Introduction
Arsenic, a naturally prevalent element, is found in
the environment and certain foods. Arsenic has two forms in the
environment: Organic and inorganic. Inorganic arsenic has two
valences: Arsenite (III) and arsenate (V) (1). Generally, inorganic arsenic is more
toxic than organic arsenic, and trivalent arsenic is more toxic
than pentavalent arsenic (2).
Arsenic trioxide (ATO) is the primary effective ingredient of white
arsenic, which is inorganic arsenic. It has been used in the
therapy of acute promyelocytic leukemia since the early 1990s. ATO
is a safe and effective anticancer drug and is not prone to drug
resistance (3). Researchers have
attempted to use ATO in the treatment of other types of tumor: ATO
has also been reported to achieve a favorable therapeutic effect in
the treatment of malignant tumors such as lymphoma, gastric
(4), esophageal (5), liver and lung cancer, neuroblastoma
(6) and breast cancer (7).
However, in the treatment of solid tumors, high
concentrations of ATO can cause serious side effects, such as
cardiotoxicity (8), hepatotoxicity
(9), fluid retention (10), alimentary symptoms (11) and rash (12). ATO induces serious cardiotoxicity
and potential cardiovascular side effects, including sudden death
due to acute toxic myocardial damage (13,14).
ATO also has a toxic effect on the liver, kidney and nervous system
(15–17). These factors limit the clinical
application of ATO. Potential mechanisms of ATO-induced
cardiotoxicity include oxidative stress, mitochondrial DNA injury,
apoptosis and functional disruption of ion channels (18). Several studies have indicated that
mitochondrial damage, caspase activation and p53 signaling are the
pathways underlying arsenic-induced apoptosis (1,18).
Mitochondria, intracellular double membrane
organelles, are considered the ‘power plant’ of eukaryotic cells
(19). Mitochondria are the
primary site of intracellular oxidative phosphorylation and
synthesis of adenosine triphosphate, which provides energy for cell
activity (20). Mitochondria also
participate in cell signaling, differentiation, proliferation and
apoptosis (21). The heart, the
most energy consuming organ, has the highest content of
mitochondria of all types of tissue (22). The heart requires efficient
oxidative metabolism and derives almost all of its energy from
mitochondrial oxidative phosphorylation (23). Therefore, mitochondria are
important for myocardial development as well as healthy function.
Beyond their role as a cellular powerhouse, mitochondria produce
reactive oxygen species (ROS) (24), which lead to oxidative injury and
regulate cardiomyocyte apoptosis. Hence, mitochondrial dysfunction
and ROS production are considered key factors in cardiac disease.
Creatine kinase (CK) and lactate dehydrogenase (LDH) are vital
biomarkers for the diagnosis of myocardial injury (25). Studies have shown that ATO
increases LDH content and consequently results in cardiomyocyte
necrosis (26,27).
There are two major antioxidant systems in the body:
The enzyme antioxidant system [involving superoxide dismutase (SOD)
and catalase (CAT)] and the non-enzyme antioxidant system
(involving vitamins C and E, glutathione, carotenoid, copper and
zinc) (28). When oxidative stress
occurs, these two systems are out of balance, which leads to tissue
damage (29). Studies have shown
that excessive ROS during ATO treatment leads to destruction in the
endogenous antioxidant system (30,31).
In addition, studies have suggested that high doses of ATO can
cause oxidative stress, increased ROS and inhibit enzyme and
mitochondrial activity (32,33).
Mitochondrial impairment leads to the release of
mitochondrial-associated proteins cytochrome c, second
mitochondria-derived activator of caspases (Smac) and
high-temperature requirement A2 (HtrA2); the release of cytochrome
c can activate caspase-3 (34). Moreover, the Bcl-2 family, which
serves a crucial role in the regulation of cardiomyocyte apoptosis,
is a primary regulator of cytochrome c release and caspase-3
activation (34). Arsenic exposure
increases the levels of pro-apoptotic proteins Bax (35) and Bad (36) and decreases the expression levels
of anti-apoptotic protein Bcl-2 (37). Furthermore, one study reported that
arsenic exposure activates phosphorylation of the NF-κB pathway
(36). The NF-κB pathway
participates in the inflammatory response, which results in
apoptosis (38). Thus, we
hypothesized that the potential mechanism of ATO-induced
cardiotoxicity may be associated with oxidative stress,
inflammation and mitochondrial apoptosis.
Tannic acid (TA) is found in plants and foods, such
as apples, pears, beans, tea and red wine. TA is a water-soluble
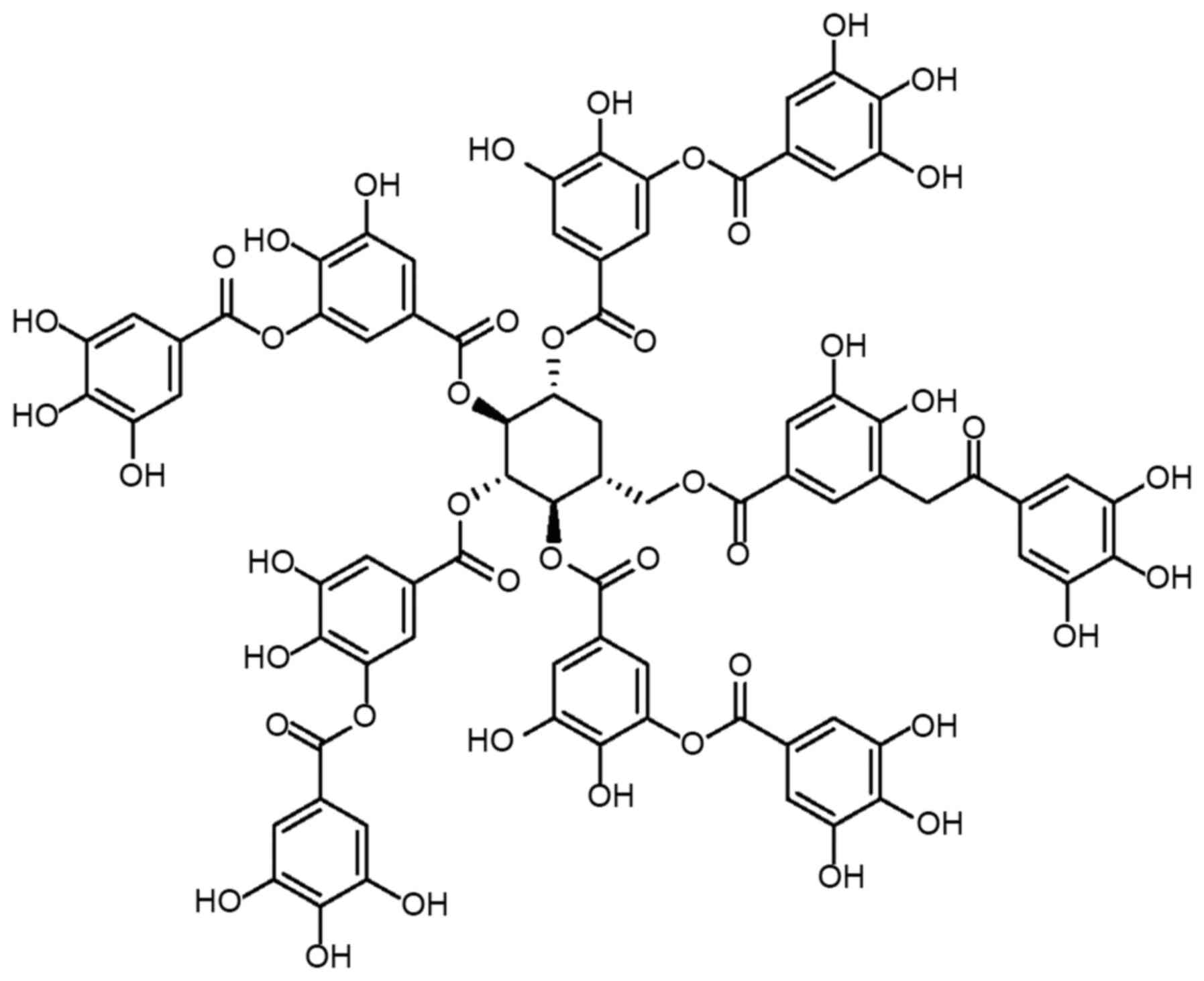
polyphenol compound with a complex chemical structure
(C76H52O46; Fig. 1), containing a glucose core
covalently linked to 3–5 gallic acid residues through the
hydrolysis of ester bonds (39).
TA has been revealed to exert antioxidant, anti-inflammatory,
anticarcinogenic, antimutagenic and antiatherogenic properties
(40). It is also capable of
protecting against drug toxicity (41).
Our previous studies have indicated that TA has
beneficial effects on cardiovascular disease (42–44);
moreover, we found that TA ameliorates ATO-induced nephrotoxicity
(45). However, the effects of TA
on ATO-induced cardiotoxicity have not yet been reported. The aim
of the present study was to evaluate whether TA can protect against
ATO-induced heart injury in rats.
Materials and methods
Chemicals and materials
TA (>98% purity) was acquired from Sigma-Aldrich
(Merck KGaA). ATO was purchased from Beijing SL Pharmaceutical Co.,
Ltd. Kits for determining total CK and LDH, as well as catalase
(CAT), malondialdehyde (MDA) and superoxide dismutase (SOD)
activity were obtained from Nanjing Jiancheng Bioengineering
Institute. All solvents were analytical grade and commercially
available.
