Introduction
Autophagy was first conceptualized in the 1960s,
with a significant contribution in 1963 when Christian de Duve's
team observed the association of lysosomes, which are integral to
cellular component degradation, with autophagosomes, leading to the
coining of the term ‘autophagy’ (1). This process, taking place in
eukaryotic cells, involves lysosomes-regulated by
autophagy-associated genes-degrading cytoplasmic proteins, damaged
organelles and intracellular pathogens. Autophagy is induced in
response to a spectrum of cellular stresses and is pivotal in
maintaining tissue homeostasis. This process involves two principal
intermediary systems, namely the proteasome and the lysosome. The
proteasome, situated in the cytoplasm and nucleus, degrades damaged
or misfolded proteins into smaller peptides (2), thus regulating essential cellular
processes such as the cell cycle and DNA repair. In cells,
lysosomes degrade not only extracellular substances and membrane
proteins, but also intracellular components, including damaged
organelles, misfolded proteins and various forms of cellular
debris, which are also targeted for lysosomal degradation. This
degradation occurs through multiple processes, including
macroautophagy (enclosing large cellular components in
autophagosomes for lysosomal delivery), microautophagy (engulfing
and digesting segments of their own cytoplasm) and
chaperone-mediated autophagy (CMA) (direct translocation of
specific proteins to lysosomes) (3,4).
Lysosomal permeases and transport proteins release amino acids and
other degradation by-products into the cytoplasm, facilitating
their reuse for macromolecule synthesis and metabolism.
Consequently, lysosomes play a pivotal role in degrading both
extracellular and intracellular materials, which is essential for
maintaining cellular homeostasis. This dual function underscores
the importance of lysosomes in cellular recycling processes and
their critical contribution to the overall metabolic efficiency of
the cell.
Autophagy, a sophisticated self-degradative process,
involves phagophore formation, interactions among autophagy-related
genes such as autophagy-related 5 (ATG5)-ATG12 and ATG16-like
(ATG16L), and is regulated by a variety of signaling pathways.
Additionally, autophagy increases energy efficiency through ATP
production, manages damage control by eliminating non-functional
proteins and organelles, and regulates cellular responses to stress
or changes in the extracellular microenvironment (5,6).
Autophagy is classified into three types: Macroautophagy,
microautophagy and CMA. This classification underscores the diverse
mechanisms and functions of autophagy in cellular maintenance and
response to environmental cues (Fig.
1).
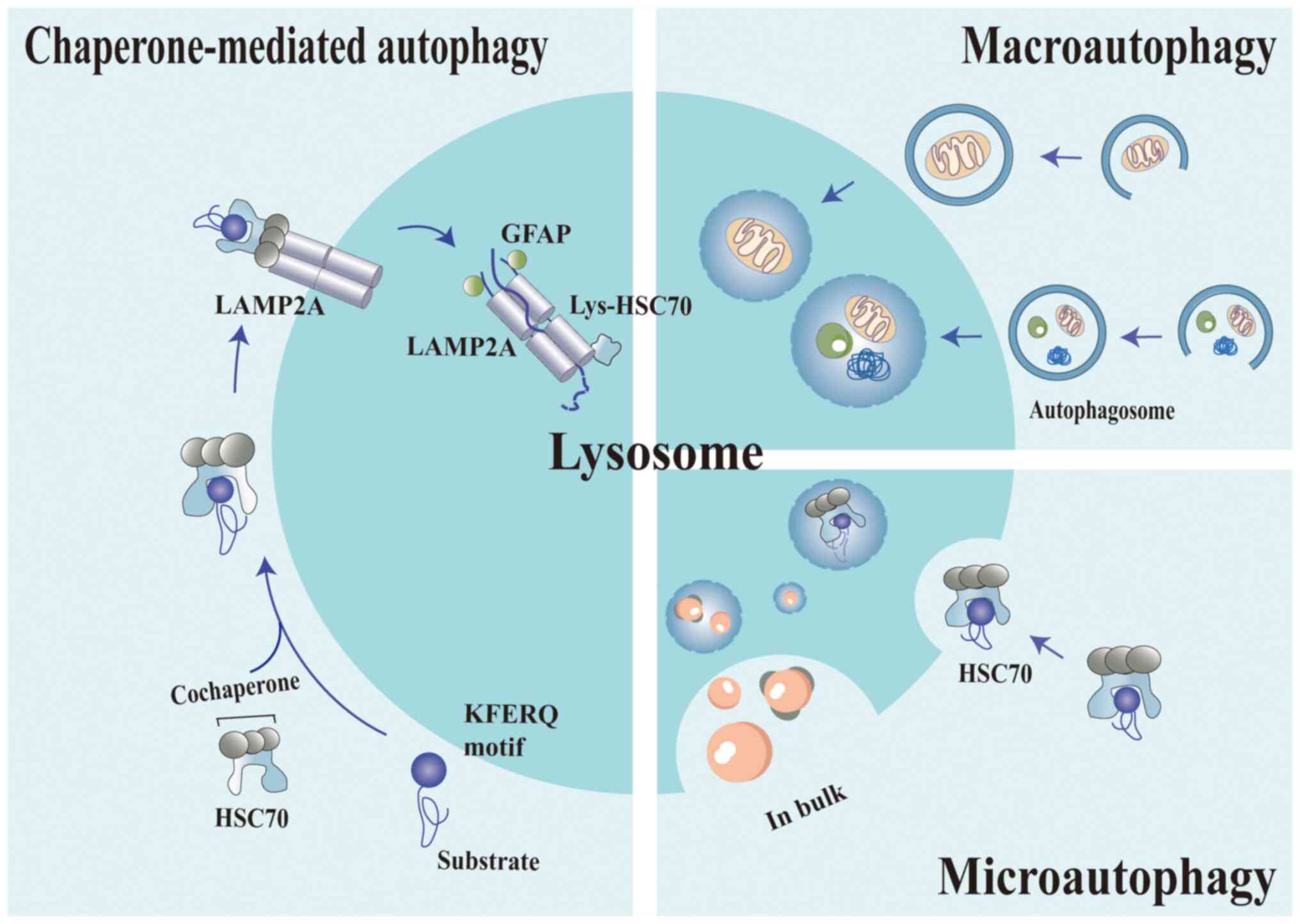 | Figure 1.Three different kinds of autophagy.
Autophagy manifests in various forms, each distinguished by its
unique characteristics and mechanisms. Macroautophagy involves the
formation of a double-membraned vesicle (an autophagosome) that
envelops cellular material destined for degradation. The
autophagosome then fuses with a lysosome, forming an autolysosome
wherein the enclosed material is degraded. Microautophagy is
characterized by the invagination of the lysosomal membrane, which
leads to the formation of a small vesicle. This vesicle then fuses
with the lysosome's interior, thereby releasing its contents for
degradation within the lysosome. Chaperone-mediated autophagy
target proteins are characterized by a distinct KFERQ-like sequence
that is recognized by chaperone proteins, notably HSC70. These
chaperone-recognized proteins subsequently interact with the LAMP2A
receptor on the lysosomal membrane. Upon binding, LAMP2A aggregates
to form multimers. Assisted by HSC70, the target protein is
gradually translocated across the lysosomal membrane into the
lysosomal lumen. Once inside the lysosome, the target protein is
rapidly degraded by acidic hydrolases. The figure was generated
using Adobe Illustrator 2021, version 25.0, by Adobe Inc. LAMP2A,
lysosome-associated membrane protein 2A; GFAP, glial fibrillary
acidic protein; HSC70, heat shock cognate 70. |
Macroautophagy
Macroautophagy, extensively studied among autophagy
forms, is differentiated from microautophagy by forming a
double-membraned vesicle structure. Cytoplasmic components,
organelles and various elements are engulfed by a phagophore that
then matures into an autophagosome. The outer membrane of the
autophagosome fuses with a lysosome, forming an autolysosome that
degrades the enclosed cytoplasmic contents (6,7).
This biogenesis begins with the activation of the Unc-51 like
autophagy activating kinase 1 (ULK1) complex, which identifies the
membranes where biogenesis occurs (8). The formation process is orchestrated
by ATG proteins, numerous of which were identified in yeast through
genetic screenings of autophagy-deficient mutants (9,10).
The ATG1 kinase complex, consisting of ATG7, ATG31, ATG29 and
ATG13, is essential for this formation (11,12).
The majority of ATG proteins are recruited to the phagophore
assembly site (PAS), where they form a pre-autophagosomal structure
(13). Early in the process, the
ULK complex, comprising ULK1 (yeast ATG1 homologue), FAK family
kinase-interacting protein of 200 kDa (FIP200; also known as
RB1CC1), ATG13 and ATG101, forms a punctate structure near the
endoplasmic reticulum (ER) membrane, acting as a scaffold for
autophagosome formation (14). In
yeast, compartments featuring ATG9 protein clusters, composed of
vesicles and tubules, are instrumental in PAS formation (15). In mammals, under conditions of
starvation, the ULK1 complex, a pivotal initiator of autophagy,
targets and associates with the autophagy isolation membrane.
