Introduction
An increasing amount of evidence has indicated that
macrophage inflammation is an important pathological manifestation
of inflammatory vascular diseases (1,2). The
gut microbiota metabolite trimethylamine N-oxide (TMAO) has been
shown to increase the incidence of major cardiovascular diseases
(3). TMAO has been reported to
promote activation of the NLR family pyrin domain containing 3
(NLRP3) inflammasome signaling pathway in endothelial cells
(4) and vascular smooth muscle
cells (VSMCs) (5), which can
promote vascular inflammation and atherosclerosis. Notably,
inhibition of TMAO production can attenuate atherosclerosis
development in mice (6). However,
the specific mechanistic link between TMAO and macrophage
inflammation has not yet been elucidated. Therefore, investigating
the specific mechanisms underlying TMAO-induced inflammation in
macrophages is essential for the development of new
therapeutics.
In mammalian cells, endogenous hydrogen sulfide
(H2S) is synthesized by enzymatic catalysis.
Cystathionine γ-lyase (CSE), cystathionine β-synthase (CBS) and
3-mercaptopyruvate sulfurtransferase (3-MST) are the major enzymes
involved in catalyzing H2S synthesis (7). These enzymes that catalyze
H2S synthesis are distributed in different tissues: CSE
is predominantly expressed in the cardiovascular system, CBS is
mainly expressed in the central nervous system, and 3-MST is
expressed in human renal tissue, cardiomyocytes, neurons and the
gastrointestinal tract (7,8). Previous studies have shown that
H2S has a wide range of cardiovascular protective
effects, including the inhibition of foam cell formation,
endothelial inflammation, VSMC proliferation and platelet
aggregation (9). CSE knockdown has
been reported to result in decreased endogenous H2S
production and increased atherosclerotic plaque formation (10). Furthermore, in a previous study,
CSE expression significantly reduced plaque size in
ApoE−/− mice (11).
Whether H2S is involved in TMAO-induced macrophage
inflammation remains unclear, despite significant progress in
understanding the role of H2S in cardioprotection
(12).
Sirtuin 1 (SIRT1) is an NAD+-dependent
class III histone deacetylase that is widely expressed in various
human organs. SIRT1 has numerous biological activities, including
anti-inflammatory, anti-aging and antioxidative activities
(13,14). Our previous studies indicated that
SIRT1 is involved in H2S-mediated cardiovascular
protection (15–17). SIRT1 activators E123 and
1evogliptin has been shown to protect against experimental
atherosclerosis by lowering plasma cholesterol and triglycerides
(18), and inhibiting the vascular
inflammatory response (19). By
contrast, SIRT1 inhibitor sirtinol has been shown to block the
protective effects of H2S in foam cell formation and
atherosclerosis (20,21). H2S has also been
reported to act as a novel SIRT1 activator that can directly induce
SIRT1 sulfhydration (22).
Therefore, H2S may exert a broad protective effect
against atherosclerosis by inducing SIRT1 sulfhydration. Based on
the aforementioned studies, the present study investigated the
modifying effects of H2S on SIRT1 sulfhydration and its
regulatory role in TMAO-induced macrophage inflammation.
Materials and methods
Cell culture
RAW264.7 macrophages (American Type Culture
Collection) were cultured in RPMI 1640 medium (HyClone; Cytiva)
supplemented with 10% fetal bovine serum (FBS; Shanghai ExCell
Biology, Inc.), 100 µg/ml streptomycin and 100 U/ml penicillin
(both Beyotime Institute of Biotechnology) in an incubator at 37°C
with 5% CO2. Before treatment, the macrophages were
seeded in 100-mm dishes at a density of 2×106 cells/dish
and were cultured with medium containing 10% FBS for 24 h, followed
by starvation in 0.5% FBS-containing RPMI 1640 medium for 24 h at
37°C. RAW264.7 macrophages was used as the control. Subsequently,
the macrophages were pretreated with 25 µM resveratrol
(Sigma-Aldrich; Merck KGaA) for 24 h before TMAO (100 mM,
Sigma-Aldrich) stimulation for 24 h. For certain experiments, the
macrophages were pretreated with 40 mM SIRT1 inhibitor nicotinamide
(Calbiochem; Merck KGaA) or DL-dithiothreitol (DTT; 1 mM, Phygene,
China) for 60 min before sodium hydrosulfide (NaHS, 100 µmol/l,
Sigma-Aldrich, USA) stimulation for 24 h at 37°C. To stimulate
SIRT1 expression, macrophages were incubated with NaHS for 12, 24
or 48 h.
Modified biotin switch assay
The SIRT1 S-sulfhydration assay was performed as
previously reported (22). The
macrophages were lysed in HENS buffer [250 mM HEPES (pH 7.7), 1 mM
EDTA, 0.1 mM Neocuproine, and 1% SDS] supplemented with protease
inhibitors. The cell lysates were blocked with HEN blocking buffer
[250 mM HEPES (pH 7.7), 1 mM EDTA, 0.1 mM Neocuproine] containing
20 mM methyl methanethiosulfonate (MMTS, Sigma-Aldrich) for 20 min
at 50°C. Subsequently, MMTS was eliminated by precipitating with
acetone for 20 min at −20°C. Thereafter, leaving a small part of
the proteins as control (input SIRT1), the remaining proteins were
resuspended in HENS buffer, and biotin-HPDP (4 mM, Thermo
Scientific) was added for 4 h at 37°C. Subsequently, the remaining
proteins were pulled down by streptavidin agarose (MilliporeSigma)
at 4°C overnight under gentle shaking conditions. After
centrifugation, biotinylated proteins (SHY-SIRT1) were resuspended
in a protein loading buffer. Finally, the samples were analyzed by
western blotting. Biotinylated proteins (SHY-SIRT1) were blotted
together with total lysates (input SIRT1) and subjected to SIRT1
immunoblotting to assess SIRT1 sulfhydration.
H2S concentration
measurement
As described previously (23), the supernatant of macrophages in
control and TMAO group was collected and centrifuged at 12,000 g
for 10 min at 4°C. H2S concentration measurement was
performed according to the methylene blue method. Briefly, 800 µl
supernatant was mixed sequentially with 8 µl NaOH (1 M), 80 µl 20
mM N, N-dimethyl-p-phenylenediamine sulfate in 7.2 M HCl, and 80 µl
30 mM FeCl3 in 1.2 M HCl. Subsequently, the mixtures
were incubated at 37°C for 20 min. Finally, the formed methylene
blue was detected at 668 nm using a microplate reader (ELx800;
BioTek Instruments, Inc.). H2S concentration was
quantified using a standard curve of NaHS.
Western blot analysis
The macrophages were lysed using ice-cold RIPA Lysis
Buffer (cat. no. P0013B; Beyotime Institute of Biotechnology),
containing a protease and phosphatase inhibitor cocktail (cat. no.
P1045; Beyotime Institute of Biotechnology). Protein concentrations
were measured using a BCA protein assay kit (cat. no. P0010S;
Beyotime Institute of Biotechnology). Equal amounts of protein (80
µg) were separated by SDS-PAGE on 10 or 12% gels and were
transferred onto PVDF membranes. The membranes were then blocked
with 5% non-fat dry milk containing Tris-buffered saline −0.1%
Tween 20 at 37°C for 2 h and were incubated with the following
primary antibodies at 4°C overnight: CSE (1:1,000; cat. 60234-1-AP;
Proteintech Group, Inc.), SIRT1 (1;750; cat. no. 13161-1-AP;
Proteintech Group, Inc.), p65 NF-κB (1:800; cat. no. 8242; Cell
Signaling Technology, Inc.), phosphorylated (p)-p65 NF-κB (1:750;
cat. no. 3033; Cell Signaling Technology, Inc.), IL-1β (1:500; cat.
no. WL00891; Wanleibio Co., Ltd.), IL-6 (1:600; cat. no.
21865-1-AP; Proteintech Group, Inc.), TNF-α (1:750; cat. no.