Animals and experimental protocol
A total of 50 adult male Sprague-Dawley rats (age,
6–8 weeks; weight, 180–220 g) were obtained from Hebei Medical
University. Male rats were raised under standard conditions
(22-25°C and 45–55% relative humidity with a 12-h light/dark
cycle), with ad libitum access to pellet food and water.
Animal experiments were performed in accordance with the Animal
Care and Ethics Committee of Hebei University of Chinese Medicine
(Shijiazhuang, China). The Animal Care and Ethics Committee of
Hebei University of Chinese Medicine approved all animal protocols
(approval no. DWLL2018038).
Male rats were stochastically separated into five
groups: i) Control (Control, 0.1 ml/kg/day); ii) ATO-alone (ATO, 5
mg/kg/day); iii) ATO + low-dose TA (ATO + L-TA, 20 mg/kg/day); iv)
ATO + high-dose TA (ATO + H-TA, 40 mg/kg/day); and v) TA-alone (TA,
40 mg/kg/day). Rats were intraperitoneally (i.p.) injected with ATO
(5 mg/kg) to establish an ATO-induced cardiotoxicity model. Dose
selection was determined according to previous literature (46,47).
Studies have reported that the median lethal dose of ATO is 14.98
mg/kg body weight in rats (48,49).
The Control group received isovolumic normal saline. The ATO + L-TA
and ATO + H-TA groups underwent intragastric administration of TA
(20 and 40 mg/kg/day, respectively) every morning and were
intraperitoneally injected with ATO (5 mg/kg/day) every afternoon
(40,42,45).
After 7 days, sodium pentobarbital (40 mg/kg, i.p.; Sigma-Aldrich;
Merck KGaA) was used to anesthetize rats, and the heart was removed
and measured.
Histopathological examination
Cardiac specimens were fixed in 4% paraformaldehyde
at room temperature for 48 h. Following fixation, all
paraffin-embedded samples were sectioned at 4 µm and stained at
room temperature with 0.1% hematoxylin (Hebei Bohai Biological
Engineering Development Co., Ltd.) for 5 min and 0.5% eosin for 3
min (50). Finally, pathological
changes in myocardial tissue structure were examined under a light
microscope at ×400 magnification (Leica DM4000B; Leica Microsystems
GmbH).
Measurement of cardiac marker
enzymes
Rat serum was separated by centrifugation at 1,500 ×
g for 10 min at 4°C and the activity of CK and LDH were detected
using CK (cat. no. A032) and LDH (cat. no. A020-1) assay kits (both
Nanjing Jiancheng Bioengineering Institute), respectively. For CK
assay, serum samples (20 µl) and mixed reagent were added into
tubes according to the manufacturer's protocol, then vortex mixed
and incubated at 37°C for 20 min. Then, R6 solution from the kit
was added and the mixed solution was centrifuged at 3,500 × g for
10 min at room temperature. Next, the tubes were heated in a water
bath at 45°C for 15 min and the absorbance was detected at 660 nm
using a microplate reader (Varioskan LUX; Thermo Fisher Scientific,
Inc.). The activity of CK was calculated according to the formula
provided in the manufacturer's protocol.
For the LDH assay, serum samples (20 µl; 1:50),
buffer solution and coenzyme I solution were added into tubes,
vortex mixed and incubated at 37°C for 15 min, according to the
manufacturer's protocol. Then, 2,4-dinitrophenylhydrazine solution
was added before being vortex mixed and incubated at 37°C for 15
min. Next, NaOH solution was added and incubated at room
temperature for 3 min, and the absorbance was detected at 440 nm
using a microplate reader (as aforementioned). The amount of LDH
was assessed by measuring the levels of pyruvic acid.
Measurement of ROS
The fluorescent probe dihydroethidine (DHE) was used
to measure the content of ROS in fresh heart tissue samples using
an ROS detection kit (cat. no. KGAF019; Nanjing KeyGen Biotech Co.,
Ltd.). The specimens were embedded at optimum cutting temperature
and flash-frozen in liquid nitrogen and sectioned (thickness, 5 µm)
using a freezing microtome (Leica CM1950; Leica Microsystems GmbH).
Then, 50 µM DHE solution was added, and sections were incubated at
room temperature in a darkened incubator for 30 min. Next, sections
were washed three times with PBS (5 min/wash). Finally, the
sections were sealed using a water-soluble encapsulant and examined
using a fluorescence microscope at ×200 magnification (Leica
DM4000B; Leica Microsystems GmbH). The stained area of ROS was
quantitatively analyzed using Image Pro Plus 6.0 software (Media
Cybernetics, Inc.).
Measurement of serum levels of SOD,
CAT and MDA
Rat serum was separated by centrifugation at 1,500 ×
g for 10 min at 4°C and the serum levels of SOD, CAT and MDA were
detected using assay kits (cat. nos. A001-3, A007-1 and A003-1,
respectively; all Nanjing Jiancheng Bioengineering Institute).
According to the manufacturer's instruction, serum samples and
relevant solutions of the SOD assay kit were mixed and incubated at
37°C for 20 min, then the absorbance was measured at 450 nm using a
microplate reader (as aforementioned). The activity of SOD was
calculated as U/ml. Similarly, the activity of CAT and MDA were
analyzed following the manufacturer's instructions. The absorbance
of CAT at 405 nm and the absorbance of MDA at 532 nm were measured
using a microplate reader (as aforementioned). The activity of CAT
was calculated as U/ml and the contents of MDA were calculated as
nmol/ml.
Immunohistochemistry
Tissue sections were subjected to conventional
dewaxing to water, rehydrated in a descending series of ethanol
(100, 95, 90 and 80%), and then incubated with 3%
H2O2 for 20 min at 37°C. Sections were
incubated overnight at 4°C with primary antibodies against Bax
protein (1:80; cat. no. 50599-2-Ig), cytochrome c (1:70;
cat. no. 10993-1-AP), HtrA2 (1:80; cat. no. 15775-1-AP) and Smac
(1:70; cat. no. 10434-1-AP) (all ProteinTech Group, Inc.). Next,
sections were washed three times using PBS solution. The sections
were incubated with horseradish peroxidase-conjugated secondary
antibody (1:2,000, cat. no. PV-6001; OriGene Technologies, Inc.)
for 20 min at room temperature, and then stained using the
3,3′diaminobenzidine (DAB) substrate kit (cat. no. ZLI-9019;
OriGene Technologies, Inc.). Color development was induced with
0.5% DAB for 20 min at room temperature. Lastly, the heart tissue
samples were re-stained with 0.5% hematoxylin for 2 min at room
temperature and observed under a light microscope (magnification,
×400). Protein expression levels were measured using Image Pro Plus
6.0 software (Media Cybernetics, Inc.).
Western blotting
Frozen samples were weighed separately and
homogenized in RIPA lysis buffer (Beijing Solarbio Science &
Technology Co., Ltd.), then lysed overnight at 4°C. The heart
tissue samples were centrifuged at 12,000 × g for 10 min at 4°C,
then supernatant (total protein extract) was transferred to an EP
tube and the protein level was quantified via the bicinchoninic
acid method. Then, the protein samples (50 µg) were transferred
onto PVDF membranes using 10% SDS-PAGE gels (EMD Millipore). The
membranes were gently removed and placed in a TBST blocking buffer
(5% skimmed milk in TBS-0.1% Tween-20) for 2 h at 37°C. Next, the
proteins were incubated with anti-NF-κB (p65) (1:2,000; cat. no.
10745-1-AP; ProteinTech Group, Inc.), anti-caspase 3 (1:600; cat.
no. 19677-1-AP; ProteinTech Group, Inc.), anti-cleaved caspase-3
(1:800; cat. no. AF7022; Affinity Biosciences), anti-Bcl-2 (1:600;
cat. no. 26593-1-AP; ProteinTech Group, Inc.) and anti-β-actin
(1:1,000; cat. no. TA-09; OriGene Technologies, Inc.) overnight at
4°C. Then, proteins were incubated with the horseradish
peroxidase-labeled secondary antibody (1:3,000; cat. no. ZB-2301;
OriGene Technologies, Inc.) for 90 min at room temperature.
Membranes were washed three times and proteins were visualized
using the ECL Detection system (TransGen Biotech Co., Ltd.). After
scanning the film with a Tanon1600, the gray value of the band was
measured by Tanon Gis 1D software (Tanon Science and Technology
Co., Ltd.).
Reverse transcription-quantitative
(RT-q)PCR
Total RNA was extracted from heart tissue samples
using TRIzol (cat. no. 15596-026; Invitrogen; Thermo Fisher
Scientific, Inc.). RT was performed with TIANScript RT kit (cat.
no. KR104-02; Tiangen Biotech Co., Ltd.) according to the
manufacturer's instructions. The gene expression levels of p53 and
Bad in heart tissue were assessed via RT-qPCR using SYBR Green
(cat. no. FP205; Tiangen Biotech Co., Ltd.). β-actin was used as
the internal control. The PCR thermocycling conditions were:
Initial denaturation (95°C for 15 min), then 40 cycles of
denaturation (95°C for 10 sec), annealing (58°C for 30 sec) and
extension (72°C for 30 sec). The data was analyzed with the
2−ΔΔCq method (51).
The primers used are listed in Table
I.
 | Table I.Primer sequences of p53, Bad and
β-actin. |
Table I.
