, Cui IH and Cui H: Biallelic insertion of a transcriptional
terminator via the CRISPR/Cas9 system efficiently silences
expression of protein-coding and non-coding RNA genes. J Biol Chem.
292:5624–5633. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bolger AM, Lohse M and Usadel B:
Trimmomatic: A flexible trimmer for Illumina sequence data.
Bioinformatics. 30:2114–2120. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim D, Langmead B and Salzberg SL: HISAT:
A fast spliced aligner with low memory requirements. Nat Methods.
12:357–360. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pertea M, Pertea GM, Antonescu CM, Chang
TC, Mendell JT and Salzberg SL: StringTie enables improved
reconstruction of a transcriptome from RNA-seq reads. Nat
Biotechnol. 33:290–295. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sahraeian SME, Mohiyuddin M, Sebra R,
Tilgner H, Afshar PT, Au KF, Bani Asadi N, Gerstein MB, Wong WH,
Snyder MP, et al: Gaining comprehensive biological insight into the
transcriptome by performing a broad-spectrum RNA-seq analysis. Nat
Commun. 8:592017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Benjamini Y and Hochberg Y: Controlling
the false discovery rate: A practical and powerful approach to
multiple testing. J Royal Statistical Soc. 57:289–300. 1995.
|
|
20
|
Lobo I: Basic local alignment search tool
(BLAST). J Mol Biol. 215:403–410. 2012.
|
|
21
|
Tafer H and Hofacker IL: RNAplex: A fast
tool for RNA-RNA interaction search. Bioinformatics. 24:2657–2663.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Boyle EI, Weng S, Gollub J, Jin H,
Botstein D, Cherry JM and Sherlock G: GO: TermFinder-open source
software for accessing Gene Ontology information and finding
significantly enriched gene ontology terms associated with a list
of genes. Bioinformatics. 20:3710–3715. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kanehisa M: The KEGG database. Novartis
Found Symp. 247:101–103, 119–128, 244–252.. 2002.
|
|
24
|
Kohl M, Wiese S and Warscheid B:
Cytoscape: Software for visualization and analysis of biological
networks. Met Mol Biol. 696:291–303. 2011. View Article : Google Scholar
|
|
25
|
Schmittgen TD: Analysis of relative gene
expression data using real-time quantitative PCR and the 2(-Delta
Delta C(T)) method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang P, Fu H, Cui J and Chen X:
Differential lncRNA-mRNA co-expression network analysis revealing
the potential regulatory roles of lncRNAs in myocardial infarction.
Mol Med Rep. 13:11952016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang Y, Yang H, Han L, Li F, Zhang T,
Pang J, Feng X, Ren C, Mao S and Wang F: Long noncoding RNA
expression profile changes associated with dietary energy in the
sheep testis during sexual maturation. Sci Rep. 7:51802017.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tuorto F, Liebers R, Musch T, Schaefer M,
Hofmann S, Kellner S, Frye M, Helm M, Stoecklin G and Lyko F: RNA
cytosine methylation by Dnmt2 and NSun2 promotes tRNA stability and
protein synthesis. Nat Struct Mol Biol. 19:900–905. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yang X, Yang Y, Sun BF, Chen YS, Xu JW,
Lai WY, Li A, Wang X, Bhattarai DP, Xiao W, et al: 5-methylcytosine
promotes mRNA export-NSUN2 as the methyltransferase and ALYREF as
an m5C reader. Cell Res. 27:606–625. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Vance KW and Ponting CP: Transcriptional
regulatory functions of nuclear long noncoding RNAs. Trends Genet.
30:348–355. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Nguyenngoc KV, Cheung KJ, Brenot A, Shamir
ER, Gray RS, Hines WC, Yaswen P, Werb Z and Ewald AJ: ECM
microenvironment regulates collective migration and local
dissemination in normal and malignant mammary epithelium. Proc Natl
Acad Sci USA. 109:2595–2604. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Armstrong SJ, Wiberg M, Terenghi G and
Kingham PJ: ECM molecules mediate both Schwann cell proliferation
and activation to enhance neurite outgrowth. Tissue Eng.
13:2863–2870. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cheresh DA and Stupack DG: Regulation of
angiogenesis: Apoptotic cues from the ECM. Oncogene. 27:6285–6298.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Steelman LS, Chappell WH, Abrams SL, Kempf
RC, Long J, Laidler P, Mijatovic S, Maksimovic-Ivanic D, Stivala F,
Mazzarino MC, et al: Roles of the Raf/MEK/ERK and
PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity
to therapy-implications for cancer and aging. Aging (Albany NY).
3:1922011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Peltier J, O'Neill A and Schaffer DV:
PI3K/Akt and CREB regulate adult neural hippocampal progenitor
proliferation and differentiation. Dev Neurobiol. 67:1348–1361.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zheng H, Fu G, Dai T and Huang H:
Migration of endothelial progenitor cells mediated by stromal
cell-derived factor-1alpha/CXCR4 via PI3K/Akt/eNOS signal
transduction pathway. J Cardiovasc Pharmacol. 50:2742007.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Brunet A, Datta SR and Greenberg ME:
Transcription-dependent and -independent control of neuronal
survival by the PI3K-Akt signaling pathway. Curr Opin Neurobiol.
11:297–305. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fang J, Ding M, Yang L, Liu L and Jiang B:
PI3K/PTEN/AKT signalling regulates prostate tumor angiogenesis.