17590-1-AP; Proteintech Group, Inc.) and β-actin (1:5,000; cat. no.
20536-1-AP; Proteintech Group, Inc.). Subsequently, the membranes
were incubated with a horseradish peroxidase-linked secondary
antibody (1:5,000; cat. no. A0208 or A0216; Beyotime Institute of
Biotechnology) for 2 h at 37°C. Eventually, images were acquired
using a chemiluminescence reagent kit (WBKlS0100, MilliporeSigma)
and the densities of the bands were analyzed using ImageJ 1.47i
software (National Institutes of Health) for
semi-quantification.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was isolated from the cells using
TRIzol® reagent (cat. no. 15596-026; Invitrogen; Thermo
Fisher Scientific, Inc.) and RT was performed with a high-capacity
cDNA synthesis kit (cat. no. RR037A; Takara Bio, Inc.). The RT
protocol was as follows: 37°C for 15 min, followed by 85°C for 5
sec, and finished at 4°C. qPCR analysis was performed using the
SYBR Green PCR Mix kit (cat. no. DRR041A; Takara Bio, Inc.) and the
ABI7500 System (Applied Biosystems; Thermo Fisher Scientific,
Inc.). The thermocycling conditions were as follows: One cycle at
95°C for 30 sec, followed by 40 cycles at 95°C for 15 sec and 60°C
for 30 sec. The PCR finished with extension at 60°C for 5 min and
holding at 4°C. The relative fold changes in gene expression were
normalized to the mRNA expression levels of β-actin and were
analyzed using the 2−ΔΔCq method (24). The specific primer sequences are
listed in Table I.
 | Table I.Quantitative PCR primer sequences
used in the present study. |
Table I.
Quantitative PCR primer sequences
used in the present study.
| Mouse gene
name | Forward
(5′-3′) | Reverse
(5′-3′) |
|---|
| β-actin |
GTGACGTTGACATCCGTAAAGA |
GCCGGACTCATCGTACTCC |
| MCP-1 |
GTCTGTGCTGACCCCAAGAAG |
TGGTTCCGATCCAGGTTTTTA |
| IL-1β |
GAAATGCCACCTTTTGACAGTG |
TGGATGCTCTCATCAGGACAG |
| IL-6 |
TTCCATCCAGTTGCCTTCTTG |
TTGGGAGTGGTATCCTCTGTGA |
| IL-10 |
GCTCTTACTGACTGGCATGAG |
CGCAGCTCTAGGAGCATGTG |
| IL-4 |
GGTCTCAACCCCCAGCTAGT |
GCCGATGATCTCTCTCAAGTGAT |
| TNF-α |
GCGACGTGGAACTGGCAGAAG |
GCCACAAGCAGGAATGAGAAGAGG |
| CSE | CTTGCTGCCACCA
TTACG |
TTCAGATGCCACCCTCCT |
Statistical analysis
The data are presented as the mean ± SD. The
significant differences between two groups were evaluated using an
unpaired Student's t-test. Differences among multiple groups were
analyzed by one-way analysis of variance with Tukey's post hoc
test. All statistical analyses were performed using SPSS software
(version 18; IBM Corp.). All experiments were performed with at
least three independent biological samples. P<0.05 was
considered to indicate a statistically significant difference.
Results
TMAO downregulates CSE and increases
inflammatory cytokine levels
Macrophages were treated for 24 h with 100 mM TMAO
to evaluate the effects of TMAO on CSE expression. In the present
model, TMAO significantly decreased the mRNA and protein expression
levels of CSE in macrophages (Fig. 1A
and B) compared with the control group. In addition, TMAO
treatment resulted in significantly lower levels of H2S
measured in cell supernatants (Fig.
1C). TMAO upregulated the mRNA expression levels of IL-6, IL-1β
and monocyte chemoattractant protein-1 (MCP-1), and downregulated
IL-4 and IL-10 (Fig. 1D).
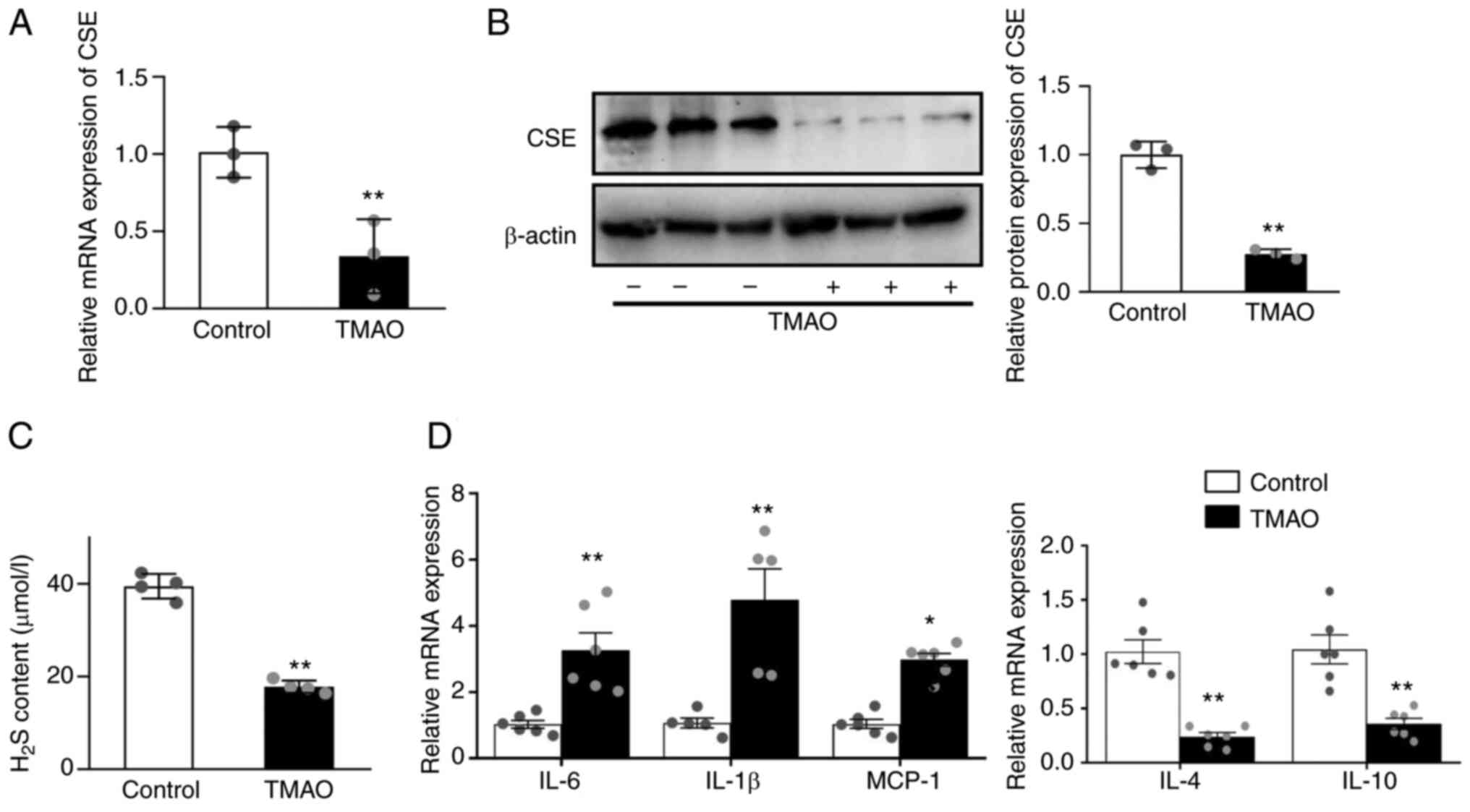 | Figure 1.TMAO treatment decreases the
expression of CSE and causes inflammation. (A) mRNA expression
levels of CSE in the macrophages after TMAO treatment, as measured
by RT-qPCR. (B) Representative western blots of CSE in the
macrophages after TMAO treatment. (C) H2S concentration
was detected in the supernatant of macrophages after TMAO
treatment. (D) mRNA expression levels of IL-1β, IL-6, MCP-1, IL-4
and IL-10 in the macrophages after TMAO treatment, as measured by
RT-qPCR (n≥4). *P˂0.05, **P˂0.01 vs. control group. CSE,
cystathionine γ-lyase; H2S, hydrogen sulfide; MCP-1, monocyte
chemoattractant protein-1; RT-qPCR, reverse
transcription-quantitative PCR; TMAO, trimethylamine N-oxide. |
NaHS upregulates SIRT1 levels and
reduces inflammation in TMAO-stimulated macrophages
H2S-induced SIRT1 has been reported to
confer a protective effect on atherosclerosis (22). NaHS is a donor of H2S.