Primer sequences of p53, Bad and
β-actin.
| Gene | Primer sequence
(5′→3′) | Fragment size,
bp |
|---|
| p53 | Forward:
CCCCAGGATGTTGCAGAGTTG | 150 |
|
| Reverse:
TTGAGAAGGGACGGAAGATGAC |
|
| Bad | Forward:
GAGTCGCCACAGTTCGTACC | 156 |
|
| Reverse:
TCAAATTCATCGCTCATTCTTC |
|
| β-actin | Forward:
CCTAGACTTCGAGCAAGAGA | 140 |
|
| Reverse:
GGAAGGAAGGCTGGAAGA |
|
Data analysis
Data are presented as the mean ± SEM of three
independent repeats. Statistical comparisons between groups were
measured using one-way ANOVA followed by Bonferroni's test. The
Bonferroni correction was used as a post hoc test to eliminate
false positives in multiple comparisons. Origin 7.5 (OriginLab) and
SPSS 15.0 (SPSS, Inc.) statistical analysis software were used.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Effects of TA on heart
histopathology
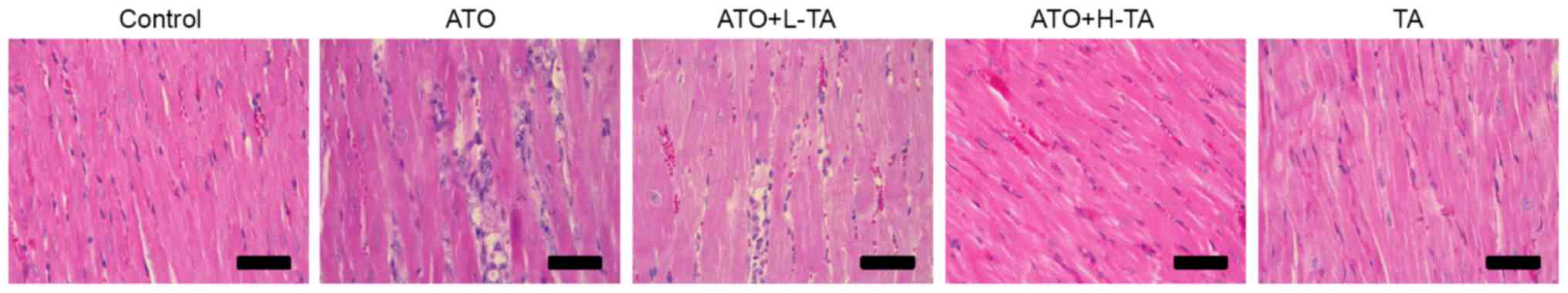
Histological changes of rat heart samples were
investigated by H&E staining. Heart tissue exhibited a normal
myocardium structure and regular myocardial cell distribution in
the Control group (Fig. 2).
However, in the ATO group, notable myocardial tissue injury,
disordered arrangement of cardiomyocytes, cell nucleus pyknosis and
degeneration, increased eosinophils and focal inflammatory cell
infiltration were observed. The ATO + L-TA and ATO + H-TA groups
retained an almost normal myocardial tissue structure (myocardial
cells arranged closely, rich cytoplasm, regular nucleus and normal
cardiac muscle bundles). In addition, there was no difference in
myocardial structure between the TA and Control groups.
Effects of TA on cardiotoxicity
indices
CK and LDH levels in the ATO group were
significantly improved compared with the Control group (Fig. 3), revealing that the experimental
model was successfully established. The CK and LDH levels in the
ATO + L-TA and ATO + H-TA groups were significantly decreased
compared with the ATO group.
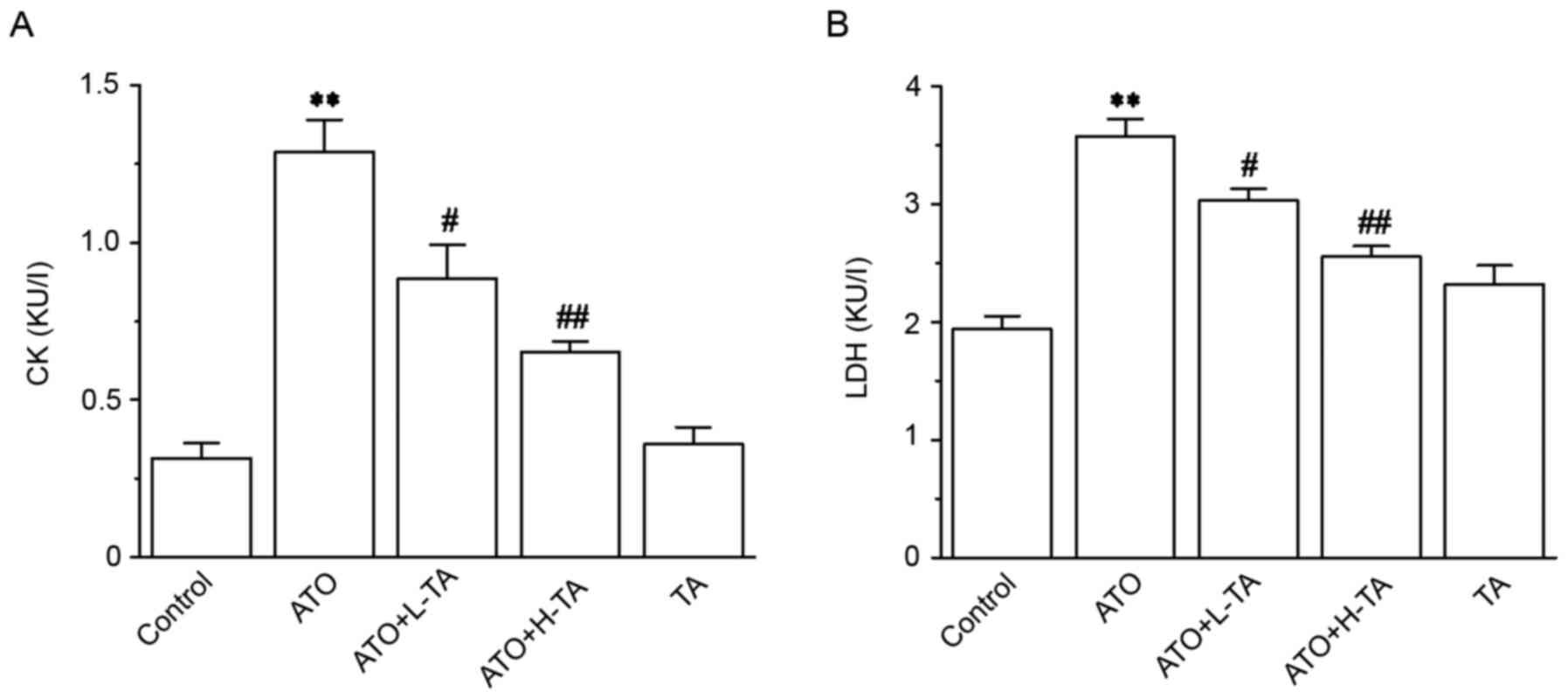 | Figure 3.Effects of TA on the activity of CK
and LDH. Activity of (A) CK and (B) LDH was measured in serum using
commercial detection kits. Serum was collected from the Control,
ATO, ATO + L-TA, ATO + H-TA, and TA groups. Data are presented as
the mean ± SEM (n=6). **P<0.01 vs. Control;
#P<0.05 and ##P<0.01 vs. ATO. CK,
creatine kinase; LDH, lactate dehydrogenase; TA, tannic acid; ATO,
arsenic trioxide; L, low dose; H, high dose. |
Effects of TA on oxidative stress
markers
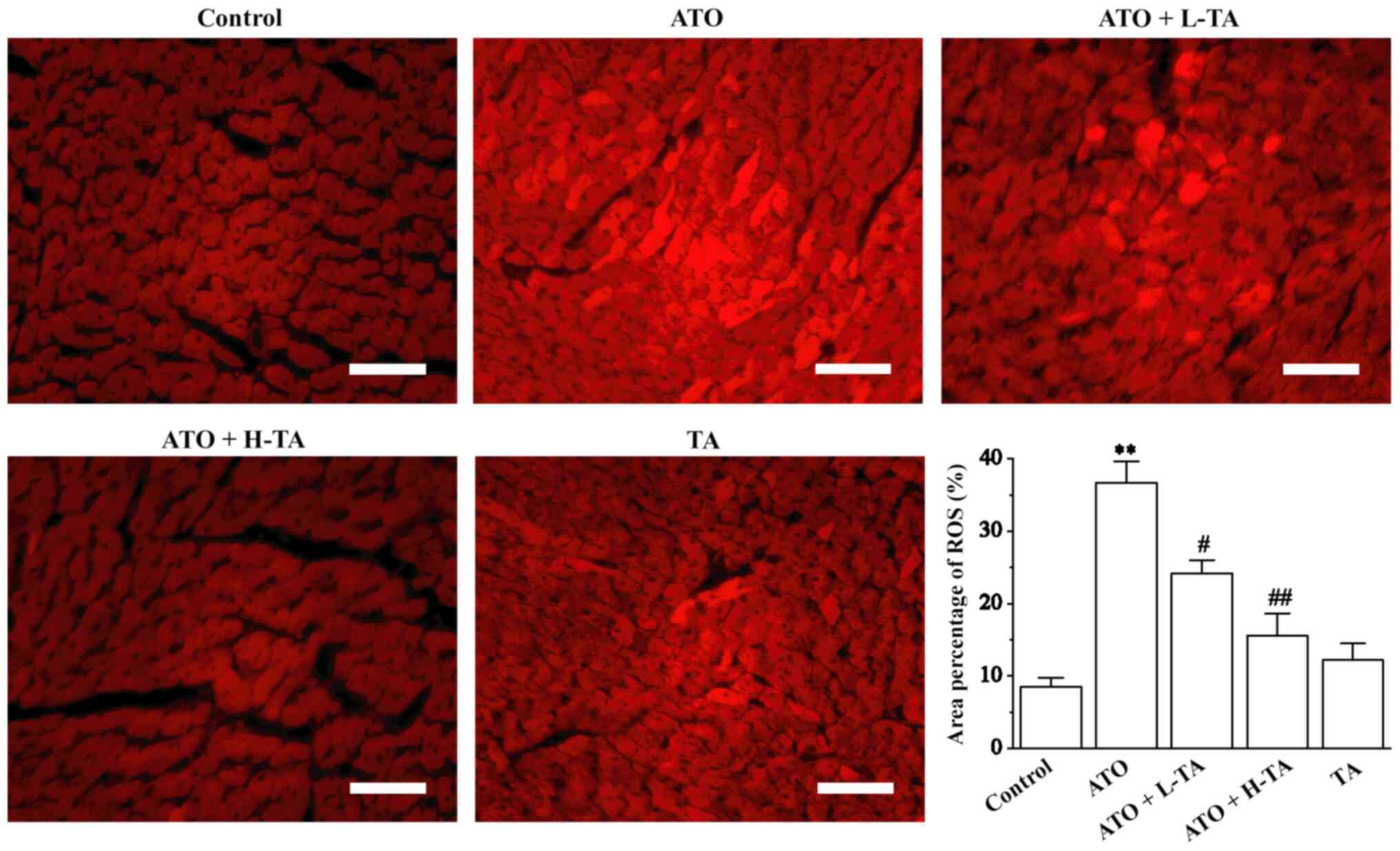
Fluorescence intensity was significantly enhanced in
the ATO group, suggesting that the level of ROS was higher in the
heart tissue compared with the Control group (Fig. 4). Following TA treatment,
fluorescence intensity significantly weakened. These results
indicated that TA protection against ATO-induced heart damage may
be associated with decreasing oxidative stress and ROS
production.