Notably, the induction of autophagy is compromised in the absence
of the FIP200 protein, underscoring its critical role in this
process (16). Mammalian target of
rapamycin (mTOR) serves as a primary negative regulator of
autophagy, directly influencing the ULK1-ATG13-FIP200 complex in
response to nutrient availability (17). The membrane at the PAS either
expands directly or engulfs cytoplasmic components through
vesicular expansion. In both yeast and mammals, the ubiquitin-like
protein system, comprising ATG8 and ATG12, facilitates phagophore
expansion (18).
Macroautophagy is instrumental in maintaining
cellular homeostasis by selectively targeting components such as
peroxisomes and mitochondria. For instance, macroautophagy degrades
excessive peroxisomes in mouse livers (19), and ULK1, an ATG1 homologue, plays a
key role in the targeted degradation of mitochondria and ribosomes
in erythrocytes (20). This
underlines the specificity and efficiency of macroautophagy in
cellular quality control, highlighting its essential function in
the removal of redundant or damaged organelles.
Microautophagy
Microautophagy involves lysosomes or vacuoles
engulfing and digesting segments of their own cytoplasm, including
proteins and organelles. The specialized autophagic tube structure
subsequently severs the vesicle membrane, delivering substrates
into the vacuolar space. Morphological variations in lysosomes have
led to identifying three distinct types of microautophagy in yeast
and mammalian cells: Type I, protruding lysosomes or vacuoles; type
II, invaginating lysosomes or vacuoles; and type III, endocytosed
invaginations (21). During
nutrient restriction-induced autophagy, early invaginations form
‘invagination tubes’ with the vacuolar transporter chaperone (VTC)
complex playing a key role in accumulating on the vacuolar membrane
(22). Furthermore, this complex
potentially serves as an activation site for calmodulin (23). Increased lipid accumulation on the
autophagic tube and depletion of integral proteins promote vesicle
formation (24). Pre-vesicular
structures, formed by enzymes, oscillate within lysosomes and the
VTC complex are essential for vesicle scission. Following
detachment from the autophagic tube, lipase Aut5p is activated amid
autophagic component interactions, compromising vacuolar integrity
and facilitating autophagosome degradation (25). Meanwhile, ATG22, an osmolytic
enzyme, recycles amino acids, contributing to nutrient and energy
cycling (26).
Microautophagy performs crucial biological functions
such as maintaining cell size, aiding in membrane formation, and
fostering cell proliferation by degrading and recycling nuclei
(27). For instance, apical
vacuoles in visceral endodermal cells, organelles typical of
lysosomes, rely on the endocytosis pathway-regulated CTP-binding
protein RAB7. They undergo microautophagy to ensure stability
during the early stages of mouse embryogenesis (28). This highlights the essential role
of microautophagy in developmental processes and cellular
maintenance, illustrating its relevance in fundamental biological
activities.
CMA
Contrary to other autophagy types, CMA does not
necessitate the transportation of substrates to the lysosome via
vesicles and membrane invagination, but rather through
translocation complexes on the lysosomal membrane (29). In 1978, Dice et al (30) found that protein degradation in
diabetes differs from healthy conditions, revealing that lysosomes
in diabetic models facilitate protein degradation and introducing
the concept of selective protein targeting by lysosomes. In 1986,
Backer and Dice (31) identified a
20-amino-acid sequence in RNase S protein that recognizes and
accelerates the degradation of RNase A protein, providing a new
perspective on selective degradation. In 1986, further research
established that attaching RNase S protein to other proteins
enhances degradation metabolism during starvation. RNase S protein,
serving as a unique sequence, activates the transport of
cytoplasmic proteins to lysosomes in serum-free conditions
(30). Subsequently, the
KFERQ-like sequence for protein targeting to lysosomes was
identified (31), and it was
confirmed that heat shock cognate 70 (HSC70) binds to this region,
targeting substrates to lysosomes for ATP-dependent degradation,
initiating research into chaperone recognition for selective
lysosomal degradation (32). In
1996, the identification of the fact that overexpressing LGP96
enhances the selective lysosomal proteolytic pathway in ovarian
cells established LGP96′s role on the lysosomal membrane as a
receptor for substrate protein degradation (33,34).
This type of autophagy, characterized by the formation of a protein
translocation complex through the recognition of specific substrate
proteins by HSC70 and translocation across the lysosomal membrane
via lysosome-associated membrane protein 2A (LAMP2A), is formally
defined as CMA. The activity of CMA and the levels of HSC70 protein
were determined in rat liver-derived lysosomes, showing a decrease
in activity and HSC70 levels with aging (35).
Molecular composition of CMA
Heat shock protein 70 (HSP70) binds to
CMA substrate proteins
The specific binding of substrate proteins to the
lysosomal membrane is crucial for CMA. A fundamental requirement
for CMA is HSC70′s recognition of the KFERQ-like pentapeptide motif
in substrate proteins. During serum withdrawal, mutation of this
pentapeptide sequence leads to reduced cellular degradation
(36). A fluorescence reporting
system measuring CMA activity revealed that insertion of an
11-amino-acid sequence containing KFERQ (KFERQ-PS-CFP2) into
non-CMA substrate proteins still results in their lysosomal
degradation (37). Thus, it is the
motif's nature that determines HSC70 binding, not any specific
amino acid. The KFERQ motif, essential for all CMA targeting,
consists of: i) One or two positively charged residues, namely K
and/or R; ii) one or two hydrophobic residues (I, L, V and/or F);
iii) one negatively charged residue (D and/or E); and iv) one
glutamine (Q) on either side of the pentapeptide (38). While canonical motifs exist in
unmodified protein sequences, post-translational modifications such
as ubiquitination, acetylation and phosphorylation can create
equivalent targeting sequences, thereby enhancing the diversity of
CMA substrate proteins (39).
However, the degradation rate remains unchanged even when multiple
KFEQR sequences exist in a protein (38).
HSC70 participates in cytoplasmic and luminal
pathways. Initially, HSC70 attaches to organelle membranes,
promoting peptide unfolding through the ATP hydrolysis cycle,
thereby facilitating entry into organelles (40). HSC70, operating in an ATP-dependent
manner, binds on both sides of the lysosome, thus breaking down
polymeric complexes. Concurrently, Hsp90 maintains the
conformational stability of LAMP2A complexes (41,42).
Luminal HSC70, characterized by a highly acidic isoelectric point,
modulates CMA in response to lysosomal acidification (43).
HSC70′s targeting function for KFERQ is contingent
upon the structural properties of the protein in which the motif is
located, and the adaptability of post-translational modifications.
The ADP-bound form of HSC70 exhibits the highest affinity for CMA
substrate proteins (33).
Cytoplasmic HSC70 (cyt-HSC70) recognizes peptide sequences,
including the KFERQ motif in CMA substrate proteins, aiding their
transport to lysosomal receptors. Cyt-HSC70, when docked on
lysosomal membranes, facilitates protein unfolding, a prerequisite
for lysosomal entry (33). Other
co-chaperones interact with HSC70 and modulate its activity. For
instance, Hsp40 activates the ATPase activity of HSC70, which
promotes substrate binding, and Hsp70-interacting protein (Hip)
stimulates the assembly of HSC70, Hsp40 and protein substrates
(44). Hsp90 is also found in the
lysosomal lumen, associated with the luminal side of the lysosomal
membrane. This protein stabilizes the fundamental components of
translocation complexes when organized into oligomeric structures
(41). The HSC70-Hsp90 organizing
protein acts as an adaptor between HSC70 and Hsp90, identifying
unfolded regions in proteins and preventing the aggregation of
substrate proteins. Within the HSP family, the ATPase of Hsp90
activates its ATPase activity, thereby stimulating protein binding
and release (44). Cell division
cycle 48 (Cdc48) enhances the activity of the HSC70-Hsp40 complex.
B-cell lymphoma 2 (Bcl-2)-associated athanogene 1 (BAG-1), an
Hsp70-related protein, exhibits unique properties as a negative
regulator of Hsp70. This protein decouples the ATPase cycle from
substrate binding, inducing conformational changes in Hsp70 and
enhancing the resistance of its substrate-binding domain to
proteolysis (45).
LAMP2A mediates the translocation of
HSC70-protein complexes to lysosomes
When the HSC70 chaperone complex translocates to the
lysosomal membrane, molecular chaperones within the lysosomal lumen
facilitate the translocation of the substrate complex across the
membrane, wherein LAMP2A plays a pivotal receptor role. Even though
LAMP2A receptors are found in all lysosome types, it is important
to note that not every lysosome is competent for CMA. LAMP2A is one
of the three splice variants of the LAMP2 gene (46), with LAMP-2B being involved in
macroautophagy (47) and LAMP-2C
in the uptake and degradation of ribonucleic acid (RNA) molecules
by lysosomes (48,49). They share the same luminal region
but differ in their transmembrane and cytosolic domains.