NaHS downregulated the mRNA expression levels of IL-6, IL-1β and
MCP-1, and upregulated the expression levels of IL-4 and IL-10
(Fig. 2A) compared with that in
the TMAO group. In addition, NaHS significantly increased the
protein expression levels of SIRT1 when RAW264.7 macrophages were
challenged with TMAO (Fig. 2B).
Furthermore, NaHS induced SIRT1 expression in a time-dependent
manner (Fig. 2C) after 12, 24 and
48 h of incubation.
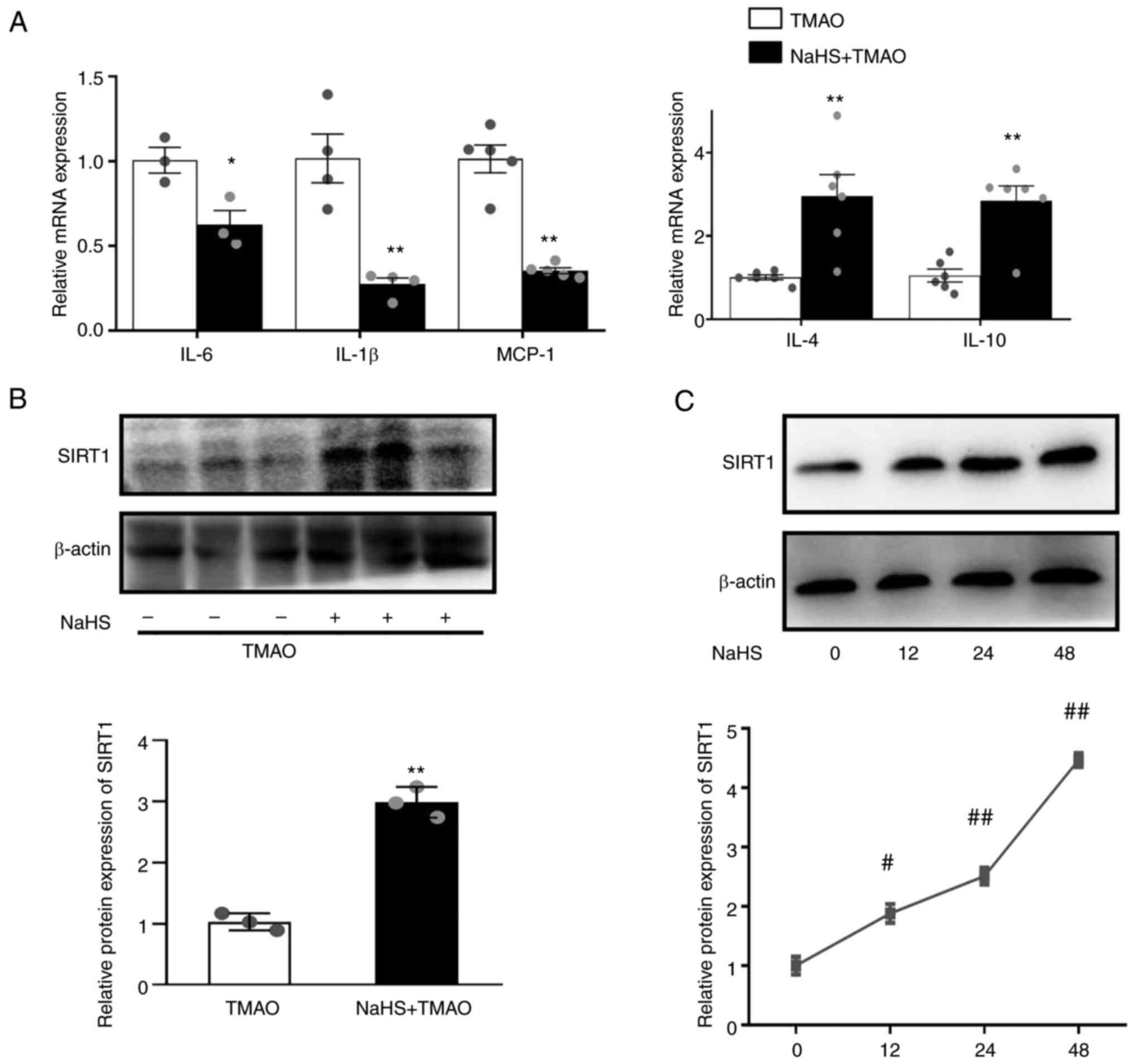 | Figure 2.Hydrogen sulfide ameliorates
TMAO-induced inflammation and SIRT1 expression. (A) mRNA expression
levels of IL-1β, IL-6, MCP-1, IL-4 and IL-10 in the macrophages
after NaHS treatment, as measured by reverse
transcription-quantitative PCR. (B) Representative western blots of
SIRT1 in the macrophages after NaHS and TMAO treatment. (C)
Representative western blots of SIRT1 in the macrophages after NaHS
treatment for different durations (n=3). *P˂0.05, **P˂0.01 vs. TMAO
group; #P˂0.05, ##P˂0.01 vs. control group.
MCP-1, monocyte chemoattractant protein-1; NaHS, sodium
hydrosulfide; SIRT1, sirtuin 1; TMAO, trimethylamine N-oxide. |
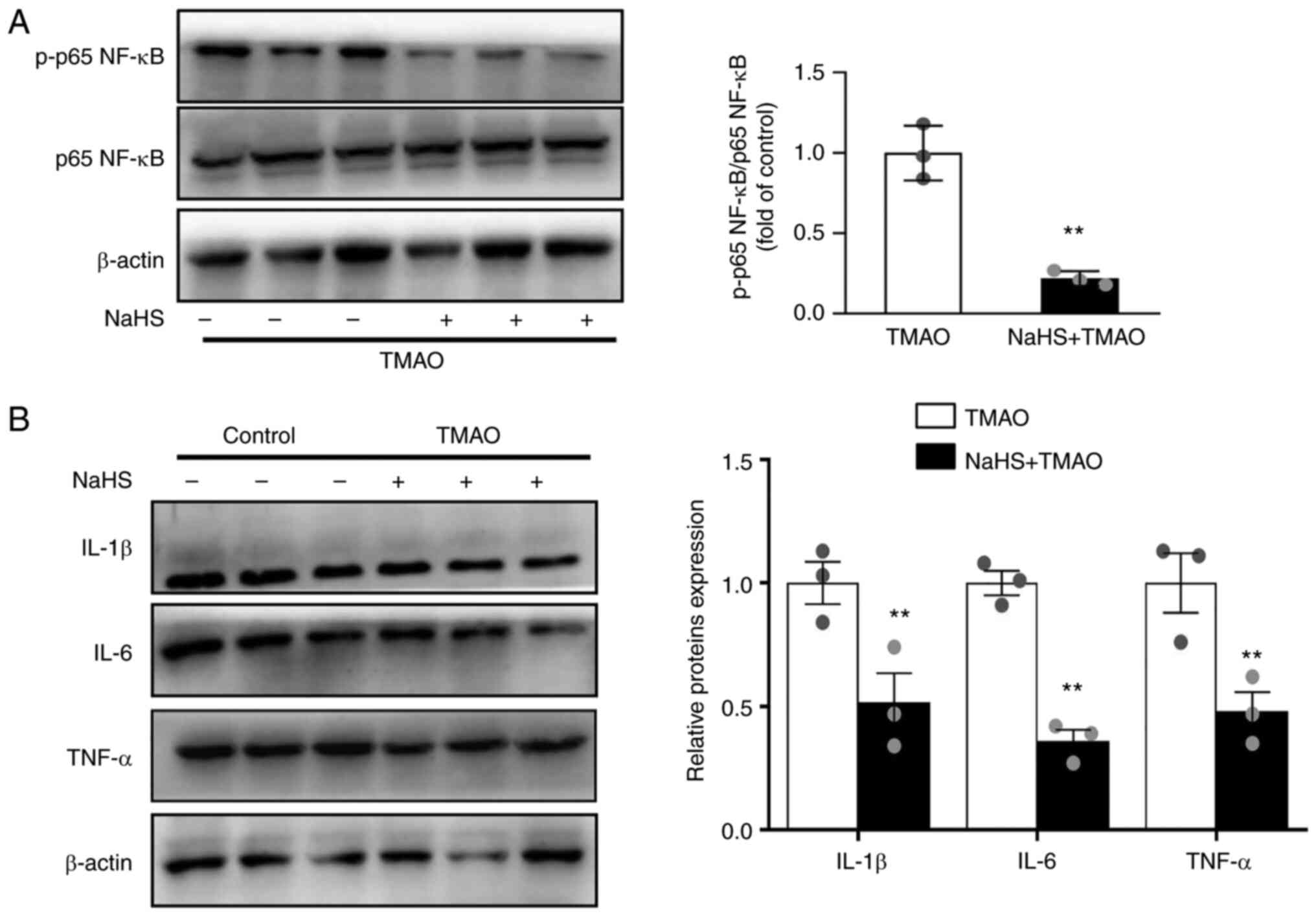
NaHS ameliorates TMAO-induced NF-κB
activation
Recent studies have shown that TMAO activates NF-κB
signals, resulting in the release of inflammatory cytokines
(5,25). The present study examined the
effects of H2S on TMAO-induced NF-κB activation. As
shown in Fig. 3A, the TMAO-induced
phosphorylation of p65 NF-κB was reversed by NaHS. In addition,
NaHS decreased the expression levels of the pro-inflammatory
cytokines IL-1β, IL-6 and TNF-α in TMAO-stimulated macrophages
(Fig. 3B).