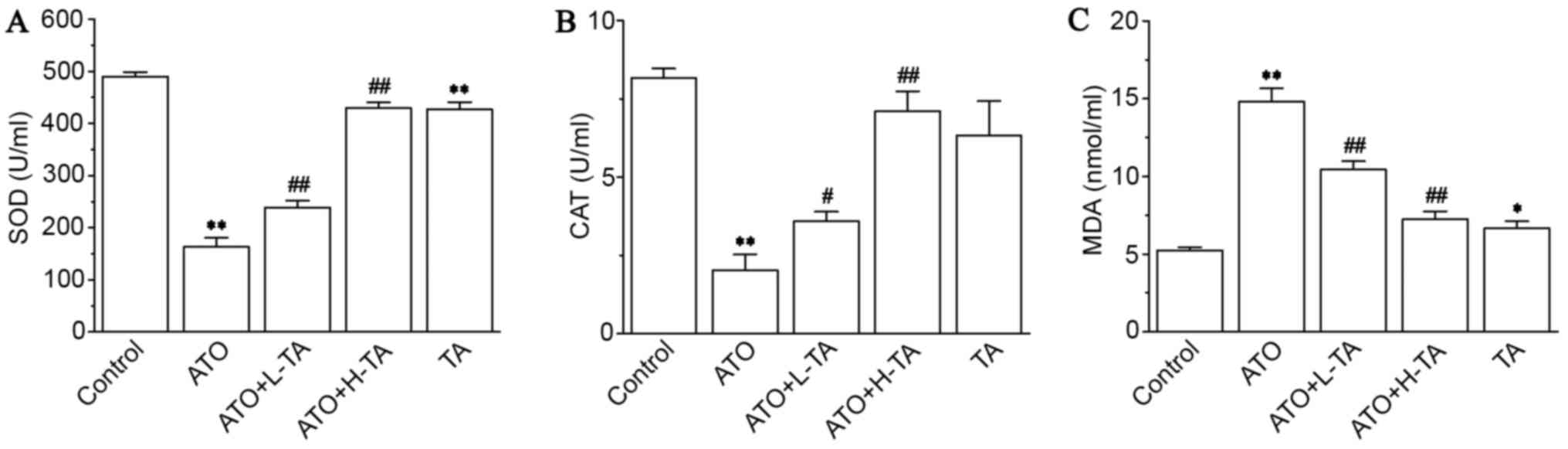
Effects of TA on levels of SOD, CAT,
and MDA
Serum analysis revealed that SOD (Fig. 5A) and CAT (Fig. 5B) levels in the ATO group were
significantly decreased compared with the Control group. Compared
with the ATO group, SOD and CAT activity were increased in the ATO
+ L-TA and ATO + H-TA groups. Moreover, MDA (Fig. 5C) levels increased following ATO
exposure compared with the Control group. Following, TA
administration, the levels of MDA were lower in the ATO + L-TA and
ATO + H-TA groups.
Effects of TA on cytochrome c, Smac,
HtrA2 and Bax expression levels
Immunohistochemistry was used to measure the
expression levels of cytochrome c, Smac, HtrA2 and Bax in
the heart tissue samples. Cytochrome c, Smac, HtrA2 and Bax
expression levels were significantly increased in the ATO group
compared with the Control (Fig.
6). Administration of 20 or 40 mg/kg/day TA decreased the
expression levels of cytochrome c, Smac, HtrA2 and Bax,
indicating that the cardiac protection of TA is a result of its
anti-mitochondrial apoptosis effect.
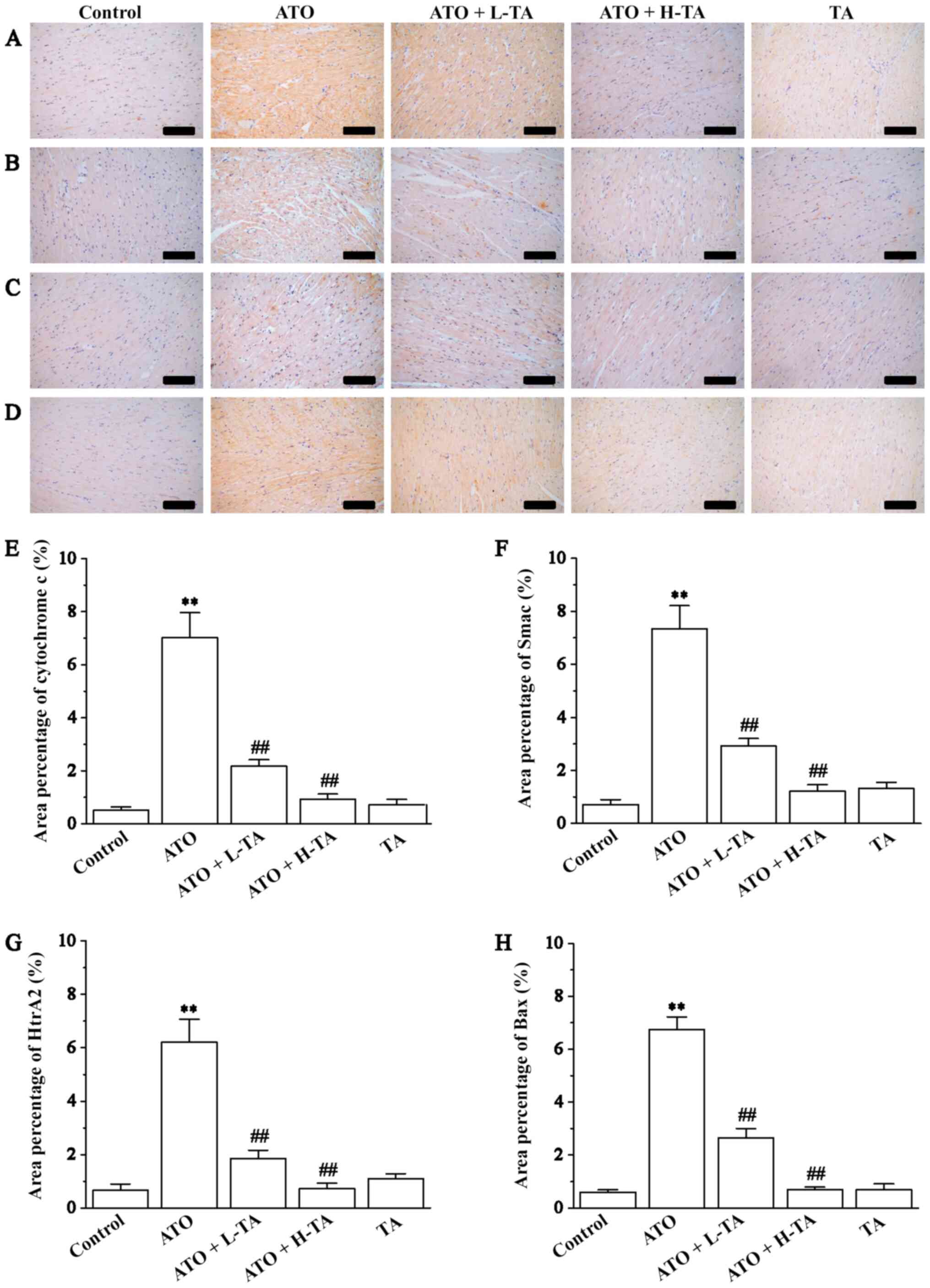 | Figure 6.Effects of TA on the expression
levels of cytochrome c, Smac, HtrA2 and Bax. Morphological
orientation of the expression levels of (A) cytochrome c,
(B) Smac, (C) HtrA2 and (D) Bax in myocardial tissue, as measured
by immunohistochemistry. Magnification, ×200; scale bar, 50 µm.