LAMP2A is cleaved from the lysosome and subsequently
released into the lysosomal matrix for degradation, in a form
truncated by metalloproteases and serine proteases (50). Pro-protein convertase A cleaves
LAMP2A at its serine active site, which is located between the
transmembrane and luminal domains, thus indirectly inducing
metalloprotease cleavage at the C-terminal region and thereby
accelerating substrate degradation rates (51). Under stress conditions, the
degradation of LAMP2A is diminished, resulting in an increased
presence of the receptor in the lysosomal membrane. The transport
of substrate proteins is associated with the internalization of
LAMP2A from the membrane into the lysosomal matrix (50). The interaction of HSP70 chaperones
and their co-chaperones with the lysosomal membrane occurs through
the four positive residues in the cytosolic tail of LAMP2A.
Specific antibodies that inhibit tail binding not only obstruct
substrate uptake and degradation in the lysosome (33), but also lead to a significant
accumulation of autophagic vesicles in tissues (52).
LAMP2A potentially influences HSC70 function,
undergoing rapid cycling to assemble a 700-kDa protein complex on
the lysosomal membrane. LAMP2A monomers on this membrane are
capable of receiving substrate proteins, and this interaction
initiates the formation of the required polymeric complex for
substrate translocation. Once the substrate protein reaches the
lysosomal lumen, LAMP2A disassembles from the polymeric complex,
thereby enabling successive rounds of substrate binding (41). Chaperone proteins located on either
side of the lysosomal membrane can regulate LAMP2A lateral
mobility. Upon the complex's transmembrane passage, Lys-HSC70
induces the disassembly of LAMP2A from the 700-kDa complex.
It has been demonstrated that LAMP2A mediates the
transport of soluble proteins with KFERQ sequence-related amino
acid sequences to newly formed exosomes on endocytosed limiting
membranes (53). Furthermore,
previous research has revealed that specific lipid (containing
cholesterol and glycosphingolipids) and protein-containing
microstructural regions with LAMP2A are present on the lysosomal
membrane. These microstructural regions facilitate the interaction
of LAMP2A with proteolytic enzymes, resulting in reduced LAMP2A in
lysosomes. This dynamic distribution of LAMP2A, as it enters and
exits lysosomal membrane microstructural areas, impacts the CMA
process (54).
Signaling pathways regulating CMA
activity
CMA is a distinct form of autophagy characterized by
its precise targeting and degradation of specific cytosolic
proteins. This process relies on recognizing specific KFERQ-like
motifs in proteins, which, once identified by chaperones, are
directly translocated across the lysosomal membrane for degradation
(55). The seamless functioning of
this mechanism is crucial for maintaining cellular homeostasis.
However, it does not operate in isolation. The efficiency and
regulation of CMA are closely linked with diverse signaling
pathways that either enhance or inhibit its activity (Fig. 2). A deeper exploration of these
signaling pathways offers a more comprehensive understanding of
CMA's role in health and disease (55).
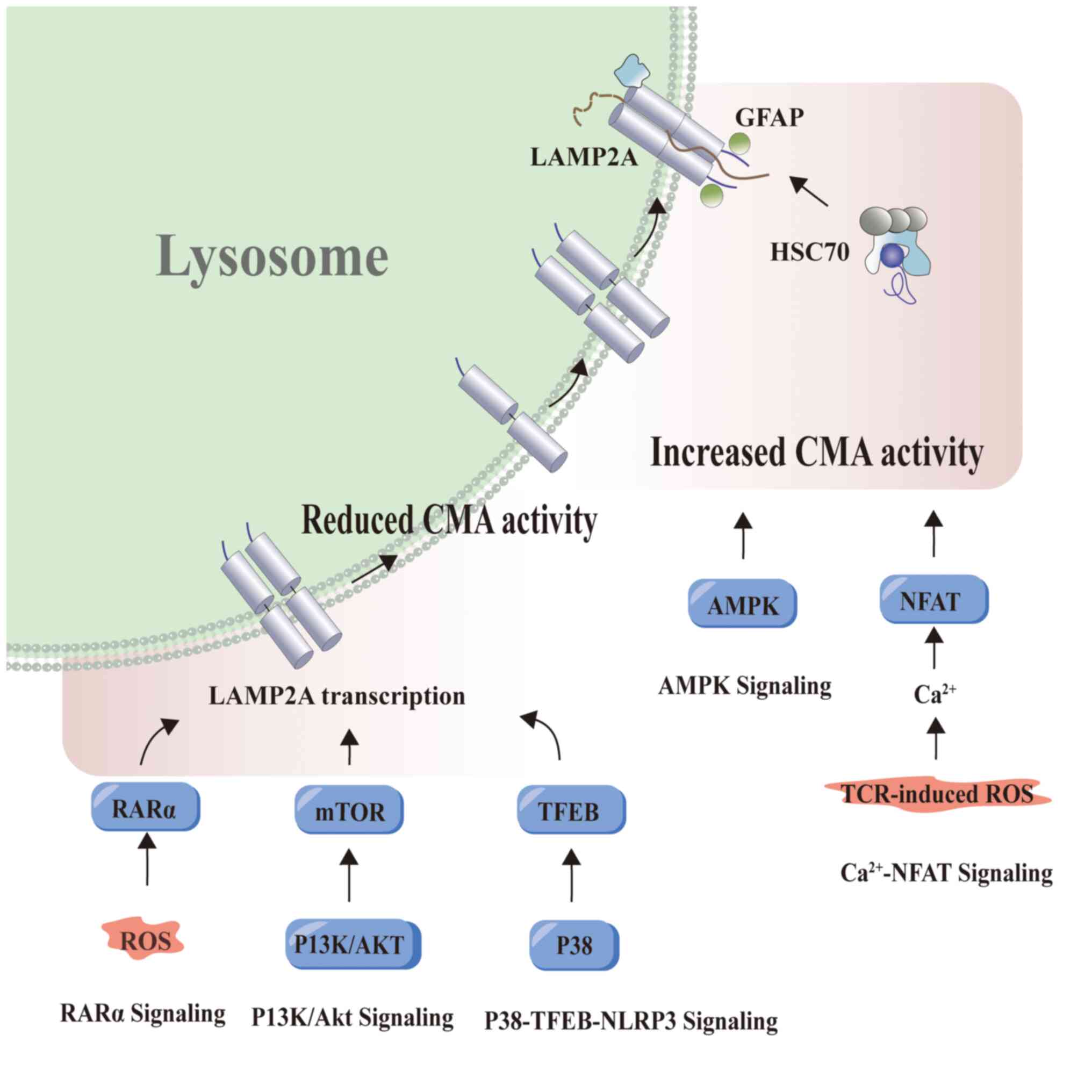 | Figure 2.Signaling pathways regulating CMA
activity. The calcineurin-NFAT pathway was initially reported to be
capable of inducing CMA activation in T cells. Furthermore, this
pathway also regulates CMA activity during T cell activation and
proliferation. In eukaryotic cells, AMP-activated protein kinase
not only regulates autophagy but also induces CMA, which leads to
the breakdown of lipid droplets by reducing cytoplasmic lipase
activity. In the context of p38-TFEB signaling, p38, which is a
serine/threonine kinase of the mitogen-activated protein kinase
family, phosphorylates TFEB, thereby inhibiting its activity. This
suppression of TFEB activity inhibits NLRP3 inflammasome
degradation mediated by CMA, thereby promoting glial cell
activation. AKT phosphorylates a downstream substrate of mTOR,
which in turn activates mTOR. mTOR is known to negatively regulate
autophagic activity. RARα reduces the levels of lysosomal proteins,
thereby affecting the interaction between lysosomal proteins Rab11
and Rab, and subsequently limiting CMA activity. The figure was
generated using Adobe Illustrator 2021, version 25.0, by Adobe Inc.
LAMP2A, lysosome-associated membrane protein 2A; GFAP, glial
fibrillary acidic protein; HSC70, heat shock cognate 70; CMA,
chaperone-mediated autophagy; AMPK, AMP-activated protein kinase;
NFAT, nuclear factor of activated T cells; RARα, retinoic acid
receptor alpha; mTOR, mammalian target of rapamycin; TFEB,
transcription factor EB; ROS, reactive oxygen species; PI3K/AKT,
phosphoinositide 3-kinases/protein kinase B. |
Retinoic acid receptor (RAR)α
In mammalian systems, RAR signaling is categorized
into three distinct types based on gene encoding: RARα, RARβ and
RARγ, with RARα being the most ubiquitously expressed (56). Inhibition of RARα significantly
upregulates the CMA pathway, leading to an increase in lysosomal
degradation. However, when the RARα signaling pathway is blocked
and simultaneously supplemented with all-trans retinoic acid (ATRA;
an effective activator of RARα signaling), it does not lead to a
suppression of autophagic degradation, which indicates that ATRA
does not activate macroautophagy via RARα. Consequently, retinoic
acid derivatives that modulate by inhibiting RARα's suppression of
CMA were studied, without affecting other autophagic pathways or
the transcriptional program of RARα (57). CMA activators in vivo
activate the RARα transcriptional pathway, stabilize the
N-CoR1/RARα interaction and ameliorate retinal lesions in a mouse
model of retinal pigment degeneration (58). It has been demonstrated that RARα
receptor activation attenuates neuroinflammation by promoting
phenotypic polarization in microglial cells and modulating the
Mafb/Msr1/PI3K-AKT/NF-κB pathway (59). The interaction between CMA and RARα
offers potential therapeutic insights for various diseases.