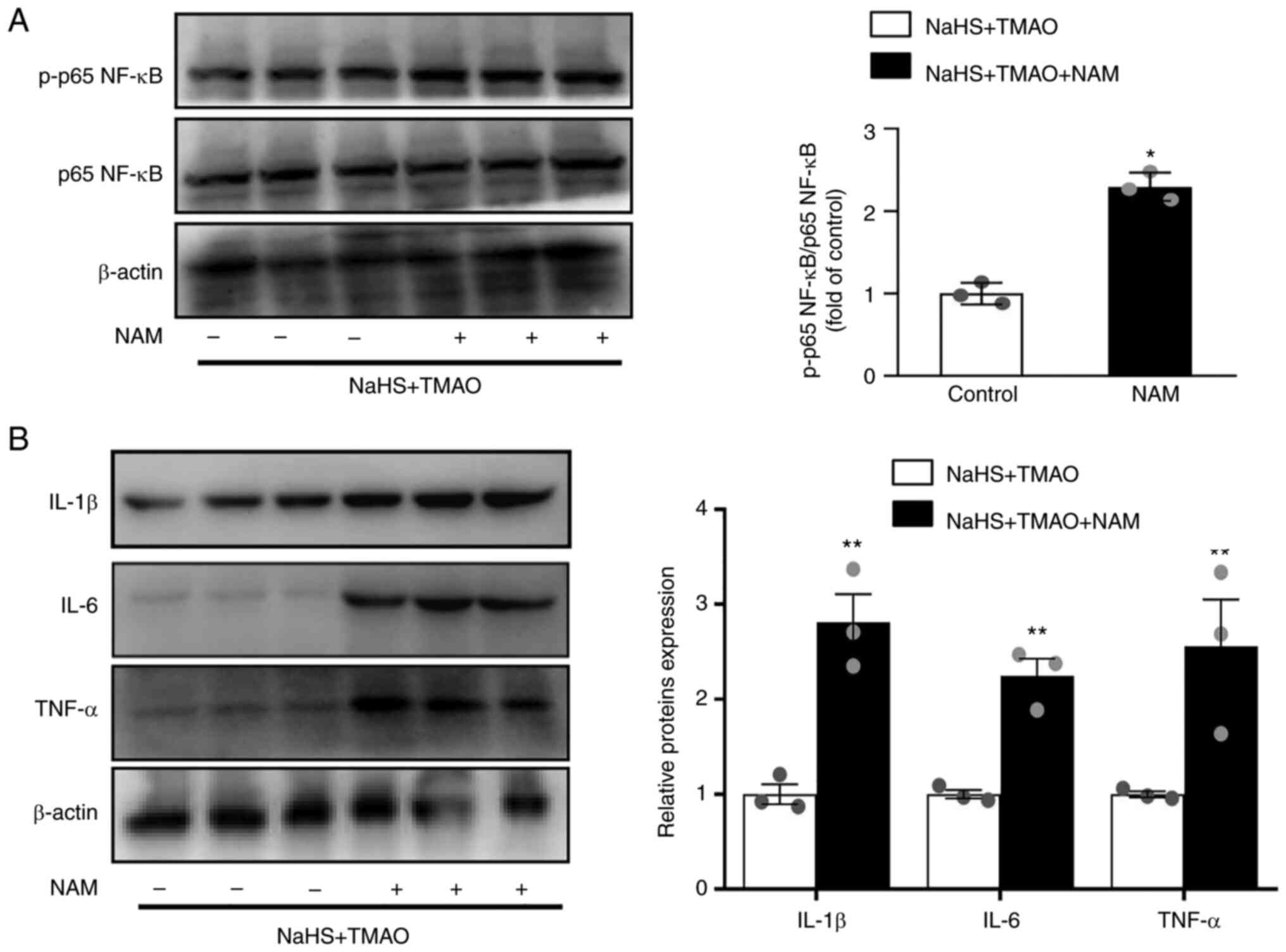
NaHS reduces inflammation via enhanced
SIRT1 activity
Macrophages were treated with the SIRT1 inhibitor
nicotinamide to investigate whether SIRT1 mediated the protective
effect of H2S on TMAO-induced NF-κB activation. The
results showed that phosphorylation of p65 NF-κB was significantly
increased by nicotinamide compared with that in the NaHS+ TMAO
group (Fig. 4A). Furthermore, the
protein expression levels of IL-1β, IL-6, and TNF-α were also
increased by nicotinamide (Fig.
4B).