Area percentage content of (E) cytochrome c, (F) Smac, (G)
HtrA2 and (H) Bax was calculated in the Control, ATO, ATO + L-TA,
ATO + H-TA and TA groups. Data are presented as the mean ± SEM
(n=6). **P<0.01 vs. Control; ##P<0.01 vs. ATO.
Smac, second mitochondria-derived activator of caspases; HtrA2,
high-temperature requirement A2; TA, tannic acid; ATO, arsenic
trioxide; L, low dose; H, high dose. |
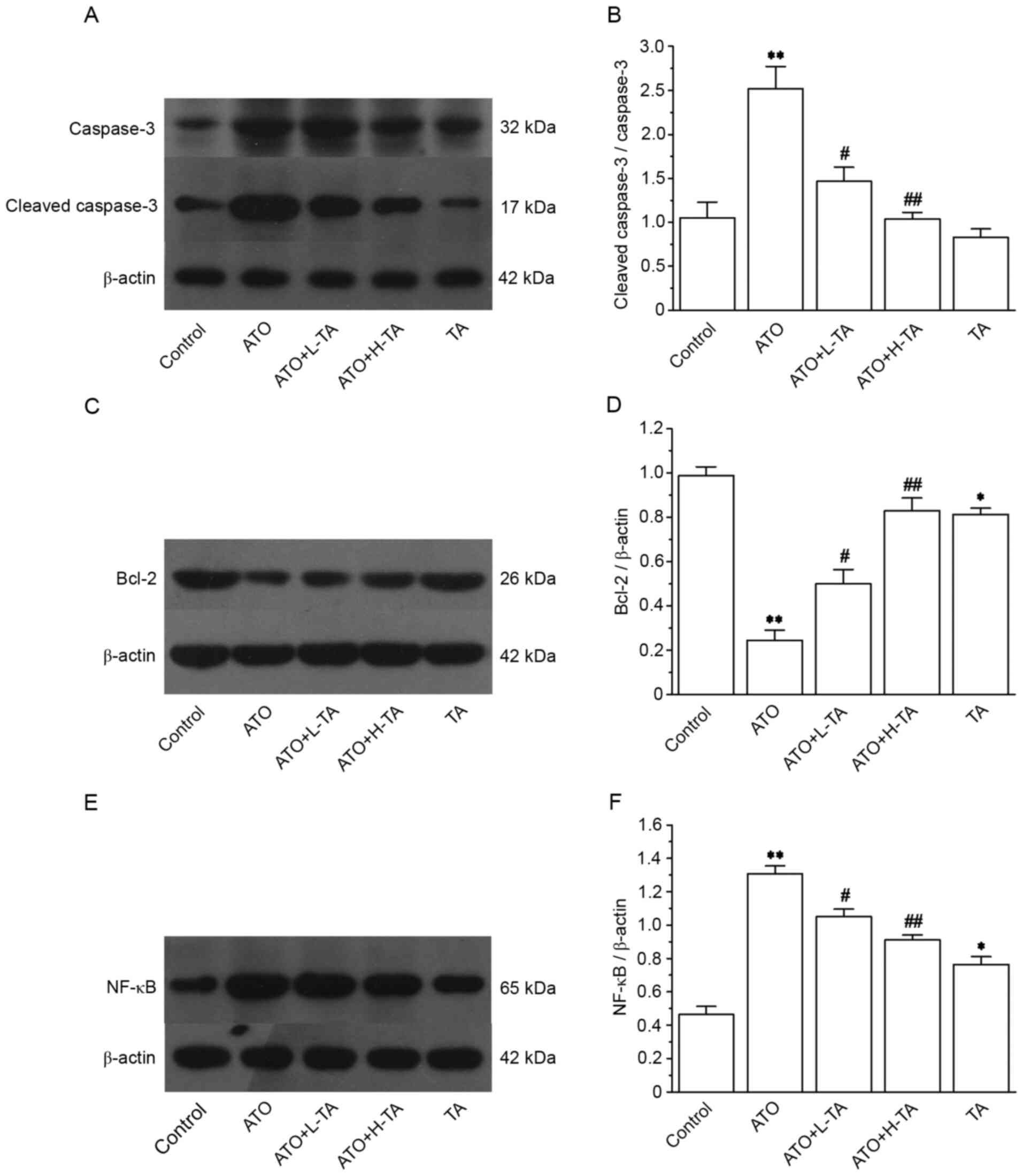
Effects of TA on the expression levels
of caspase-3, cleaved caspase-3, Bcl-2 and NF-κB (p65)
Expression levels of caspase-3, cleaved caspase-3
and NF-κB (p65) were markedly increased in the ATO group (Fig. 7A and E). However, the expression
levels of Bcl-2 significantly decreased (Fig. 7C). The ratio of cleaved
caspase-3/caspase-3 and NF-κB (p65) was significantly upregulated
in the ATO group compared with the Control (Fig. 7B and F). However, compared with the
Control group, the Bcl-2 (Fig. 7D)
expression levels were significantly lower in the ATO group.
Following TA treatment, the ratio of cleaved caspase-3/caspase-3
was significantly decreased, and the expression levels of NF-κB
(p65) were downregulated, whereas Bcl-2 expression levels were
significantly increased.
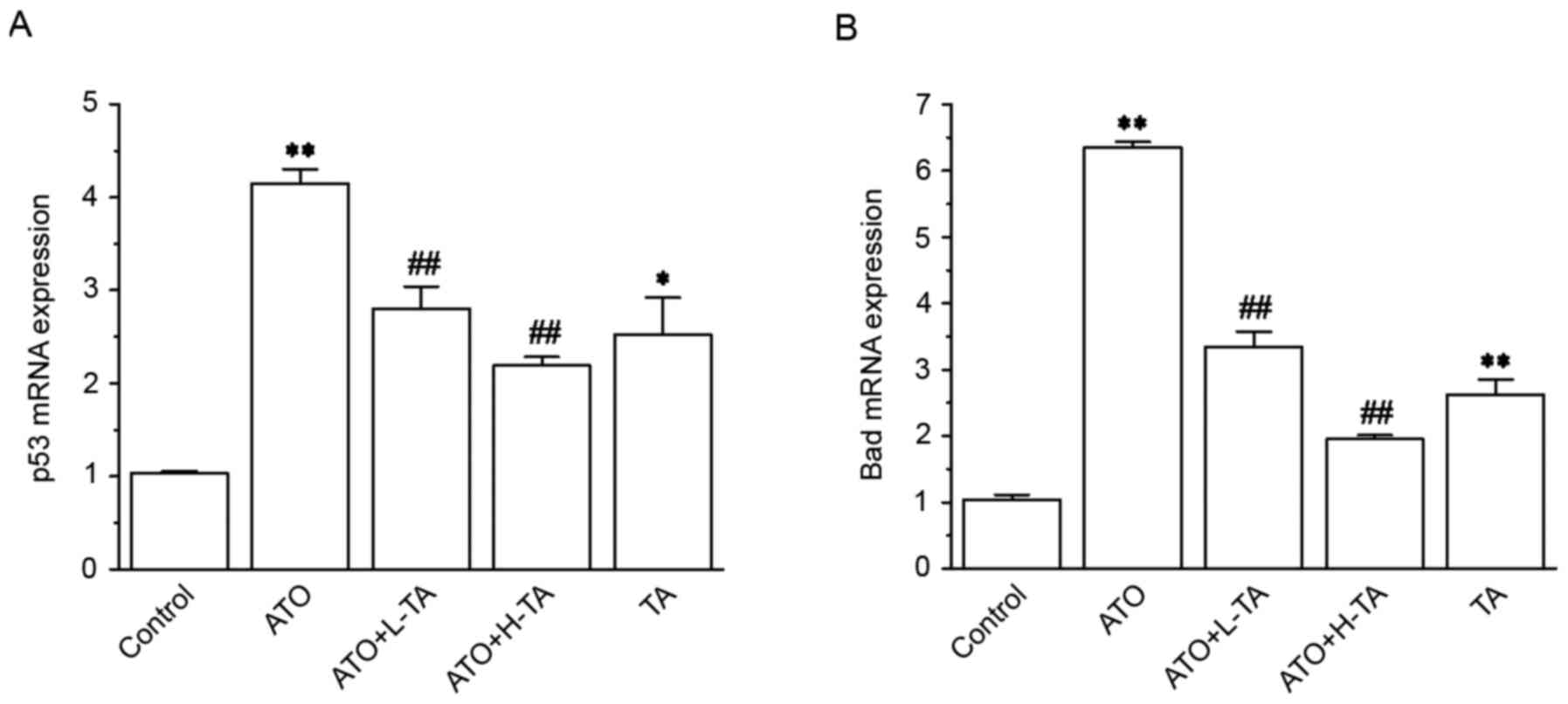
Effects of TA on the expression levels
of p53 and Bad
The expression levels of p53 and Bad in the ATO
group were significantly increased compared with the Control group
(Fig. 8). Subsequent TA
administration caused a significant decrease in p53 and Bad
expression levels.
Discussion
There are an increasing number of studies on the
cardiotoxicity of ATO, which limits its wide clinical application
(13,14). The present study established a
cardiotoxicity model in rats by intraperitoneal injection with ATO
(5 mg/kg). In addition, high-dose TA-alone was administered to
evaluate whether it has a toxic effect on the heart. The results
demonstrated that the cardiotoxicity induced by ATO was primarily
characterized by histopathological changes. In the ATO group,
myocardial cells were swollen, the cytoplasm exhibited vacuolation
and myocardial fiber was abnormal (swelling of myocardial fiber,
interstitial oedema and myofibrillar loss. Serum increase of CK and
LDH enzymes was also detected. These findings demonstrated that ATO
had a toxic effect on the myocardium. Administration of TA
significantly ameliorated ATO-induced pathological changes in
myocardial tissue. In addition, there was no difference in
myocardial structure between the TA and Control groups. Compared
with the Control group, the levels of CK and LDH in the TA group
were not significantly different. These results indicated that
high-dose TA alone hardly induced cardiotoxicity.