AMP-activated protein kinase
(AMPK)
In eukaryotic cells, AMPK functions as a critical
regulator of autophagy, orchestrating cellular energy balance and
systemic energy metabolism (60).
The cellular function of AMPK depends on ATP levels; its activation
increases the rate of catabolic pathways (ATP production) while
reducing that of anabolic pathways (ATP consumption) (61,62).
Activation of AMPK prompts CMA to trigger the breakdown of lipid
droplets. This mechanism entails either reducing cytoplasmic lipase
degradation or initiating lipolysis through macroautophagy.
Notably, lipid droplet-associated proteins, perilipin 2 (PLIN2) and
PLIN3, act as substrates for CMA degradation and interact with
HSC70 during lipid droplet-induced lipolysis (63,64).
Sirtuin 3 (SIRT3) regulates the acetylation state and activity of
substrates involved in energy metabolism, and activates
macroautophagy in lipotoxic hepatocytes via the AMPK-ULK1 pathway
(65). Similarly, overexpression
of SIRT3 enhances LAMP2A expression, stimulates CMA activity and
reduces the accumulation of the lipid-associated protein PLIN2
(65,66).
P38-TEEB-NLRP3
The mitogen-activated protein kinase (MAPK) pathway
is involved in numerous cellular regulatory processes. P38 MAPK is
a key member of the MAPK family, involved in cell cycle, apoptosis,
development, differentiation, senescence and tumorigenesis, and
also functions as a specific serine/threonine kinase in modulating
inflammatory responses. P38 directly phosphorylates the CMA
receptor LAMP2A at threonine residues, resulting in its membrane
accumulation and conformational activation, and subsequent
activation of the CMA pathway (67). Activation of the NLR family pyrin
domain containing 3 (NLRP3) inflammasome is important in various
pathological processes; the presence of the KFERQ targeting
sequence within its amino acid sequence interacts with HSC70, and
the sequence becomes unrecognizable to CMA when these two amino
acids mutate to proline. Starvation activates the CMA process in
glial cells, leading to the degradation of NLRP3 through
S-palmitoylation transferase-mediated palmitoylation of NLRP3
(68). Inhibition of the p38
pathway significantly reduces neuroinflammation caused by the
accumulation of α-synuclein, particularly the activation of the
NLRP3 inflammasome and increases the levels of the CMA receptor
LAMP2A, thereby enhancing the degradation of NLRP3 (69). Transcription factor EB (TFEB)
serves as a principal regulator of the autophagosome-lysosome
pathway (70), and it
downregulates the NLRP2 inflammasome through upregulation of
LAMP3A. Inhibiting p38 leads to TFEB nuclear translocation,
decreases its phosphorylation level and initiates TFEB-mediated
autophagy in Parkinson's models (69). Upon TFEB knockdown, the effects of
p38 inhibitors are completely abolished. The p38-TFEB pathway not
only inhibits CMA-mediated NLRP3 inflammasome degradation but also
promotes glial cell activation; thus, CMA-mediated processes may
represent a novel therapeutic target for Parkinson's disease (PD)
(69).
Calcium signaling-NFAT
The calcineurin-NFAT pathway was one of the first
mechanisms identified as capable of inducing CMA activation in
T-cells. T-cells are found throughout the body's immune organs,
performing immune functions within tissues. Activation of the T
cell receptor (TCR) leads to upregulation of LAMP2A, thereby
initiating the CMA pathway in CD2T cells. In mice with LAMP2A
knockout, there is a compromise in both immune tissues and T-cell
functionality, resulting in decreased induced proliferation and a
significant reduction in cytokine levels (71). Upon T-cell activation, the
production of reactive oxygen species (ROS) occurs. TCR activates
nuclear factor of activated T cells (NFAT), which in turn modulates
LAMP-4A expression in CD2T cells, playing a role in CMA activation.
Inhibitors of TCR signaling, including the E3 ubiquitin ligase Itch
and the regulator of calcineurin 1 (RCAN1), feature CMA target
sequences and are concentrated on the lysosomes of activated
T-cells. CMA supports effective T-cell activation by degrading both
Itch and Rcan-1 (71). During
T-cell activation, the ROS generated prompt the nuclear
translocation of NFAT1. NFAT2 directly binds to the proximal
promoter region of LAMP2A, including the presumed NFAT1 binding
site, thereby promoting LAMP2A mRNA expression and enhancing CMA
activation (71). Inhibition of
calcineurin through cyclosporin A administration or blockade of ROS
production results in the abolishment of CMA activation in these
cells. This provides a novel insight into the oxidative stress
response linked to CMA activation.
P13K/AKT
PI3K-AKT pathway mediation occurs through serine or
threonine phosphorylation of downstream substrates in response to
extracellular signals. This cascade is instrumental in facilitating
a variety of cellular processes, including metabolism,
proliferation, cell survival, growth and angiogenesis. Central to
this pathway are the enzymes PI3K and protein kinase B (AKT), the
latter also being referred to as protein kinase B (72). AKT has the capability of
phosphorylating substrates downstream of mTOR, thereby activating
mTOR. Autophagic activity is negatively modulated by mTOR (73). In models of acute liver failure in
rats, or in cells infected with the hepatitis virus, activation of
the PI3K/AKT/mTOR pathway is observed, accompanied by significant
elevations in HSC70 protein and mRNA levels (74). Molecular inhibitors that target the
PI3K/AKT/mTOR axis not only promote cellular proliferation and
inhibit apoptosis but also exert protective effects against acute
liver failure by activating CMA. Consequently, the PI3K/AKT/mTOR
axis may play a pivotal role as an upstream regulator of CMA
(74). At the lysosomal membrane,
CMA inhibition occurs due to the AKT-dependent phosphorylation of
the CMA regulatory factor glial fibrillary acidic protein (GFAP).
Lysosomes isolated from mice treated with PI3K inhibitors exhibited
enhanced CMA activity and reduced lysosomal GFAP phosphorylation,
with no changes observed in macroautophagy (75). Additionally, the PI3K/AKT-dependent
CMA mechanism significantly promotes osteoblastic differentiation
in rat bone marrow stromal stem cells (74). Vitamin D plays a critical role in
stimulating the differentiation of osteoblasts from bone marrow
stromal stem cells. Leptin, acting synergistically with the vitamin
D metabolite 25-hydroxyvitamin D3, induces osteoblastic
differentiation. Leptin upregulates methionine expression by
activating the PI3K/AKT signaling pathway, resulting in increased
HSC70 expression levels. Furthermore, inhibiting leptin expression
can modify its inhibitory effect on CMA. Additionally, leptin
suppresses CMA activity through the activation of vitamin D
metabolites, thereby further enhancing osteoblastic differentiation
(76).
CMA in cell physiology: From
homeostasis to aging
In the context of CMA, the complex interplay of
signaling pathways not only determines its regulatory mechanisms
but also highlights its adaptability in cellular processes. Serving
as conduits for both external and internal signals, these pathways
finely adjust CMA's activity, influencing a wide range of cellular
functions. Delving into the role of CMA in cellular senescence and
metabolic regulation reveals that this type of autophagy is more
than a cellular housekeeper; it is a vital mediator of cellular
aging and metabolic adaptation (77). This dual function underscores CMA's
significance in preserving cellular vitality and in orchestrating
the intricate dynamics of aging and metabolic processes.
Additionally, CMA plays a crucial role in cell cycle regulation
(78). Alterations in specific
signaling pathways can affect CMA activity, thus indirectly
modulating different stages of the cell cycle. Metabolically, CMA
aids in maintaining cellular equilibrium by ensuring a consistent
energy supply within cells. Within the realm of protein
homeostasis, CMA guarantees the timely and precise degradation and
recycling of cellular proteins, thereby ensuring cellular stability
(79,80). In the forthcoming discussion, these
functions and their physiological implications associated with CMA
are further described (Fig.
3).