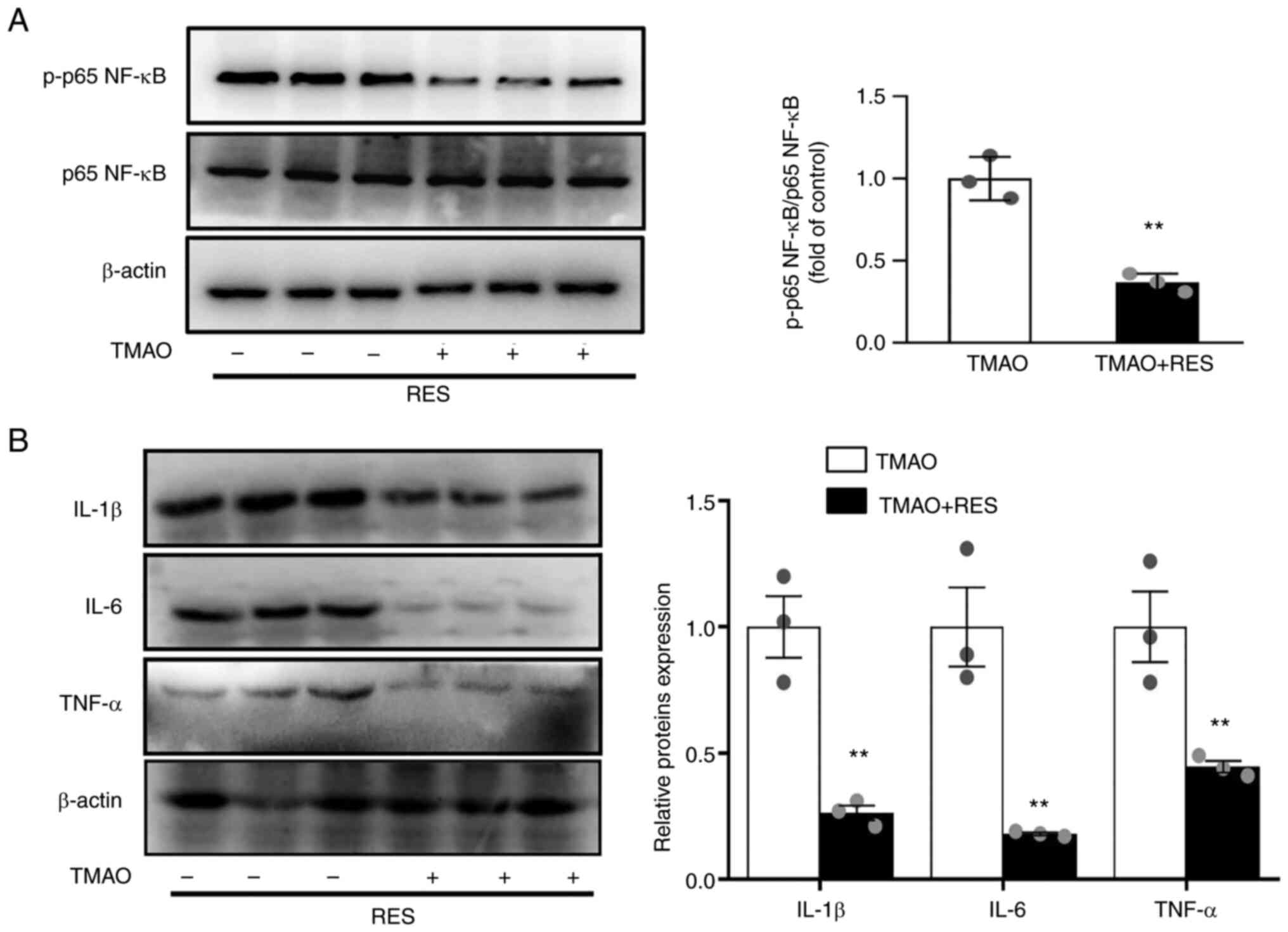
Resveratrol counteracts TMAO-induced
NF-κB activation via SIRT1
Resveratrol has been identified as a SIRT1
activator, which can modulate the inflammatory response in
endothelial cells by inhibiting NF-κB activation (26). Resveratrol reversed the
TMAO-induced phosphorylation of p65 NF-κB (Fig. 5A). In addition, resveratrol
significantly ameliorated the TMAO-induced expression of IL-1β,
IL-6 and TNF-α (Fig. 5B).
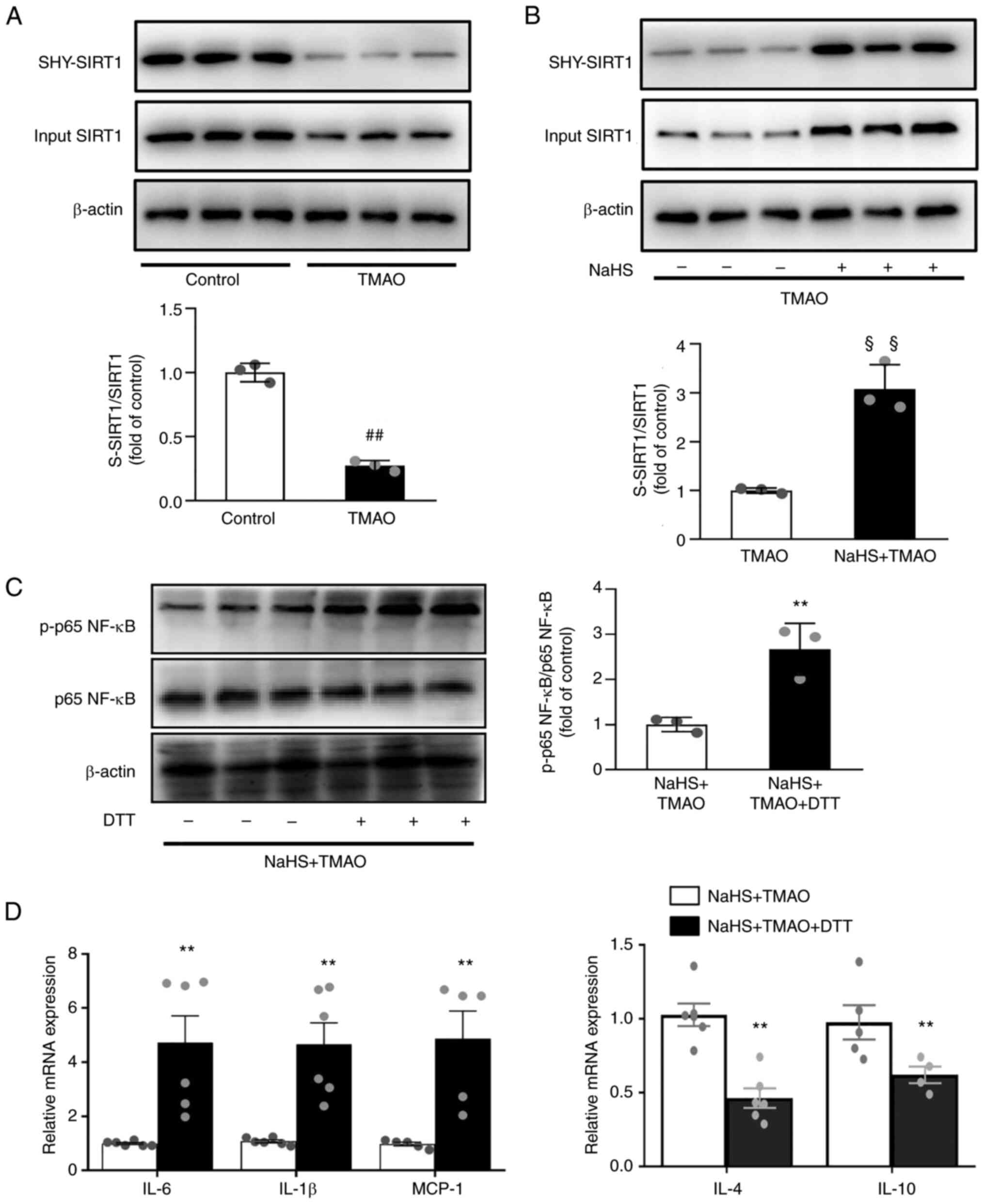
H2S inhibits NF-κB
activation via S-sulfhydrated SIRT1
S-sulfhydration refers to the formation of
hydropersulfide (−SSH) from H2S by attaching sulfur to thiol (−SH)
groups of cysteines (27). The
results revealed that TMAO significantly decreased SIRT1
sulfhydration (Fig. 6A). By
contrast, SIRT1 sulfhydration was induced after NaHS treatment in
TMAO-stimulated macrophages (Fig.
6B). Given that S-sulfhydrated SIRT1 can reduce inflammation
(28), the present study evaluated
its effects on NF-κB activation. Following pre-treatment with the
thiol-reducing agent DTT in macrophages, the inhibitory effect of
NaHS on NF-κB activation was significantly reversed (Fig. 6C). DTT also upregulated the mRNA
expression levels of IL-1β, IL-6 and MCP-1, and downregulated IL-4
and IL-10 (Fig. 6D) compared with
that in the NaHS +TMAO group. These data indicated that
H2S may attenuate TMAO-induced inflammatory signaling in
macrophages by inducing SIRT1 sulfhydration.