Candidate mechanisms for the cardiotoxicity induced
by ATO include changes of cardiac ion channels, oxidative stress
injury and cardiomyocyte apoptosis (51). Compared with other cells,
cardiomyocytes are more susceptible to oxidative stress due to weak
antioxidant defenses (52) and
enrichment of mitochondria (30).
Oxidative stress is considered to be an imbalance
between generation of ROS and the activity of antioxidant defenses.
Oxidative stress is a negative consequence of the in vivo
production of radicals, and it is considered to be an important
factor leading to apoptosis (34).
Multiple studies have reported that high a concentration of arsenic
can cause oxidative stress and increase ROS (53,54).
Excessive ROS cause injury to numerous types of macromolecules,
including DNA, lipid and protein (34,55).
ROS alters cell signaling processes, such as gene expression level
changes, transcription factor activation and apoptosis (56). ATO has a high affinity with
sulfhydryl groups (57). ATO can
penetrate the cell membrane and reach the cytoplasm via diffusion,
resulting in cytotoxicity by increasing ROS (58). ROS are eliminated by the catalytic
deactivation of SOD and CAT in vivo. Therefore, these
enzymes can protect the body from radicals (59). In myocardial cells, the electron
transport chain, situated in the internal membrane of the
mitochondria, is the primary source of ROS (60). Myocardial cells provide energy
essential for cellular survival and function. In addition, CK and
LDH serve important roles in energy metabolism in vivo. CK
and LDH are also important parameters for the diagnosis of
myocardial injury (25). In the
present study, following ATO administration, CAT and SOD activity
markedly decreased, and the MDA levels markedly increased in serum.
This result suggested that the cardiotoxicity of ATO was associated
with oxidative stress injury and that TA significantly improved
this phenomenon.
Mitochondria have long been considered to serve a
considerable role in cell growth. However, the important function
of mitochondria in programmed cell death was not recognized until
the mid-1990s (61). Mitochondria
are not only an important site generating cellular energy, but also
the primary source of ROS and free radicals (23). Concurrently, the mitochondria are
the target of multiple apoptotic signals, which contribute to
apoptosis (62). Studies have
shown that polyphenols affect mitochondrial function and structure
by modulating biosynthesis (mitogenesis), dynamics (fission,
fusion), transport and autophagic cleavage of damaged mitochondria
(mitophagy) (63,64). Wang et al (65) revealed that curcumin downregulated
PI3K/AKT/mTOR and mTOR/p70S6K signaling pathways and activated
autophagy, thus demonstrating neuroprotection in APP/presenilin 1
double transgenic mice. Apoptosis or programmed death is the
mechanism of cell evolutionary conservation, which selectively
removes aged, damaged and other unnecessary cells. This is a
crucial part of numerous normal physiological processes, such as
embryonic development, normal tissue growth and immunoreaction
(66). Apoptosis is mediated by
caspase activation (67). Caspase
is the effector of apoptosis; it can bind to multiple types of
substrate, resulting in specific biochemical and morphological
changes in apoptotic cells, including changes in mitochondrial
membrane permeability, cytoskeleton reorganization, exposure of
phosphatidylserine and DNA fragmentation (68). Endogenous apoptosis is caused by
mitochondrial activation of oxidative stress. Certain mitochondrial
proteins, such as pro-apoptotic factor, cytochrome c
apoptosis-inducing factor, Smac, HtrA2 and endonuclease G, serve a
regulatory role in apoptosis (63). The first released proteins are
cytochrome c, Htr2A and Smac in the mitochondrial apoptosis
pathway, and cytochrome c, released into the cytoplasm, is
combined with apoptotic protease-activating factor to form
associated apoptotic bodies. This promotes the self-activation of
the caspase-9 precursor, then initiates the caspase-3 precursor and
cleaves caspase-3, resulting in apoptosis (69). In the present study, the ratio of
cleaved caspase-3/caspase-3 was increased, which suggested
activation of the caspase apoptosis pathway. Moreover, Du et
al (70) revealed that
Smac/DIABLO enhances cytochrome c-mediated caspase-3
activity. The present results demonstrated an increase in
expression levels of cytochrome c, Smac and Htr2A, as well
as the ratio of cleaved caspase-3/caspase-3, in the ATO group,
confirming that ATO induced apoptotic events. The present study
showed that TA treatment markedly downregulated the expression
levels of the aforementioned apoptosis-associated genes and
decreased the number of apoptotic cells in the ATO + L-TA and ATO +
H-TA groups. In addition, the TA group exhibited no significant
difference in these protein expression levels.
Bcl-2 family proteins include anti-apoptotic genes
Bcl-2 and apoptosis stimulating proteins, such as Bad and Bax
(71). These regulate the release
of cytochrome c and the activation of caspase-3, which play
an essential role in the control of mitochondrial apoptosis
(72). The p53 protein is a tumor
suppressor and serves a crucial role in the regulation of
mitochondrial apoptosis, cell cycle and senescence (69). p53 signaling activates the
transcription of Bad and Bax (73,74)
and suppresses the transcription of Bcl-2 (75,76).
A previous study demonstrated that low levels of p53 contribute to
maintaining mitochondrial activity and function (77). Additionally, studies have reported
that arsenic exposure causes the phosphorylation of the NF-κB (p65)
pathway (36,78). Activation of the NF-κB pathway
triggers an inflammatory response, which induces apoptosis. NF-κB
exhibits pro-inflammatory properties (79), and apoptosis is an indispensable
mechanism that inhibits prolonged inflammation (80). In accordance with published
studies, we observed that ATO exposure increased the expression
levels of p53 protein, Bax, Bad and NF-κB (p65) and decreased those
of Bcl-2 in rat heart. These results demonstrated that ATO induced
myocardial apoptosis via mitochondria dysfunction and inflammation.
Furthermore, following administration of TA, the expression levels
of p53 protein, Bax, Bad and NF-κB (p65) were suppressed and Bcl-2
protein expression levels were promoted.
Based on the findings obtained from the present
study, we hypothesize that the beneficial protective effect of TA
may be achieved via ameliorating ATO-induced injury of
cardiomyocytes and inhibiting the release of cardiac marker enzymes
from the myocardium. In the present study, TA significantly
improved ATO-induced oxidative stress injury and mitochondrial
function due to ROS scavenging properties. As such, TA ameliorates
ATO-induced cardiomyocyte apoptosis via decreasing the release of
mitochondrial-associated proteins cytochrome c, Smac and
Htr2A and inhibition of caspase-3 activation. In addition, the
present study indicated that TA plays an anti-inflammatory role by
inhibiting the activation of the NF-κB pathway; similarly, TA has a
significant suppression of ATO-induced activation of the p53
pathway resulting in decreased release of Bax and increasing the
release of Bcl-2. This protected the cardiomyocytes from
ATO-induced cell death.
The present study investigated the potential effects
and mechanisms of TA against ATO-induced cardiotoxicity. It has
recently been reported that combination of TA and antitumor agent
cisplatin may exert synergistic anticancer effects and may be a
novel adjuvant treatment for liver cancer (81). Our next study will investigate
whether TA has synergistic or antagonistic effects when combined
with ATO to treat malignant tumors.
In conclusion, the present data suggested that
mitochondrial dysfunction contributed to the cardiotoxicity of ATO,
as well as to an oxidative stress reaction and inflammatory
response. The present study demonstrated that TA administration
effectively improved ATO-induced cardiotoxicity. The protective
functions of TA may be associated with suppression of activation of
the mitochondrial apoptosis pathway. Collectively, the present
findings revealed that TA provided effective protection against
ATO-induced cardiotoxicity. Based on these results, TA may have a
potential defense mechanism in ATO clinical therapy and diminish
its cardiotoxic effects. However, further investigation is required
before its clinical application.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Research
Foundation of Administration of Traditional Chinese Medicine of
Hebei Province, China (grant no. 2020188).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YucX, ML, XC and LC conceived and designed the
study. YucX, ML, YurX, WJ and XH performed the experiments. YucX
and LC contributed to the writing of the original draft. ML
contributed to data collection and interpretation. YurX and WJ
provided help for analyzing the data. XH and JZ supervised the
experiments and interpreted data. XC and ZL provided guidance for
software and figures. YucX, ZL and LC revised the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Animal experiments were performed at Hebei Medical
University of Chinese Medicine were in accordance with the Animal
Care and Ethics Committee of Hebei Medical University of Chinese
Medicine (Shijiazhuang, China) under the provisions of the UK
Animals (Scientific Procedures) Act 1986. The Animal Care and
Ethics Committee of Hebei Medical University of Chinese Medicine
approved all animal protocols (approval no. DWLL2018038).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Alamolhodaei NS, Shirani K and Karimi G:
Arsenic cardiotoxicity: An overview. Environ Toxicol Pharmacol.
40:1005–1014. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Manna P, Sinha M and Sil PC:
Arsenic-induced oxidative myocardial injury: Protective role of
arjunolic acid. Arch Toxicol. 82:137–149. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mathews V, Chendamarai E, George B,
Viswabandya A and Srivastava A: Treatment of acute promyelocytic
leukemia with single-agent arsenic trioxide. Mediterr J Hematol
Infect. 3:e20110562011. View Article : Google Scholar
|
|
4
|
Zhang TC, Cao EH, Li JF, Ma W and Qin JF:
Induction of apoptosis and inhibition of human gastric cancer
MGC-803 cell growth by arsenic trioxide. Eur J Cancer.