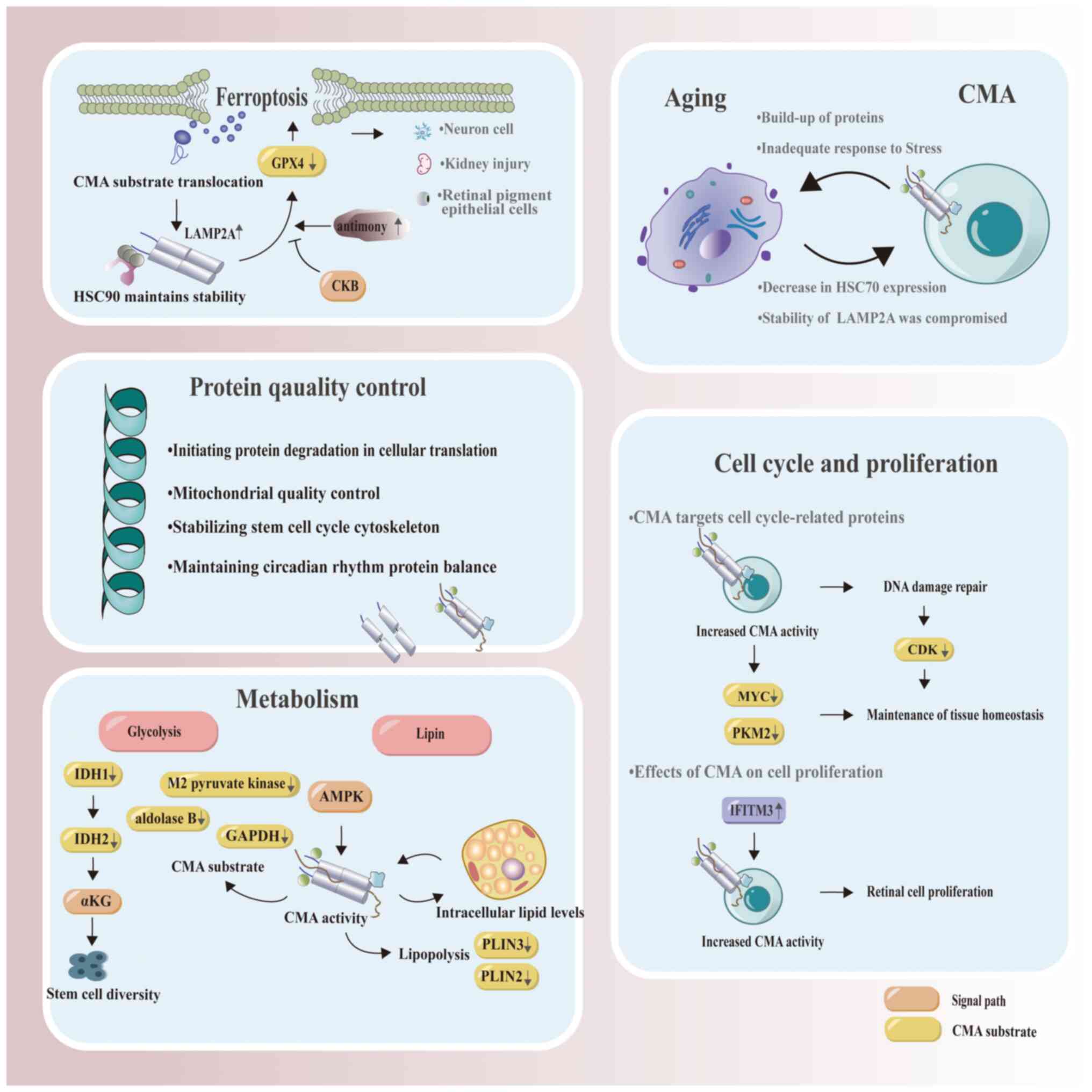 | Figure 3.Physiological roles of CMA. In cells
affected by ferroptosis, CMA activation not only leads to an
increase in LAMP2A levels but also induces CMA in an
HSP90-dependent manner, which in turn mediates GPX4 degradation.
CKB-mediated GPX4 phosphorylation serves to prevent CMA-mediated
degradation of GPX4, while Sb is known to induce neurotoxicity.
Upon Sb increase, CMA-related proteins (HSC70 and LAMP2A, which
exhibit increased levels) bind to GPX4, thereby forming a chaperone
complex that accelerates GPX4 degradation and ultimately leads to
ferroptosis in neurons. The complex interplay between ferroptosis
and CMA has undergone extensive investigation in various
conditions, including renal injury, neuronal injury and impairments
of retinal function. CMA plays a crucial role in maintaining
metabolic homeostasis, especially in glucose and lipid metabolism.
This process influences stem cell diversity by degrading IDH1 and
IDH2, subsequently regulating intracellular αKG levels.
Furthermore, the activation of AMPK stimulates CMA to initiate the
breakdown of lipid droplets, and these intracellular lipid levels
in turn influence CMA activity. CMA exhibits a close association
with aging, characterized by mutual interaction. As aging
progresses, the functionality of CMA becomes impaired and its
activity reduces, leading to an accelerated aging process
attributable to the accumulation of oxidized or misfolded proteins.
CMA plays a crucial role in maintaining cellular homeostasis by
regulating key cell cycle proteins such as MYC and PKM2. CMA
responds to DNA damage by timely degradation of CDKs, thereby
facilitating entry into the cell cycle after DNA repair. The
protein IFITM3 promotes retinal cell proliferation through the
activation of CMA. The figure was generated using Adobe Illustrator
2021, version 25.0, by Adobe Inc. Sb, antimony; CMA,
chaperone-mediated autophagy; GPX4, glutathione peroxidase 4; CKB,
creatine kinase B; HSC70, heat shock cognate 70; LAMP2A,
lysosome-associated membrane protein 2A; IDH, isocitrate
dehydrogenase; αKG, alpha-ketoglutarate; GAPDH, glyceraldehyde
3-phosphate dehydrogenase; AMPK, AMP-activated protein kinase;
PLIN, perilipin; MYC, MYC proto-oncogene, BHLH transcription
factor; PKM2, pyruvate kinase M2; CDK, cyclin-dependent kinase;
IFITM3, interferon-induced transmembrane protein 3. |
CMA regulates ferroptosis
Ferroptosis represents a dependency-driven form of
programmed cell death. Under the influence of divalent iron or
lipoxygenase, ferroptosis catalyzes the overexpression of
polyunsaturated fatty acids on the cell membrane, resulting in
lipid peroxidation and subsequent cell death (81). CMA is implicated in the onset of
ferroptosis, with glutathione peroxidase 4 (GPX4) degradation being
pivotal in inducing lipid peroxidation. The triterpenoid compound
2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid (CDDO) inhibits
ferroptosis, induced by erastin/glutamate, by blocking GPX4
(82). CDDO acts upstream of lipid
peroxidation, suppressing the end-product malondialdehyde without
altering GPX4 mRNA levels. Additionally, CDDO suppresses HSP90, a
shared regulator of necroptotic death and ferroptosis (82). In ferroptosis, fluorescently tagged
CMA substrates translocate from the cytoplasm to the lysosome,
indicating CMA activation. Knockdown of LAMP2A and HSC70
significantly alleviates erastin-induced ferroptosis. HSP90
maintains LAMP2A stability, interacts with HSC70 and regulates
ferroptosis by modulating CMA. During the ferroptotic activation
process, elevated LAMP2A levels and HSP90-dependent CMA induction
mediate GPX4 degradation (82).
Creatine kinase B mediates GPX4 phosphorylation at the serine 104
residue, which prevents CMA-mediated GPX4 degradation (83). Prolonged exposure to antimony (Sb),
which is prevalent in agriculture and industry, induces
neurotoxicity. Increased levels of Sb enhance the binding of
CMA-associated proteins (HSC70 and LAMP2A) to GPX4, forming a
chaperone complex that accelerates GPX4 degradation and neuronal
cell ferroptosis (84). In studies
examining legumain's nephroprotective role, interactions between
HSC70, HSP90 and GPX4 were observed to mediate the lysosomal
transport of GPX4, leading to ferroptosis (85). In hyperglycemic environments,
excessive secretion of mature neuroglial factor-β in the vitreous
body impairs the acidification and activation of retinal pigment
epithelial cell lysosomes, thereby hampering autophagolysosomal
degradation. Disrupted lysosomal homeostasis leads to elevated
levels of the ferroptotic marker acyl-CoA synthetase long-chain
family member 4 (ACSL4). ACSL4 has also been identified as a
substrate for CMA by the receptor HSC70 (86). CMA, when activated during cellular
ferroptosis, maintains autophagolysosomal balance. Both the CMA
activator QX77 and ferroptosis inhibitors reduce neuropathy and
enhance retinal physiological function (86).
Mitochondrial changes typifying ferroptosis include
mitochondrial shrinkage, cristae loss, increased mitochondrial
membrane density and outer mitochondrial membrane rupture.
Ferroptosis and apoptosis are intertwined processes, with apoptosis
potentially transitioning into ferroptosis under specific
conditions, thus augmenting cellular sensitivity to apoptosis. CMA,
which is possibly activated alongside apoptosis under stress, can
result in cell death. BCL2 binding component 3 (BBC3), a
pro-apoptotic member of the BCL-2 family, orchestrates various
apoptotic responses to cellular insults and is subject to
CMA-dependent degradation. In the process of CMA, HSPA8 recruits
BBC3 to the lysosome for degradation. Direct interactions between
HSPA8 and BBC3 result in the formation of a protein complex, with
BBC3 possessing the KFERQ motif (87). Tumor necrosis factor (TNF) elicits
a range of cellular responses, including survival, proliferation
and apoptosis. Prolonged TNF signaling activates inhibitor of
nuclear factor kappa b kinase subunit beta (IKBKB), leading to the
phosphorylation of BBC30 at Ser3, which is subsequently suppressed
by CMA-mediated degradation (87).