Discussion
It has been reported that increased TMAO levels are
associated with the development of metabolic diseases, such as
diabetes and atherosclerosis (29). To the best of our knowledge, the
present study demonstrated for the first time that H2S
was negatively associated with TMAO-induced macrophage
inflammation. Mechanistically, it was revealed that H2S
promoted SIRT1 sulfhydration and decreased SIRT1 proteasomal
degradation, reducing the phosphorylation of P65 NF-κB, and
inhibiting inflammation (Fig. 7).
Thus, these results indicated that NaHS attenuated TMAO-induced
macrophage inflammation, suggesting that targeting H2S
may be useful in developing novel therapeutic strategies for
TMAO-induced cardiovascular events.
TMAO, an intestinal microbial metabolite, has been
associated with cardiovascular disease (30). Clinical studies have shown that
high TMAO levels are significantly associated with an increased
risk of major adverse cardiovascular events (31). TMAO exposure has also been shown to
promote endothelial cell inflammation via activation of the NLRP3
inflammasome (32). The present
study showed that TMAO induced the expression of inflammatory
cytokines in macrophages.
Our previous studies indicated that H2S
has powerful antioxidative (33)
and cytoprotective effects (16,23,34)
on cardiovascular systems. In addition, H2S exhibits a
significant ability to protect against inflammation (1,35).
H2S can inhibit the TLR4/NF-κB pathway (35,36),
scavenge excess reactive oxygen species (37,38)
and suppress HMGB1 (39), which
are related to NLRP3 activation (40,41)
and ferroptosis (42,43). Recently, H2S has been
proven to relieve myocardial infarction by promoting M2 macrophage
polarization, macrophage migration and infiltration (1). CSE is the main enzyme for
H2S production in the cardiovascular system. In the
present study, it was revealed that TMAO significantly decreased
CSE expression and increased expression of pro-inflammatory M1
cytokines, resulting in increased M1 polarization and inflammatory
response in macrophages. Notably, the endogenous production of
H2S is largely dependent on the activity of CSE and is
less dependent on the activity of CBS or 3-MST in macrophages
(44). Previous studies have
identified that TMAO significantly decreases plasma H2S
levels (40,45). However, the present study did not
investigate whether TMAO influenced the expression levels of CBS
and 3-MST in macrophages; therefore, this study has some
limitations, which require further study in the future. Moreover,
the present data revealed that H2S promotes a shift from
the M1 to M2 macrophage phenotype by decreasing the expression of
pro-inflammatory M1 cytokines and by increasing anti-inflammatory
M2 cytokines in TMAO-stimulated macrophages, which is consistent
with an earlier report (46).
Thus, H2S may be considered a potentially promising
therapeutic approach to the treatment of TMAO-induced macrophage
inflammation.
SIRT1 is important in the regulation of inflammation
and atherosclerosis (47). SIRT1
activation has been shown to ameliorate endothelial function,
reduce foam cell formation (48)
and attenuate atherosclerotic plaque formation (49). Specific knockdown of SIRT1 in
endothelial cells (50) or VSMCs
(51) can significantly increase
atherosclerotic plaque area. Our previous study indicated that
H2S induced SIRT1 activation in human umbilical vein
endothelial cells (17). The
present study demonstrated that NaHS increased SIRT1 expression in
TMAO-exposed macrophages, indicating the importance of
H2S in SIRT1 signaling activation. A number of studies
have revealed that SIRT1 serves as a potential mediator of NLRP3
inflammasome activation and ferroptosis in chronic inflammation
(52,53). NF-κB activity is regulated by
post-translational modifications, such as acetylation and
phosphorylation (54,55). Notably, SIRT1 signaling has been
shown to alleviate NF-κB activity in macrophages after stimulation
of the cells with lipopolysaccharide (56). Activating SIRT1 can also
deacetylate NF-κB p65 (52) or p53
(53) to alleviate NLRP3
inflammasome activation and ferroptosis in macrophages.
Furthermore, SIRT1 defects may lead to NLRP3 activation in
hepatocytes (57) or ferroptosis
in H9c2 cardiomyocyte cells (58),
and thereby SIRT1 signaling relieves inflammation. The present
study further verified that TMAO induced activation of the NF-κB
signaling pathway by increasing the phosphorylation of p65 NF-κB.
Moreover, the SIRT1 inhibitor nicotinamide significantly increased
the phosphorylation of p65 NF-κB and impaired the anti-inflammatory
actions of H2S. Conversely, the SIRT1 activator
resveratrol attenuated the phosphorylation of p65 NF-κB and
inflammation in macrophages. Accordingly, SIRT1 mediates the
protective effect of H2S on TMAO-induced NF-κB
activation in macrophages. A previous study also revealed that
SIRT1 directly acetylates P65 and promotes inflammation (59). However, the present study did not
exclude the effect of SIRT1-induced P65 acetylation on
inflammation. Nevertheless, inhibition of P65 phosphorylation by
SIRT1 is involved in the protective effects of H2S
against TMAO-induced inflammation in macrophages. The present
findings suggested that H2S inhibited NF-κB p65 mediated
inflammatory activation via enhanced SIRT1 activation.
S-sulfhydration is a post-translational modification
that increases SIRT1 protein stability and activity (22). NaHS promotes SIRT1 expression and
activity by sulfhydrating SIRT1, thereby lowering its
ubiquitin-dependent degradation (22). In addition, SIRT1 sulfhydration
reduces acetylation and phosphorylation of P65 NF-κB, ultimately
inhibiting inflammation (22,28).
The present study discovered that NaHS induced SIRT1 sulfhydration
to enhance SIRT1 stability and protein levels, which is similar to
the findings of a previous study (22). Furthermore, SIRT1 sulfhydration by
H2S decreases the phosphorylation of p65 NF-κB, which
downregulates pro-inflammatory cytokine expression in TMAO-exposed
macrophages. However, DTT blocked its action, suggesting that SIRT1
sulfhydration mediated the effects of H2S. Therefore,
the present study indicated that SIRT1 sulfhydration mediated the
protective effects of H2S on TMAO-induced M1
polarization and inflammation in macrophages.
In conclusion, the present study demonstrated that
H2S can attenuate TMAO-induced inflammatory signaling in
macrophages via the upregulation and sulfhydration of SIRT1. The
present study not only provided information on the mechanisms
underlying H2S-mediated anti-inflammatory effects, but
also provided evidence that SIRT1 is involved in the protective
effects of H2S on TMAO-associated macrophage
inflammation.
Acknowledgements
Not applicable.
Funding
This study was supported by grants from the Guiding Technology
Plan of Ganzhou City (grant no. GZ2022ZSF186), the Jiangxi
Provincial Natural Science Foundation (grant no. 20224BAB206020),
the Science and Technology Plan Project of Jiangxi Provincial
Health Commission (grant no. 202210091/SKJP_220217891), and the
Science and Technology Plan of Jiangxi Provincial Administration of
Traditional Chinese Medicine (grant no. 2022A337).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MHL collected and analyzed the data and wrote the
manuscript. MHL and LLX performed the experiments. XLL contributed
to the study design and data analyses and revised the manuscript.
All authors read and approved the final manuscript. MHL and LLX
confirm the authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no known
competing interests.
References
|
1
|
Zhang H, Du J, Huang Y, Tang C and Jin H:
Hydrogen sulfide regulates macrophage function in cardiovascular
diseases. Antioxid Redox Signal. 38:45–56. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Susser LI and Rayner KJ: Through the
layers: How macrophages drive atherosclerosis across the vessel
wall. J Clin Invest. 132:e1570112022. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tang WH and Hazen SL: The contributory
role of gut microbiota in cardiovascular disease. J Clin Invest.