35:1258–1263. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shen ZY, Zhang Y, Chen JY, Chen MH, Shen
J, Luo WH and Zeng Y: Intratumoral injection of arsenic to enhance
antitumor efficacy in human esophageal carcinoma cell xenografts.
Oncol Rep. 11:155–159. 2004.PubMed/NCBI
|
|
6
|
Akao Y, Nakagawa Y and Akiyama K: Arsenic
trioxide induces apoptosis in neuroblastoma cell lines through the
activation of caspase 3 in vitro. FEBS Lett. 455:59–62. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tai S, Xu LF, Xu M, Zhang LG, Zhang YY,
Zhang KP, Zhang L and Liang CZ: Combination of arsenic trioxide and
everolimus (Rad001) synergistically induces both autophagy and
apoptosis in prostate cancer cells. Oncotarget. 8:11206–11218.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bao ZY, Han ZB, Zhang B, Yu Y, Xu ZH, Ma
WY, Ding FZ, Zhang L, Yu MX, Liu SZ, et al: Arsenic trioxide
blocked proliferation and cardiomyocyte differentiation of human
induced pluripotent stem cells: Implication in cardiac
developmental toxicity. Toxicol Lett. 309:51–58. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu YS, Liang YR, Zheng B, Chu L, Ma DL,
Wang HF, Chu X and Zhang JP: Protective Effects of crocetin on
arsenic trioxide-induced hepatic injury: Involvement of suppression
in oxidative stress and inflammation through activation of Nrf2
signaling pathway in rats. Drug Des Devel Ther. 14:1921–1931. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Unnikrishnan D, Dutcher JP, Garl S,
Varshneya N, Lucariello R and Wiernik PH: Cardiac monitoring of
patients receiving arsenic trioxide therapy. Br J Haematol.
124:610–617. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mu MY, Zhao HJ, Wang Y, Liu JJ, Fei DX and
Xing MW: Arsenic trioxide or/and copper sulfate co-exposure induce
glandular stomach of chicken injury via destruction of the
mitochondrial dynamics and activation of apoptosis as well as
autophagy. Ecotoxicol Environ Saf. 185:1096782019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Badarkhe GV, Sil A, Bhattacharya S, Nath
UK and Das NK: Erythema multiforme due to arsenic trioxide in a
case of acute promyelocytic leukemia: A diagnostic challenge.
Indian J Pharmacol. 48:216–218. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Roboz GJ, Ritchie EK, Carlin RF, Samuel M,
Gale L, Provenzano-Gober JL, Curcio TJ, Feldman EJ and Kligfield
PD: Prevalence, management, and clinical consequences of QT
interval prolongation during treatment with arsenic trioxide. J
Clin Oncol. 32:3723–3728. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hai JJ, Gill H, Tse HF, Kumana CR, Kwong
YL and Siu CW: Torsade de pointes during oral arsenic trioxide
therapy for acute promyelocytic leukemia in a patient with heart
failure. Ann Hematol. 94:501–503. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang Y, Wei ZK, Liu WJ, Wang JJ, He XX,
Huang HL, Zhang JL and Yang ZT: Melatonin protects against arsenic
trioxide-induced liver injury by the upregulation of Nrf2
expression through the activation of PI3K/AKT pathway. Oncotarget.
8:3773–3780. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang XN, Zhao HY, Shao YL, Wang P, Wei YR,
Zhang WQ, Jiang J, Chen Y and Zhang Z: Nephroprotective effect of
astaxanthin against trivalent inorganic arsenic-induced renal
injury in wistar rats. Nutr Res Pract. 8:46–53. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cheng Y, Xue J, Jiang H, Wang M, Gao L, Ma
D and Zhang Z: Neuroprotective effect of resveratrol on arsenic
trioxide-induced oxidative stress in feline brain. Hum Exp Toxicol.
33:737–747. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ahamed M, Akhtar MJ and Alhadlaq HA:
Co-exposure to SiO2 nanoparticles and arsenic induced
augmentation of oxidative stress and mitochondria-dependent
apoptosis in human cells. Int J Environ Res Public Health.
16:31992019. View Article : Google Scholar
|
|
19
|
Sabbah HN: Targeting the mitochondria in
heart failure: A translational perspective. JACC Basic Transl Sci.
5:88–106. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chistiakov DA, Shkurat TP, Melnichenko AA,
Grechko AV and Orekhov AN: The role of mitochondrial dysfunction in
cardiovascular disease: A brief review. Ann Med. 50:121–127. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Roy D, Felty Q, Narayan S and Jayakar P:
Signature of mitochondria of steroidal hormones-dependent normal
and cancer cells: Potential molecular targets for cancer therapy.
Front Biosci. 12:154–173. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Brown DA, Perry JB, Allen ME, Sabbah HN,
Stauffer BL, Shaikh SR, Cleland JG, Colucci WS, Butler J, Voors AA,
et al: Expert consensus document: Mitochondrial function as a
therapeutic target in heart failure. Nat Rev Cardiol. 14:238–250.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pohjoismäki JL and Goffart S: The role of
mitochondria in cardiac development and protection. Free Radic Biol
Med. 106:345–354. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Peoples JN, Saraf A, Ghazal N, Pham TT and
Kwong JQ: Mitochondrial dysfunction and oxidative stress in heart
disease. Exp Mol Med. 51:1–13. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Abdelrahman RS, El-Awady MS, Nader MA and
Ammar EM: Hydrogen sulfide ameliorates cardiovascular dysfunction
induced by cecal ligation and puncture in rats. Hum Exp Toxicol.
34:953–964. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gill C, Mestril R and Samali A: Losing
heart: The role of apoptosis in heart disease-a novel therapeutic
target? FASEB J. 16:135–146. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yu XJ, Wang ZY, Shu ZP, Li ZQ, Ning Y, Yun
KL, Bai HN, Liu RH and Liu WL: Effect and mechanism of Sorbus
pohuashanensis (Hante) Hedl. Flavonoids protect against arsenic
trioxide-induced cardiotoxicity. Biomed Pharmacother. 88:1–10.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
McDonough KH: The role of alcohol in the
oxidant antioxidant balance in heart. Front Biosci. 4:D601–D606.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang Y, Wu YP, Wang YY, Xu H, Mei XQ, Yu
DY, Wang YB and Li WF: Antioxidant properties of probiotic
bacteria. Nutrients. 9:5212017. View Article : Google Scholar
|
|
30
|
Vineetha VP, Soumya RS and Raghu KG:
Phloretin ameliorates arsenic trioxide induced mitochondrial
dysfunction in H9c2 cardiomyoblasts mediated via alterations in
membrane permeability and ETC complexes. Eur J Pharmacol.
754:162–172. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lindskog M, Gleissman H, Ponthan F, Castro
J, Kogner P and Johnsen JI: Neuroblastoma cell death in response to
docosahexaenoic acid: Sensitization to chemotherapy and
arsenic-induced oxidative stress. Int J Cancer. 118:2584–2593.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen H, Liu G, Qiao N, Kang Z, Hu L, Liao
J, Yang F, Pang C, Liu B, Zeng Q, et al: Toxic effects of arsenic
trioxide on spermatogonia are associated with oxidative stress,
mitochondrial dysfunction, autophagy and metabolomic alterations.
Ecotoxicol Environ Saf. 190:1100632020. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Patlolla AK and Tchounwou PB: Serum acetyl
cholinesterase as a biomarker of arsenic induced neurotoxicity in
sprague-dawley rats. Int J Environ Res Public Health. 2:80–83.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Vineetha VP and Raghu KG: An overview on
arsenic trioxide-induced cardiotoxicity. Cardiovasc Toxicol.
19:105–119. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Amini-Khoei H, Hosseini MJ, Momeny M,
Rahimi-Balaei M, Amiri S, Haj-Mirzaian A, Khedri M, Jahanabadi S,
Mohammadi-Asl A, Mehr SE and Dehpour AR: Morphine attenuated the
cytotoxicity induced by arsenic trioxide in H9c2 cardiomyocytes.
Biol Trace Elem Res. 173:132–139. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ghosh J, Das J, Manna P and Sil PC:
Taurine prevents arsenic-induced cardiac oxidative stress and
apoptotic damage: Role of NF-kappa B, p38 and JNK MAPK pathway.
Toxicol Appl Pharmacol. 240:73–87. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang JY, Sun GB, Luo Y, Wang M, Wang W,
Du YY, Yu YL and Sun XB: Salvianolic acid a protects H9c2 cells
from arsenic trioxide-induced injury via inhibition of the MAPK
signaling pathway. Cell Physiol Biochem. 41:1957–1969. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhu S, Wang Y, Liu H, Wei W, Tu Y, Chen C,
Song J, Xu Z, Li J, Wang C and Sun S: Thyroxine affects
lipopolysaccharide-induced macrophage differentiation and
myocardial cell apoptosis via the NF-κB p65 pathway both in vitro
and in vivo. Mediators Inflamm. 2019:20989722019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liu X, Kim J, Li Y, Li J, Liu F and Chen
X: Tannic acid stimulates glucose transport and inhibits adipocyte
differentiation in 3T3-L1 cells. J Nutr. 135:165–171. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hemmati AA, Olapour S, Varzi HN, Khodayar
MJ, Dianat M, Mohammadian B and Yaghooti H: Ellagic acid protects
against arsenic trioxide-induced cardiotoxicity in rat. Hum Exp
Toxicol. 37:412–419. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ashafaq M, Sharma P, Khatoon S, Haque D,
Tabassum H and Parvez S: Heavy metal-induced systemic dysfunction
attenuated by tannic acid. J Environ Pathol Toxicol Oncol.