Depletion of CMA stabilizes BBC3, thereby compromising cell
viability in both the presence and absence of genotoxic
insults.
CMA maintains protein homeostasis
CMA plays a crucial role in homeostasis of cellular
proteins. Under injury conditions, accumulation of oxidized and
aggregated proteins occurs in cells and organs. CMA regulates
proteostasis under a variety of stress conditions, thereby
preventing protein aggregation. For instance, within oxidized
cytoplasmic proteins, ~30% of substrate proteins possessing
targeting sequences are degraded through the CMA pathway (88). During toxic exposure, CMA becomes
activated, selectively degrading proteins altered by toxic
compounds (89). When aging and
oxidative stress diminish CMA activity, protein damage intensifies,
leading to increased aggregation and elevated oxidation levels
(90). Cells regulate the
production and degradation of proteins in a coordinated manner to
maintain cellular homeostasis and adapt to the constantly changing
environmental conditions throughout the day. This equilibrium
ensures optimal functioning of fundamental cellular processes even
amidst fluctuations in external factors. Diurnal rhythm in cellular
proteomics delicately balances protein synthesis with degradation.
The CMA sub-proteome varies across different diurnal timepoints.
Proteins involved in translation and endocytosis undergo
degradation at night, while repair-related proteins degrade during
the day. Additionally, Golgi regulatory proteins are subjected to
CMA degradation irrespective of diurnal preference (91). The majority of proteins regulated
diurnally possess KFERQ targeting sequences, which are recognizable
by LAMP2A. Disruption of CMA in mice leads to central and
peripheral diurnal rhythm disorders, offering insights into diurnal
rhythm abnormalities in the elderly (91).
Proteomic analyses of isolated lysosomes in cancer
cells undergoing CMA activation reveal that CMA selectively targets
specific cellular processes. Differential gene enrichment analysis
comparing CMA activation and LAMP2A knockout indicates significant
enrichment of cytoplasmic translation initiation factors during CMA
activation (92). Changes in the
expression of translation initiation proteins are correlated with
malignant transformation in various cancer types. For example,
eukaryotic translation initiation factor 4A1 (EIF4A1), EIF4H and
DEAD-box helicase 3 X-linked (DDX3X), three translation initiation
factors, exhibit elevated expression in primary tumors and undergo
degradation through CMA pathways owing to their targeting
sequences. Activation of CMA significantly diminishes these
translation initiation factor proteins in various cell lines,
including ovarian, lung and breast cancer, highlighting CMA's role
in protein degradation related to cellular translation initiation
(92).
CMA plays a regulatory role in hematopoietic stem
cell renewal and metabolic balance. It stimulates the metabolism of
long-chain fatty acids, catering to the demands of differentiating
hematopoietic stem cells, by timely degrading acetylated marker
proteins. Proteomic analysis indicates that a CMA defect results in
global proteome remodeling, leading to inability to regulate the
cell cycle and skeletal functions, failure to balance protein
levels with other protein components, and impaired hematopoietic
stem cell function (93).
Regulation of metabolic diversity
through CMA
During the early phases of autophagy research,
glycolytic enzymes, including those containing the KFERQ motif
found in most glycolytic enzymes such as GAPDH, were identified as
substrates for CMA This list also includes pyruvate kinase
(94) and isoform M2 of aldolase B
(95). In LAMP2A knockout mouse
livers, enzymes involved in glucose catabolism within lysosomes are
not timely degraded. As a result, gluconeogenesis decreases,
glycolysis increases, leading to liver glycogen depletion and
hepatocyte steatosis (96). Under
normal nutritional conditions, enzymes from both glycolysis and the
tricarboxylic acid cycle (TCA) cycle are degraded via CMA (96), suggesting a pivotal role for CMA in
metabolic adaptation. CMA also plays a role in stem cell
self-renewal and differentiation by influencing metabolic products
in stem cells, promoting embryonic stem cell self-renewal with high
activity, enhancing differentiation and maintaining embryonic stem
cell viability through the mediation of pluripotency factors such
as octamer-binding transcription factor 4 (OCT4) and SRY-box
transcription factor 2 (SOX2) (97). Intracellular α-ketoglutarate (αKG),
a product of the TCA cycle and an essential cofactor for
demethylation, plays a key role in sustaining undifferentiated
pluripotent stem cells (98,99).
Following reduced LAMP2A activity, αKG emerges as one of the most
abundant metabolites in cells. Conversely, αKG levels decrease with
overexpression of LAMP2A. αKG is produced by various enzymes in the
TCA cycle, serine biosynthesis pathway and amino acid metabolism.
Sequencing of αKG generation enzymes confirms that both isocitrate
dehydrogenase 1 (IDH1) and IDH2 possess CMA-targeting sequences.
The expression levels of IDH1 and IDH2 in cells correlate with αKG
levels when CMA activity is altered. Thus, by degrading IDH1 and
IDH2, CMA regulates intracellular αKG levels, consequently
affecting stem cell diversity (97).
CMA, in its role of maintaining metabolic
homeostasis, acts as an upstream regulator for lipid autophagy and
lipid metabolism. The selective degradation of key enzymes by CMA
induces alterations in carbohydrate and lipid metabolism. CMA
promotes lipid metabolism through the degradation of lipogenic
enzymes and the selective loss of lipid droplet proteins (100). Lipid carriers and protein
lipogenic enzymes, both integral to lipid metabolism, have been
identified as CMA substrates (63). CMA selectively degrades proteins
surrounding lipid droplets, thereby increasing cytoplasmic
lipolysis and activating macroautophagy, initiating lipid breakdown
(63). Moreover, CMA orchestrates
proteins that regulate cell cycle arrest, glycolysis and specific
lipid synthesis, thereby driving adipocyte differentiation
(101). Rab7, a small GTPase,
plays an essential role in the degradation of lipid droplets in
starved liver cells. Rab7 modulates the interactions between lipid
droplets and vesicles with LAMP1 lysosomes. Depletion of Rab7 leads
to significant morphological changes in lysosomes and
autophagosomes, suggesting an increased probability of CMA
activation (102). Intracellular
lipid levels additionally influence CMA activity. Long-term
high-fat diets or acute high cholesterol intake reduce CMA's
ability to transport lysosomal cytoplasmic substrates into the
lumen, decrease LAMP2A receptor levels and disrupt lysosomal
membrane structural stability (103).
CMA in cell cycle and
proliferation
CMA specifically targets cell cycle-associated
proteins, thereby regulating tissue homeostasis. MYC
proto-oncogene, BHLH transcription factor (MYC), a member of the
helix-loop-helix leucine zipper family of transcription factors,
modulates a multitude of target genes both positively and
negatively, stimulating the cell cycle through various mechanisms
(104). A deficiency in CMA
results in the accumulation of MYC protein. Notably, although MYC,
containing two CMA targeting sequences, is not directly degraded,
its phosphorylated form during cellular transformation is crucial
for CMA-mediated indirect regulation (105). Pyruvate kinase M2 (PKM2)
stimulates MYC and cyclin D, thereby advancing the G1
phase of the cell cycle. Acetylated PKM2, undergoing a
conformational change, interacts more effectively with HSC70, thus
facilitating its CMA-mediated degradation (94). Rho family GTPase 3 (RND3), a
regulator of the cell cycle, suppresses MYC expression and its
transcriptional activity, thereby affecting several critical genes,
including cyclins, cyclin-dependent kinases (CDKs) and the EF2
transcription factor (105). Its
recognition by CMA is crucial for the proliferation of gastric
cancer cells (106). Checkpoint
kinase 1 (CHK1) mediates both normal and DNA damage-induced cell
cycle arrest (107). Within
cells, CHK1 mediates the S and M phases. During the S phase, CHK1
induces a reduction in CDK2 activity through the phosphorylation
and proteasomal degradation of Cdc25A, resulting in slowed or
blocked DNA replication. In the presence of DNA damage, CHK1 blocks
the G1/M transition via CHK2 regulatory phosphorylation,
leading to CDK1 inactivation (107). In response to DNA damage,
upregulated CMA aids in cell cycle re-entry post DNA repair by
facilitating the timely degradation of cell cycle CHK1 (108).
Hypoxia-inducible factor 1 (HIF-1), a transcription
factor, orchestrates the adaptive response to hypoxia. HIF-1α
interacts with CDK1 and CDK2, playing a key role in cellular
processes. CDK1 activity hinders lysosomal degradation of HIF-1α,
thereby enhancing its protein stability and transcriptional
activity. During the G1/S phase transition, CMA-mediated
degradation of HIF-1α is stimulated by CDK2 (109). Within the ubiquitin-proteasome
system, substrate recognition is marked by ubiquitination (110). The ubiquitin ligase STIP1
homology and U-box containing protein 1 (STUB1) ubiquitinates
lysine 63 (K63), which then binds and degrades HIF1A in the
lysosome, in conjunction with CMA proteins (111,112). The interaction of CMA with
immune-associated proteins plays a significant regulatory role in
cell proliferation. Interferon-induced transmembrane protein 3
(IFITM3), a transmembrane protein, modulates the
interferon-mediated innate immune response. IFITM3 regulates viral
infections through autophagy, and its knockdown activates CMA,
resulting in decreased retinal progenitor cell proliferation,
notwithstanding the lack of significant mTOR pathway activation
(113). The activation of CMA by
IFITM3 plays a pivotal role in cell proliferation, although
excessive CMA can lead to cell death (113). Chromosome 11 open reading frame
54 (C11orf54), a cytoplasmic esterase, influences cell
proliferation and apoptosis. Transcriptomic analysis of
C11orf54-knocked down and control cells demonstrated a marked
inhibition of HIF1A signaling, which promoted HSC70 and HIF1A
binding (114).