124:4204–4211. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sun X, Jiao X, Ma Y, Liu Y, Zhang L, He Y
and Chen Y: Trimethylamine N-oxide induces inflammation and
endothelial dysfunction in human umbilical vein endothelial cells
via activating ROS-TXNIP-NLRP3 inflammasome. Biochem Biophys Res
Commun. 481:63–70. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang X, Li Y, Yang P, Liu X, Lu L, Chen
Y, Zhong X, Li Z, Liu H, Ou C, et al: Trimethylamine-N-oxide
promotes vascular calcification through activation of NLRP3
(nucleotide-binding domain, leucine-rich-containing family, pyrin
domain-containing-3) inflammasome and NF-κB (nuclear factor κB)
signals. Arterioscler Thromb Vasc Biol. 40:751–765. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang W, Miikeda A, Zuckerman J, Jia X,
Charugundla S, Zhou Z, Kaczor-Urbanowicz KE, Magyar C, Guo F, Wang
Z, et al: Inhibition of microbiota-dependent TMAO production
attenuates chronic kidney disease in mice. Sci Rep. 11:5182021.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Casin KM and Calvert JW: Harnessing the
benefits of endogenous hydrogen sulfide to reduce cardiovascular
disease. Antioxidants (Basel). 10:3832021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lv B, Chen S, Tang C, Jin H, Du J and
Huang Y: Hydrogen sulfide and vascular regulation-an update. J Adv
Res. 27:85–97. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu HT, Zhou ZX, Ren Z, Yang S, Liu LS,
Wang Z, Wei DH, Ma XF, Ma Y and Jiang ZS: EndMT: Potential target
of H2S against atherosclerosis. Curr Med Chem.
28:3666–3680. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mani S, Li H, Untereiner A, Wu L, Yang G,
Austin RC, Dickhout JG, Lhoták Š, Meng QH and Wang R: Decreased
endogenous production of hydrogen sulfide accelerates
atherosclerosis. Circulation. 127:2523–2534. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cheung SH, Kwok WK, To KF and Lau JYW:
Anti-atherogenic effect of hydrogen sulfide by over-expression of
cystathionine gamma-lyase (CSE) gene. PLoS One. 9:e1130382014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao
J, Cheng L, Li J, Wang X, Wang F, et al: Single-cell landscape of
bronchoalveolar immune cells in patients with COVID-19. Nat Med.
26:842–844. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Winnik S, Auwerx J, Sinclair DA and Matter
CM: Protective effects of sirtuins in cardiovascular diseases: From
bench to bedside. Eur Heart J. 36:3404–3412. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lin XL, Liu Y, Liu M, Hu H, Pan Y, Fan XJ,
Hu XM and Zou WW: Inhibition of hydrogen peroxide-induced human
umbilical vein endothelial cells aging by allicin depends on
sirtuin1 activation. Med Sci Monit. 23:563–570. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Luo XY, Qu SL, Tang ZH, Zhang Y, Liu MH,
Peng J, Tang H, Yu KL, Zhang C, Ren Z and Jiang ZS: SIRT1 in
cardiovascular aging. Clin Chim Acta. 437:106–114. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang Y, Tang ZH, Ren Z, Qu SL, Liu MH,
Liu LS and Jiang ZS: Hydrogen sulfide, the next potent preventive
and therapeutic agent in aging and age-associated diseases. Mol
Cell Biol. 33:1104–1113. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Suo R, Zhao ZZ, Tang ZH, Ren Z, Liu X, Liu
LS, Wang Z, Tang CK, Wei DH and Jiang ZS: Hydrogen sulfide prevents
H2O2 induced senescence in human umbilical
vein endothelial cells through SIRT1 activation. Mol Med Rep.
7:1865–1870. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Feng T, Liu P, Wang X, Luo J, Zuo X, Jiang
X, Liu C, Li Y, Li N, Chen M, et al: SIRT1 activator E1231 protects
from experimental atherosclerosis and lowers plasma cholesterol and
triglycerides by enhancing ABCA1 expression. Atherosclerosis.
274:172–181. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nguyen PA, Won JS, Rahman MK, Bae EJ and
Cho MK: Modulation of Sirt1/NF-κB interaction of evogliptin is
attributed to inhibition of vascular inflammatory response leading
to attenuation of atherosclerotic plaque formation. Biochem
Pharmacol. 168:452–464. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li X, Zhang KY, Zhang P, Chen LX, Wang L,
Xie M, Wang CY and Tang XQ: Hydrogen sulfide inhibits
formaldehyde-induced endoplasmic reticulum stress in PC12 cells by
upregulation of SIRT-1. PLoS One. 9:e898562014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Stein S, Lohmann C, Schäfer N, Hofmann J,
Rohrer L, Besler C, Rothgiesser KM, Becher B, Hottiger MO, Borén J,
et al: SIRT1 decreases Lox-1-mediated foam cell formation in
atherogenesis. Eur Heart J. 31:2301–2309. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Du C, Lin X, Xu W, Zheng F, Cai J, Yang J,
Cui Q, Tang C, Cai J, Xu G and Geng B: Sulfhydrated sirtuin-1
increasing its deacetylation activity is an essential epigenetics
mechanism of anti-atherogenesis by hydrogen sulfide. Antioxid Redox
Signal. 30:184–197. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Huang YE, Tang ZH, Xie W, Shen XT, Liu MH,
Peng XP, Zhao ZZ, Nie DB, Liu LS and Jiang ZS: Endogenous hydrogen
sulfide mediates the cardioprotection induced by ischemic
postconditioning in the early reperfusion phase. Exp Ther Med.
4:1117–1123. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen CY, Leu HB, Wang SC, Tsai SH, Chou
RH, Lu YW, Tsai YL, Kuo CS, Huang PH, Chen JW and Lin SJ:
Inhibition of trimethylamine N-oxide attenuates neointimal
formation through reduction of inflammasome and oxidative stress in
a mouse model of carotid artery ligation. Antioxid Redox Signal.
38:215–233. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nallasamy P, Kang ZY, Sun X, Anandh Babu
PV, Liu D and Jia Z: Natural compound resveratrol attenuates
TNF-alpha-induced vascular dysfunction in mice and human
endothelial cells: The Involvement of the NF-κB signaling pathway.
Int J Mol Sci. 22:124862021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gupta R, Sahu M, Tripathi R, Ambasta RK
and Kumar P: Protein S-sulfhydration: Unraveling the prospective of
hydrogen sulfide in the brain, vasculature and neurological
manifestations. Ageing Res Rev. 76:1015792022. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sun HJ, Xiong SP, Cao X, Cao L, Zhu MY, Wu
ZY and Bian JS: Polysulfide-mediated sulfhydration of SIRT1
prevents diabetic nephropathy by suppressing phosphorylation and
acetylation of p65 NF-κB and STAT3. Redox Biol. 38:1018132021.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fu BC, Hullar MAJ, Randolph TW, Franke AA,
Monroe KR, Cheng I, Wilkens LR, Shepherd JA, Madeleine MM, Le
Marchand L, et al: Associations of plasma trimethylamine N-oxide,
choline, carnitine, and betaine with inflammatory and
cardiometabolic risk biomarkers and the fecal microbiome in the
multiethnic cohort adiposity phenotype study. Am J Clin Nutr.
111:1226–1234. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tang WHW and Hazen SL: Microbiome,
trimethylamine N-oxide, and cardiometabolic disease. Transl Res.