Cell Signall. 19:2487–2497. 2007. View Article : Google Scholar
|
|
39
|
Karar J and Maity A: PI3K/AKT/mTOR Pathway
in Angiogenesis. Front Mol Neurosci. 4:512011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Shayesteh L, Lu Y, Kuo WL, Baldocchi R,
Godfrey T, Collins C, Pinkel D, Powell B, Mills GB and Gray JW:
PIK3CA is implicated as an oncogene in ovarian cancer. Nat Genet.
21:99–102. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bellacosa A, De Feo D, Godwin AK, Bell DW,
Cheng JQ, Altomare DA, Wan M, Dubeau L, Scambia G, Masciullo V, et
al: Molecular alterations of the AKT2 oncogene in ovarian and
breast carcinomas. Int J Cancer. 64:280–285. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Roy HK, Olusola BF, Clemens DL, Karolski
WJ, Ratashak A, Lynch HT and Smyrk TC: AKT proto-oncogene
overexpression is an early event during sporadic colon
carcinogenesis. Carcinogenesis. 23:2012002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lee KB, Byun HJ, Park SH, Park CY, Lee SH
and Rho SB: CYR61 controls p53 and NF-κB expression through
PI3K/Akt/mTOR pathways in carboplatin-induced ovarian cancer cells.
Cancer Lett. 315:86–95. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wang T, Yuan J, Feng N, Li Y, Lin Z, Jiang
Z and Gui Y: Hsa-miR-1 downregulates long non-coding RNA urothelial
cancer associated 1 in bladder cancer. Tumour Biol. 35:10075–10084.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zheng Q, Wu F, Dai WY, Zheng DC, Zheng C,
Ye H, Zhou B, Chen JJ and Chen P: Aberrant expression of UCA1 in
gastric cancer and its clinical significance. Clin Transl Onco.
17:640–646. 2015. View Article : Google Scholar
|
|
46
|
Han Y, Yang YN, Yuan HH, Zhang TT, Sui H,
Wei XL, Liu L, Huang P, Zhang WJ and Bai YX: UCA1, a long
non-coding RNA up-regulated in colorectal cancer influences cell
proliferation, apoptosis and cell cycle distribution. Pathology.
46:396–401. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wang F, Ying HQ, He BS, Pan YQ, Deng QW,
Sun HL, Chen J, Liu X and Wang SK: Upregulated lncRNA-UCA1
contributes to progression of hepatocellular carcinoma through
inhibition of miR-216b and activation of FGFR1/ERK signaling
pathway. Oncotarget. 6:78992015.PubMed/NCBI
|
|
48
|
Yang C, Li X, Wang Y, Zhao L and Chen W:
Long non-coding RNA UCA1 regulated cell cycle distribution via CREB
through PI3-K dependent pathway in bladder carcinoma cells. Gene.
496:82012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Wang W: mRNA methylation by NSUN2 in cell
proliferation. Wiley Interdiscip Rev RNA. 7:838–842. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Venkatraman A, He XC, Thorvaldsen JL,
Sugimura R, Perry JM, Tao F, Zhao M, Christenson MK, Sanchez R, Yu
JY, et al: Maternal imprinting at the H19-Igf2 locus maintains
adult haematopoietic stem cell quiescence. Nature. 500:345–349.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Gabory A, Ripoche MA, Le Digarcher A,
Watrin F, Ziyyat A, Forné T, Jammes H, Ainscough JF, Surani MA,
Journot L and Dandolo L: H19 acts as a trans regulator of the
imprinted gene networkcontrolling growth in mice. Development.
136:3413–3421. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Debaun MR, Niemitz EL and Feinberg AP:
Association of in vitro fertilization with Beckwith-Wiedemann
syndrome and epigenetic alterations of LIT1 and H19. Am J Hum
Genet. 72:156–160. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
53
|
Steenman MJ, Rainier S, Dobry CJ, Grundy
P, Horon IL and Feinberg AP: Loss of imprinting of IGF2 is linked
to reduced expression and abnormal methylation of H19 in Wilms'
tumour. Nat Genet. 7:433–439. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Hibi K, Nakamura H, Hirai A, Fujikake Y,
Kasai Y, Akiyama S, Ito K and Takagi H: Loss of H19 imprinting in
esophageal cancer. Cancer Res. 56:4801996.PubMed/NCBI
|
|
55
|
Barsytelovejoy D, Lau SK, Boutros PC,
Khosravi F, Jurisica I, Andrulis IL, Tsao MS and Penn LZ: The c-Myc
oncogene directly induces the H19 noncoding RNA by allele-specific
binding to potentiate tumorigenesis. Cancer Res. 66:53302006.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Yang Z, Lu Y, Xu Q, Tang B, Park CK and
Chen X: HULC and H19 played different roles in overall and
disease-free survival from hepatocellular carcinoma after curative
hepatectomy: A preliminary analysis from gene expression omnibus.
Dis Markers. 2015:1910292015. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Xu J, Xia Y, Zhang H, Guo H, Feng K and
Zhang C: Overexpression of long non-coding RNA H19 promotes
invasion and autophagy via the PI3K/AKT/mTOR pathways in
trophoblast cells. Biomed Pharmacother. 101:691–697. 2018.
View Article : Google Scholar : PubMed/NCBI
|