Additionally, CMA plays a crucial role in cell
differentiation processes. During the differentiation of mouse
mesenchymal stem cells into osteoblasts, LAMP2A levels increase,
which promotes the transformation into osteoblasts while
concurrently suppressing their adipogenic or chondrogenic potential
(115). Vangl2, localized in the
lysosomal differentiation-related gene of mesenchymal stem cells,
binds to LAMP2A and undergoes degradation in specific domains,
thereby interfering with CMA activity and inhibiting osteogenesis
(115). Adipocyte
differentiation, being a dynamic and multifactorial process, is
closely associated with CMA activity both in vitro and in
vivo. Blocking CMA leads to the downregulation of pre-adipocyte
differentiation markers PLIN2 and adipocyte proteins, impairing the
early stages of adipogenesis and resulting in the accumulation of
undifferentiated pre-adipocytes. Persistent cell proliferation,
dysregulated glycolysis rate and cell cycle disruption in
pre-adipocytes arise from the inability to degrade MYC post-CMA
blockage. The use of transforming growth factor beta (TGFβ)
molecular inhibitors can partially rescue pre-adipocyte degradation
following CMA deficiency, suggesting that CMA plays a pivotal role
in adipocyte differentiation by transiently inhibiting TGFβ
signaling in pre-adipocytes (101).
CMA's protective mechanism against
oxidative stress
Oxidative stress arises from an imbalance between
oxidative and antioxidative processes in the body, characterized by
elevated intracellular levels of ROS. While ROS play a role in
regulating normal cell differentiation and apoptosis, excessive ROS
can result in cellular toxicity, a key factor in aging and disease
progression. Inhibition of CMA leads to elevated ROS levels and
subsequent cell apoptosis. Kiffin et al (88) demonstrated that cytoplasmic protein
damage, induced by low concentrations of hydrogen peroxide, led to
an increase in the number of proteins binding to the lysosomal
membrane and being transported into the lysosomal lumen,
correlating with the duration of CMA substrate oxidation. CMA
responds to oxidative stress by upregulating the activity of its
lysosomal translocation complex. Nuclear factor erythroid 2 (Nrf2)
is a key antioxidant transcription factor, which is regulated by
the Keap1-E3 ligase complex. CMA is critical for the antioxidative
response mediated by Nrf2. Upon CMA activation, an increase in both
Nrf2 protein levels and its transcriptional activity is observed.
LAMP2A stabilizes Nrf2 by reducing the antioxidative levels of
Keap2, thereby decreasing Nrf2 ubiquitination, with Keap1 being
identified as a CMA substrate (116).
CMA selectively eliminates oxidized and pro-oxidant
proteins to mitigate damage. Myocyte enhancer factor 2A (MEF2A),
regulated by a critical transcription factor, protects primary
neurons from oxidative stress-induced cellular injury. Under mild
oxidative stress, MEF2A activity is notably enhanced; however,
under excessive conditions of MEF2A activation the cleavage of
histone deacetylase 4 in MEF2A is intensified, leading to the
release of lysosomal serine proteases from ruptured lysosomes in a
protein kinase-independent manner, thus targeting MEF2A for
CMA-mediated degradation (117).
Association between CMA and
mitochondrial quality control
Mitochondria, crucial for respiration-generated ROS,
can experience damage, leading to oxidative and antioxidative
imbalances. CMA combats oxidative stress by regulating
mitochondrial function. Parkinsonism associated deglycase
(PARK7/DJ-1), a crucial redox signaling intermediate, is activated
under oxidative stress conditions in the presence of ROS (118). PARK7 is a recognizable CMA
protein substrate. When cells undergo changes induced by the
mitochondrial toxin 1-methyl-4-phenylpyridinium (MPP), CMA
prioritizes the removal of oxidized and non-functional PARK7/DJ-1
by targeting it for lysosomal degradation to maintain the internal
balance. Upon LAMP2A knockdown, CMA's ability to mitigate MPP
damage diminishes, leading to more pronounced mitochondrial rupture
and further ROS increase. Additionally, PARK7 overexpression
mitigates cell death and mitochondrial dysfunction induced by MPP,
even with concurrent LAMP2A knockdown. The CMA-PARK7 pathway is
important for maintaining mitochondrial homeostasis. Dysregulation
of this pathway could lead to neuronal stress and death (119). CMA regulates oxidative stress and
mitochondrial damage under neuronal stress stimuli such as
neurotoxins, thus maintaining mitochondrial quality control and
homeostasis. E5 ubiquitin ligase MARCHF3, essential for
mitochondrial fission, is a direct CMA substrate, and CMA regulates
this ubiquitin ligase to influence mitochondrial positioning
factors (120).
CMA impacts aging
It is well-established that, during aging, CMA
functionality deteriorates and its activity diminishes, leading to
an inefficient stress response and the accumulation of oxidized or
misfolded proteins, which is a primary cause of multiple
age-related diseases. Lysosomes from elderly rat livers show
decreased transport rates of CMA substrates to the lysosomal
membrane and reduced cytosolic levels of HSC70 in human fibroblasts
(35). While the transcription
rate of LAMP2A remains consistent during aging, changes occur in
receptor dynamics and stability within the lysosomal lumen,
including alterations in proteases responsible for cleavage and
specific membrane microstructures. This observation suggests that
lysosomal membrane alterations contribute to CMA-associated
age-related changes (121).
Restoring CMA activity in aging can significantly enhance organ
function and the clearance of damaged proteins. Zhang and Cuervo
(122) maintained LAMP2A activity
in aged transgenic mice using Tet-regulators (responsive to
tetracycline or its derivative doxycycline), observing reductions
in liver damage markers, apoptotic cells and undegraded damaged
products in the lysosomal compartment. Under similar regulatory
conditions, aging exacerbates LAMP2A functional defects in mice,
leading to decreased hepatocyte vitality, glucose intolerance,
dysregulated blood sugar levels and lipid metabolic anomalies
(90). With increasing age, there
is a rise in cancer-related senile morbidity. Mice with a
deficiency in CMA activity exhibit protein homeostasis imbalances
in their livers, increased susceptibility to oxidative stress,
liver dysfunction and a higher incidence rate of spontaneous liver
tumors (90).
As aging progresses, the levels of CMA-related
molecules undergo changes. For instance, in 24-month-old rats,
HSC70 levels increase in the pons, medulla, striatum and thalamus,
thereby inhibiting cytosolic protein denaturation (123). A previous study by Calabrese
et al (124) found that
HSC70 expression increases with age in the hippocampus and
substantia nigra, and that augmenting HSP expression can enhance
the repair of oxidatively damaged proteins, thereby shielding cells
from age-related damage (124).
In female rats, the striatal levels of HSC70 and its co-chaperone
are lower (125). In age-related
macular degeneration, a leading cause of blindness in the elderly,
increased concentrations of LAMP2A and HSC70 in the retina are
observed, compensating for macroautophagy deficiencies (126). White blood cells from various age
groups exhibit distinct LAMP2A expression, with a decline in
activity as age progresses (127). In skeletal muscles of aged
(27-month-old) versus young (5-month-old) mice, LAMP2A and
HSPA8/HSC70 levels decreased. In the myocardium of aged mice,
LAMP2A levels increased, while HSPA8/HSC70 levels remained
consistent (128).
Cellular senescence, characterized as a primary
hallmark of aging, includes cell cycle arrest and impaired DNA
damage repair (129). In
senescent cells, lysosomal compartment expansion occurs alongside
upregulation of LAMP2A transcription. Both macroautophagy and CMA
levels show a positive association with aging (130,131). Knocking down LAMP2A significantly
induces senescence in primary human fibroblasts, as evidenced by
increased senescence-associated β-galactosidase activity, ROS
production and lipofuscin accumulation (132). A functional deficiency in CMA can
continuously activate CHK1, leading to DNA damage associated with
cellular senescence (108).
p21/CDKn1a, a cyclin-dependent kinase inhibitor, and HIF-1α, a
primary regulator of oxygen homeostasis, both serve as major
targets of CMA (133). In the
absence of hypoxic signals, HIF-1α upregulates p21/CDKn1a by
displacing MYC at the p21/CDKn1a promoter, inducing cell cycle
arrest (134). Thus, during
aging, impaired CMA function may induce cellular senescence by
degrading HIF-1α, which affects p21/CDKn1a.