179:108–115. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Stubbs JR, House JA, Ocque AJ, Zhang S,
Johnson C, Kimber C, Schmidt K, Gupta A, Wetmore JB, Nolin TD, et
al: Serum trimethylamine-N-oxide is elevated in CKD and correlates
with coronary atherosclerosis burden. J Am Soc Nephrol. 27:305–313.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen ML, Zhu XH, Ran L, Lang HD, Yi L and
Mi MT: Trimethylamine-N-oxide induces vascular inflammation by
activating the NLRP3 inflammasome through the SIRT3-SOD2-mtROS
signaling pathway. J Am Heart Assoc. 6:e0063472017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu MH, Lin XL, Zhang Y, He J, Tan TP, Wu
SJ, Liu J, Tian W, Chen L, Yu S, et al: Hydrogen sulfide attenuates
doxorubicin-induced cardiotoxicity by inhibiting reactive oxygen
species-activated extracellular signal-regulated kinase 1/2 in H9c2
cardiac myocytes. Mol Med Rep. 12:6841–6848. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu MH, Zhang Y, He J, Tan TP, Wu SJ, Guo
DM, He H, Peng J, Tang ZH and Jiang ZS: Hydrogen sulfide protects
H9c2 cardiac cells against doxorubicin-induced cytotoxicity through
the PI3K/Akt/FoxO3a pathway. Int J Mol Med. 37:1661–1668. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Luo ZL, Ren JD, Huang Z, Wang T, Xiang K,
Cheng L and Tang LJ: The role of exogenous hydrogen sulfide in free
fatty acids induced inflammation in macrophages. Cell Physiol
Biochem. 42:1635–1644. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang D, Wu C, Ba D, Wang N, Wang Y, Li X,
Li Q and Zhao G: Ferroptosis contribute to neonicotinoid
imidacloprid-evoked pyroptosis by activating the
HMGB1-RAGE/TLR4-NF-κB signaling pathway. Ecotoxicol Environ Saf.
253:1146552023. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Olas B: Hydrogen sulfide as a
‘double-faced’ compound: One with Pro- and antioxidant effect. Adv
Clin Chem. 78:187–196. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang Y, Yu R, Wu L and Yang G: Hydrogen
sulfide guards myoblasts from ferroptosis by inhibiting ALOX12
acetylation. Cell Signal. 78:1098702021. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhao X, Zhang L, Liu X, Zhao Z, Zhong X
and Wang Y: Exogenous hydrogen sulfide inhibits neutrophils
extracellular traps formation via the HMGB1/TLR4/p-38 MAPK/ROS axis
in hyperhomocysteinemia rats. Biochem Biophys Res Commun. 537:7–14.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bai L, Dai J, Xia Y, He K, Xue H, Guo Q,
Tian D, Xiao L, Zhang X, Teng X, et al: Hydrogen sulfide
ameliorated high choline-induced cardiac dysfunction by inhibiting
cGAS-STING-NLRP3 inflammasome pathway. Oxid Med Cell Longev.
2022:13928962022. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Qin M, Long F, Wu W, Yang D, Huang M, Xiao
C, Chen X, Liu X and Zhu YZ: Hydrogen sulfide protects against
DSS-induced colitis by inhibiting NLRP3 inflammasome. Free Radic
Biol Med. 137:99–109. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang Y, Liao S, Pan Z, Jiang S, Fan J, Yu
S, Xue L, Yang J, Ma S, Liu T, et al: Hydrogen sulfide alleviates
particulate matter-induced emphysema and airway inflammation by
suppressing ferroptosis. Free Radic Biol Med. 186:1–16. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhao Z, Li G, Wang Y, Li Y, Xu H, Liu W,
Hao W, Yao Y and Zeng R: Cytoplasmic HMGB1 induces renal tubular
ferroptosis after ischemia/reperfusion. Int Immunopharmacol.
116:1097572023. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Castelblanco M, Lugrin J, Ehirchiou D,
Nasi S, Ishii I, So A, Martinon F and Busso N: Hydrogen sulfide
inhibits NLRP3 inflammasome activation and reduces cytokine
production both in vitro and in a mouse model of inflammation. J
Biol Chem. 293:2546–2557. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Koeth RA, Lam-Galvez BR, Kirsop J, Wang Z,
Levison BS, Gu X, Copeland MF, Bartlett D, Cody DB, Dai HJ, et al:
l-Carnitine in omnivorous diets induces an atherogenic gut
microbial pathway in humans. J Clin Invest. 129:373–387. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Feng S, Chen S, Yu W, Zhang D, Zhang C,
Tang C, Du J and Jin H: H2S inhibits pulmonary arterial
endothelial cell inflammation in rats with monocrotaline-induced
pulmonary hypertension. Lab Invest. 97:268–278. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhang QJ, Wang Z, Chen HZ, Zhou S, Zheng
W, Liu G, Wei YS, Cai H, Liu DP and Liang CC: Endothelium-specific
overexpression of class III deacetylase SIRT1 decreases
atherosclerosis in apolipoprotein E-deficient mice. Cardiovasc Res.
80:191–199. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Liu Z, Han Y, Li L, Lu H, Meng G, Li X,
Shirhan M, Peh MT, Xie L, Zhou S, et al: The hydrogen sulfide
donor, GYY4137, exhibits anti-atherosclerotic activity in high fat
fed apolipoprotein E(−/-) mice. Br J Pharmacol. 169:1795–1809.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Miranda MX, van Tits LJ, Lohmann C,
Arsiwala T, Winnik S, Tailleux A, Stein S, Gomes AP, Suri V, Ellis
JL, et al: The Sirt1 activator SRT3025 provides atheroprotection in
Apoe-/- mice by reducing hepatic Pcsk9 secretion and enhancing Ldlr
expression. Eur Heart J. 36:51–59. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wen L, Chen Z, Zhang F, Cui X, Sun W,
Geary GG, Wang Y, Johnson DA, Zhu Y, Chien S and Shyy JY:
Ca2+/calmodulin-dependent protein kinase kinase β phosphorylation
of sirtuin 1 in endothelium is atheroprotective. Proc Natl Acad Sci
USA. 110:E2420–E2427. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Gorenne I, Kumar S, Gray K, Figg N, Yu H,
Mercer J and Bennett M: Vascular smooth muscle cell sirtuin 1
protects against DNA damage and inhibits atherosclerosis.
Circulation. 127:386–396. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
He S, Wang Y, Liu J, Li P, Luo X and Zhang
B: Activating SIRT1 deacetylates NF-κB p65 to alleviate liver
inflammation and fibrosis via inhibiting NLRP3 pathway in
macrophages. Int J Med Sci. 20:505–519. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Chen H, Lin X, Yi X, Liu X, Yu R, Fan W,
Ling Y, Liu Y and Xie W: SIRT1-mediated p53 deacetylation inhibits
ferroptosis and alleviates heat stress-induced lung epithelial
cells injury. Int J Hyperthermia. 39:977–986. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Breitenstein A, Stein S, Holy EW, Camici
GG, Lohmann C, Akhmedov A, Spescha R, Elliott PJ, Westphal CH,
Matter CM, et al: Sirt1 inhibition promotes in vivo arterial
thrombosis and tissue factor expression in stimulated cells.
Cardiovasc Res. 89:464–472. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Fiordelisi A, Iaccarino G, Morisco C,
Coscioni E and Sorriento D: NFkappaB is a key player in the
crosstalk between inflammation and cardiovascular diseases. Int J
Mol Sci. 20:15992019. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Yuan Q, Zhang D, Liu C, Zhang C and Yuan
D: Chikusetsusaponin V inhibits LPS-activated inflammatory
responses via SIRT1/NF-κB signaling pathway in RAW264.7 cells.
Inflammation. 41:2149–2159. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Adjei-Mosi J, Sun Q, Smithson SB, Shealy
GL, Amerineni KD, Liang Z, Chen H, Wang M, Ping Q, Han J, et al:
Age-dependent loss of hepatic SIRT1 enhances NLRP3 inflammasome
signaling and impairs capacity for liver fibrosis resolution. Aging
Cell. e138112023.(Epub ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Li D, Liu X, Pi W, Zhang Y, Yu L, Xu C,
Sun Z and Jiang J: Fisetin attenuates doxorubicin-induced
cardiomyopathy in vivo and in vitro by inhibiting ferroptosis
through SIRT1/Nrf2 signaling pathway activation. Front Pharmacol.
12:8084802022. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Guo C, Zhang Y, Ling T, Zhao C, Li Y, Geng
M, Gai S, Qi W, Luo X, Chen L, et al: Chitosan oligosaccharides
alleviate colitis by regulating intestinal microbiota and
PPARγ/SIRT1-mediated NF-κB pathway. Mar Drugs. 20:962022.
View Article : Google Scholar : PubMed/NCBI
|