35:109–120. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhang JP, Cui LJ, Han X, Zhang YY, Zhang
X, Chu X, Zhang FH, Zhang Y and Chu L: Protective effects of tannic
acid on acute doxorubicin-induced cardiotoxicity: Involvement of
suppression in oxidative stress, inflammation, and apoptosis.
Biomed Pharmacother. 93:1253–1260. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chu L, Li PY, Song T, Han X, Zhang X, Song
QT, Liu T, Zhang YY and Zhang JP: Protective effects of tannic acid
on pressure overload-induced cardiac hypertrophy and underlying
mechanisms in rats. J Pharm Pharmacol. 69:1191–1207. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhu FL, Chu X, Wang H, Zhang X, Zhang YY,
Liu ZY, Guo H, Liu HY, Liu Y, Chu L and Zhang JP: New Findings on
the effects of tannic acid: Inhibition of L-Type calcium channels,
Calcium transient and contractility in rat ventricular myocytes.
Phytother Res. 30:510–516. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Jin WY, Xue YR, Xue YC, Han X, Song QT,
Zhang JP, Li ZL, Cheng J, Guan SJ, Sun SJ and Chu L: Tannic acid
ameliorates arsenic trioxide-induced nephrotoxicity, contribution
of NF-κB and Nrf2 pathways. Biomed Pharmacother. 126:1100472020.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kumazaki M, Ando H, Sasaki A, Koshimizu
TA, Ushijima K, Hosohata K, Oshima Y and Fujimura A: Protective
effect of α-lipoic acid against arsenic trioxide-induced acute
cardiac toxicity in rats. J Pharmacol Sci. 115:244–248. 2011.
View Article : Google Scholar
|
|
47
|
Miao X, Tang ZF, Wang YG, Su GF, Sun WX,
Wei W, Li W, Miao LN, Cai L, Tan Y and Liu QJ: Metallothionein
prevention of arsenic trioxide-induced cardiac cell death is
associated with its inhibition of mitogen-activated protein kinases
activation in vitro and in vivo. Toxicol Lett. 220:277–285. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Saxena PN, Anand S, Saxena N and Bajaj P:
Effect of arsenic trioxide on renal functions and its modulation by
curcuma aromatica leaf extract in albino rat. J Environ Biol.
30:527–531. 2009.PubMed/NCBI
|
|
49
|
Liu J, Lu Y, Wu Q, Goyer RA and Waalkes
MP: Mineral arsenicals in traditional medicines: Orpiment, realgar,
and arsenolite. J Pharmacol Exp Ther. 326:363–368. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Song J, Ding WB, Liu BJ, Liu D, Xia Z,
Zhang L, Cui L, Luo Y, Jia XB and Feng L: Anticancer effect of
caudatin in diethylnitrosamine-induced hepatocarcinogenesis in
rats. Mol Med Rep. 22:697–706. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Costa VM, Carvalho F, Duarte JA, Bastos
Mde L and Remiao F: The heart as a target for xenobiotic toxicity:
The cardiac susceptibility to oxidative stress. Chem Res Toxicol.
26:1285–1311. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
James TN: Long reflections on the QT
interval: The sixth annual Gordon K. Moe Lecture. J Cardiovasc
Electrophysiol. 7:738–759. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Best PJ, Hasdai D, Sangiorgi G, Schwartz
RS, Holmes DR Jr, Simari RD and Lerman A: Apoptosis. Basic concepts
and implications in coronary artery disease. Arterioscler Thromb
Vasc Biol. 19:14–22. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Matsui M, Nishigori C, Toyokuni S, Takada
J, Akaboshi M, Ishikawa M, Imamura S and Miyachi Y: The role of
oxidative DNA damage in human arsenic carcinogenesis: Detection of
8-hydroxy-2′-deoxyguanosine in arsenic-related Bowen's disease. J
Invest Dermatol. 113:26–31. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Berridge MJ, Lipp P and Bootman MD: The
versatility and universality of calcium signalling. Nat Rev Mol
Cell Biol. 1:11–21. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Pulido MD and Parrish AR: Metal-induced
apoptosis: Mechanisms. Mutat Res. 533:227–241. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Torka P, Al Ustwani O, Wetzler M, Wang ES
and Griffiths EA: Swallowing a bitter pill-oral arsenic trioxide
for acute promyelocytic leukemia. Blood Rev. 30:201–211. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Sugden PH and Clerk A: Oxidative stress
and growth-regulating intracellular signaling pathways in cardiac
myocytes. Antioxid Redox Signal. 8:2111–2124. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Vineetha RC, Binu P, Arathi P and Nair RH:
L-ascorbic acid and alpha-tocopherol attenuate arsenic
trioxide-induced toxicity in H9c2 cardiomyocytes by the activation
of Nrf2 and Bcl2 transcription factors. Toxicol Mech Methods.
28:353–360. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Petit PX, Susin SA, Zamzami N, Mignotte B
and Kroemer G: Mitochondria and programmed cell death: Back to the
future. FEBS Lett. 396:7–13. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Lopez J and Tait SW: Mitochondrial
apoptosis: Killing cancer using the enemy within. Br J Cancer.
112:957–962. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Naoi M, Wu Y, Shamoto-Nagai M and Maruyama
W: Mitochondria in neuroprotection by phytochemicals: Bioactive
polyphenols modulate mitochondrial apoptosis system, function and
structure. Int J Mol Sci. 20:24512019. View Article : Google Scholar
|
|
64
|
Teixeira J, Deus CM, Borges F and Oliveira
PJ: Mitochondria: Targeting mitochondrial reactive oxygen species
with mitochondriotropic polyphenolic-based antioxidants. Int J
Biochem Cell Biol. 97:98–103. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Wang C, Zhang X, Teng ZP, Zhang T and Li
Y: Downregulation of PI3K/Akt/mTOR signaling pathway in
curcumin-induced autophagy in APP/PS1 double transgenic mice. Eur J
Pharmacol. 740:312–320. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Tower J: Programmed cell death in aging.
Ageing Res Rev. 23:90–100. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Moloudi K, Neshasteriz A, Hosseini A,
Eyvazzadeh N, Shomali M, Eynali S, Mirzaei E and Azarnezhad A:
Synergistic Effects of arsenic trioxide and radiation: Triggering
the intrinsic pathway of apoptosis. Iran Biomed J. 21:330–337.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Adil M, Kandhare AD, Ghosh P and Bodhankar
SL: Sodium arsenite-induced myocardial bruise in rats: Ameliorative
effect of naringin via TGF-β/Smad and Nrf/HO pathways. Chem Biol
Interact. 253:66–77. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Estaquier J, Vallette F, Vayssiere JL and
Mignotte B: The mitochondrial pathways of apoptosis. Adv Exp Med
Biol. 942:157–183. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Du CY, Fang M, Li YC, Lily L and Wang XD:
Smac, a mitochondrial protein that promotes cytochrome c-dependent
caspase activation by eliminating IAP inhibition. Cell. 102:33–42.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Boise LH, Gottschalk AR, Quintáns J and
Thompson CB: Bcl-2 and Bcl-2-related proteins in apoptosis
regulation. Curr Top Microbiol Immunol. 200:107–121.
1995.PubMed/NCBI
|
|
72
|
Tischner D, Woess C, Ottina E and
Villunger A: Bcl-2-regulated cell death signalling in the
prevention of autoimmunity. Cell Death Dis. 1:e482010. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Miyashita T and Reed JC: Tumor suppressor
p53 is a direct transcriptional activator of the human bax gene.
Cell. 80:293–299. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Oda E, Ohki R, Murasawa H, Nemoto J,
Shibue T, Yamashita T, Tokino T, Taniguchi T and Tanaka N: Noxa, a
BH3-only member of the Bcl-2 family and candidate mediator of
p53-induced apoptosis. Science. 288:1053–1058. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Shen Y and Shenk T: Relief of p53-mediated
transcriptional repression by the adenovirus E1B 19-kDa protein or
the cellular Bcl-2 protein. Proc Natl Acad Sci USA. 91:8940–8944.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Hoffman WH, Biade S, Zilfou JT, Chen J and
Murphy M: Transcriptional repression of the anti-apoptotic survivin
gene by wild type p53. J Biol Chem. 277:3247–3257. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Bensaad K and Vousden KH: p53: New roles
in metabolism. Trends Cell Biol. 17:286–291. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Mathas S, Lietz A, Janz M, Hinz M, Jundt
F, Scheidereit C, Bommert K and Dorken B: Inhibition of NF-kappaB
essentially contributes to arsenic-induced apoptosis. Blood.
102:1028–1034. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Pahl HL: Activators and target genes of
Rel/NF-kappaB transcription factors. Oncogene. 18:6853–6866. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Pace C, Dagda R and Angermann J:
Antioxidants protect against arsenic induced mitochondrial
cardio-toxicity. Toxics. 5:382017. View Article : Google Scholar
|
|
81
|
Geng NN, Zheng X, Wu MS, Yang L, Li XY and
Chen JD: Tannic acid synergistically enhances the anticancer
efficacy of cisplatin on liver cancer cells through
mitochondria-mediated apoptosis. Oncol Rep. 42:2108–2116.
2019.PubMed/NCBI
|