CMA as a driving force for disease
progression
CMA occupies a central role in numerous cellular
physiological processes, thus ensuring the balanced functioning of
the cell's intricate machinery. Central to this function is the
cell cycle, where CMA coordinates the timely degradation and
recycling of proteins, thereby aiding in the progression and
regulation of various cell cycle stages (103). Furthermore, CMA substantially
impacts cellular metabolism by maintaining a balance between energy
production and consumption, and by ensuring vital substrates are
promptly available for essential cellular reactions. However, the
significance of CMA extends beyond mere normal physiological
functions. Disturbances or malfunctions in CMA can result in
pathological implications and a myriad of diseases. In oncology,
aberrations in CMA activities are known to contribute to tumor
progression and resistance to therapies (135,136). Similarly, disruptions in CMA
affecting organs such as the lungs (137), heart (138), liver (139) and kidneys (140) can lead to debilitating
conditions, underscoring its crucial role not only in maintaining
cellular health but also in the onset and progression of diseases.
A comprehensive knowledge of CMA is pivotal, both for understanding
normal cell functions and for unraveling the complexities of
various diseases.
Cancer
Consistent upregulation of CMA has been observed in
various cancer types and cell lines, including melanoma, lung,
gastric, colorectal and uterine cancer (141). This in vivo dependency on
CMA has been confirmed using human lung cancer xenograft models
(141). The ability of CMA to
promote limitless proliferation of tumor cells, a hallmark of
cancer, is remarkable. Changes in cell cycle control are central to
malignancy, and interfering with CMA can drive tumorigenesis.
Following malignant transformation, increased CMA activity has been
shown to promote tumorigenesis (141). CMA modulates several molecules
involved in cell cycle regulation, including the tumor suppressor
protein p73 (p73), which can induce cell cycle arrest and
apoptosis. Neural growth factor receptor (NGFR) is upregulated in
various cancer types, including glioblastoma (GB) and breast
carcinoma, enhancing transformed cell survival. NGFR directly binds
to p73, inhibiting its transcriptional activity and promoting its
degradation through CMA (142).
Tumor cells exploit the LAMP2A-PRDX1/CRTC1 axis to regulate the
activation of tumor-associated macrophages, thereby promoting tumor
growth (143). Tumor cells
require a higher glucose energy supply than normal cells, and
through glycolytic metabolism, the embryonic M2 isoform of PKM2 in
these cells facilitates aerobic glycolysis. Acetylated PKM2,
degraded by CMA, promotes tumor cell proliferation when ectopically
expressed (94).
Activation of CMA in cancer cells contributes to
cancer development. Malignant cells in hepatocellular carcinoma
(HCC) commonly demonstrate defects in autophagy and are associated
with CMA activation. In patients with liver cancer, a significant
correlation has been found between LAMP2A expression and tumor size
and recurrence (144). Research
by Ding et al (144)
suggested that the growth and recurrence of HCC require CMA
activation, as demonstrated using a murine tumor xenograft model.
The dysregulated proteins yes-associated protein 1 (YAP1) and
interleukin 6 cytokine family signal transducer (IL6ST) in HCC are
recognized for promoting tumor growth. These proteins demonstrate
dependency on binding with the CMA chaperone HSP8A and accumulate
in isolated lysosomes following CMA induction (145). Immunohistochemical staining of
LAMP2A and p62 in hepatitis C virus (HCV)-infected liver tissue
samples validated the autophagy compensation mechanism between HCC
and cirrhotic areas (144).
Beclin 1 plays a crucial role in the initiation of autophagy and
the fusion of autophagosomes with lysosomes. Reduced expression of
beclin 1 leads to impaired autophagy in human HCC, and
downregulation of beclin 1 in HCC is associated with poor prognosis
(146,147). Mutations in beclin 1 accelerate
the development of pre-cancerous lesions induced by hepatitis B
virus (148). Research by Aydin
et al (149) identified
that cellular responses to HCV-induced stress inhibited autophagy
due to the activation of stress-adaptive CMA, resulting in beclin 1
degradation and autophagy-related p62 accumulation. Activation of
CMA hinders the endocytosis and degradation of epidermal growth
factor receptor (EGFR), thereby activating the EGFR signaling
pathway. This results in impaired autophagosome-lysosome fusion,
consequently activating downstream oncogenic signaling pathways
such as RAS/RAF/MEK/ERK, which further drives the progression of
cirrhosis-associated HCC (149).
In lung cancer cells, LAMP2A modulates their malignant phenotype by
regulating apoptosis through CMA. Knockdown of LAMP2A alters the
cancer phenotype and enhances drug sensitivity (150). CMA-mediated stabilization of
myeloid cell leukemia 1 (MCL1), a pro-survival protein, has been
confirmed to facilitate the survival of non-small cell lung cancer
(NSCLC) cell lines (151).
Nuclear receptor co-repressor (N-CoR) plays a pivotal role in
transcriptional control mediated by tumor suppressor proteins and
possesses a CMA targeting sequence. CMA induces the degradation of
misfolded N-CoR, thereby neutralizing ER stress in NSCLC and
indirectly promoting NSCLC cell survival (152).
Knockdown of LAMP2A in gastric cancer cells impedes
cell proliferation, resulting in increased expression of the cell
cycle-related protein RND3 and its interaction with HSP8A, leading
to degradation via CMA, thereby supporting rapid proliferation of
gastric cancer cells (106).
CMA-mediated degradation of acetyltransferase p300/CREB-binding
protein (p300/CBP) can increase the chemoresistance of colorectal
cancer to 5-fluorouracil (153).
ALL1 fused gene from chromosome 1q (AF1Q), a biomarker for myeloid
leukemia and a target substrate for CMA, undergoes lysosomal
degradation upon CMA activator treatment (154). The prostate-specific and
androgen-responsive gene, tumor protein d52 (TPD52) subtype 1,
amplified in prostate cancer, is closely associated with poor
prognosis. TPD52 activates CMA physiologically by enhancing the
transfer of CMA protein substrates in prostate cancer cells through
complex formation with HSPA8. Acetylation of TPD52 at lysine 52
plays a key role in modulating the interaction between TPD52 and
HSPA8, thereby regulating CMA activity (155). In histopathological samples from
patients with lung cancer, high expression of the CMA marker LAMP2A
is associated with the prognosis of both adenocarcinoma and
squamous cell carcinoma (156).
CMA in the tumor microenvironment is closely
related to tumor progression, being particularly activated under
conditions such as nutrient deficiency, hypoxia and elevated ROS
(88,157,158). GB is a highly lethal malignant
brain tumor for which no effective therapeutics are currently
available. Interaction between GB and peritumoral cells (PCs)
facilitates an increase in ROS, resulting in upregulation of CMA in
PCs via LAMP2A expression. This anomalous induction of CMA in GB
impacts the proteome and transcriptome of PCs, influencing the
expression of immune-suppressive and inflammatory genes (159). When the crosstalk between
CMA-deficient PCs and mouse GB cells is compromised, reduced tumor
cell viability and impaired tumorigenesis occur (159).
Although CMA activity is positively correlated with
increased expression in various cancer cells, it exhibits
antitumoral effects in non-transformed cells (160). Inhibition of CMA in fibroblasts
enhances MYC-mediated cell transformation, intensifying MYC-driven
oxygen consumption and extracellular acidification, and shifting
metabolism from oxidative phosphorylation to aerobic glycolysis,
which is closely associated with tumorigenesis (105). Although MYC degradation primarily
occurs through the proteasome, CMA indirectly mediates this
process. CMA participates in DNA damage repair, aiding in the
prevention of malignant transformation by maintaining genomic
stability and reducing oncoprotein expression to counteract cancer.
Translationally controlled tumor protein (Tpt1/TCTP) is subject to
both transcriptional and translational regulation. Acetylated TCTP,
recognized and degraded by CMA, helps to maintain cellular
homeostasis. The degradation of oncoprotein mouse double minute 2
homolog (MDM2) via CMA contributes to the inhibition of
tumorigenesis (161,162). Breast cancer represents the most
prevalent malignancy. Microrchidia family CW-type zinc finger 2, an
oncoprotein upregulated in various cancer types, interacts with
HSP8A and LAMP2A, and undergoes phosphorylation modifications to
prevent CMA-lysosomal degradation (163). TFEB and trehalose, a disaccharide
that induces TFEB activation, enhance the expression of OCT4 in
stem cells and LAMP2A. In ovarian cancer stem cells, fructose
treatment reduces spheroid formation while elevating LAMP2A
expression. CMA influences cancer stem cells through fructose
metabolism (164). Aberrant
activation of NF-κB can promote tumorigenesis. The p65 protein,
which is central to this pathway, interacts with HSC70 targeting a
basic sequence and is degraded through the CMA pathway.
Additionally, in various epithelial-mesenchymal transition (EMT)
models, diminished CMA activity is observed, and overexpression of
LAMP2A can rescue EMT impairment by degrading p65 (165). CMA offers a novel avenue for
cancer therapy; however, the underlying mechanisms of its
antitumoral function in relation to cancer cells require further
exploration (Fig. 4).
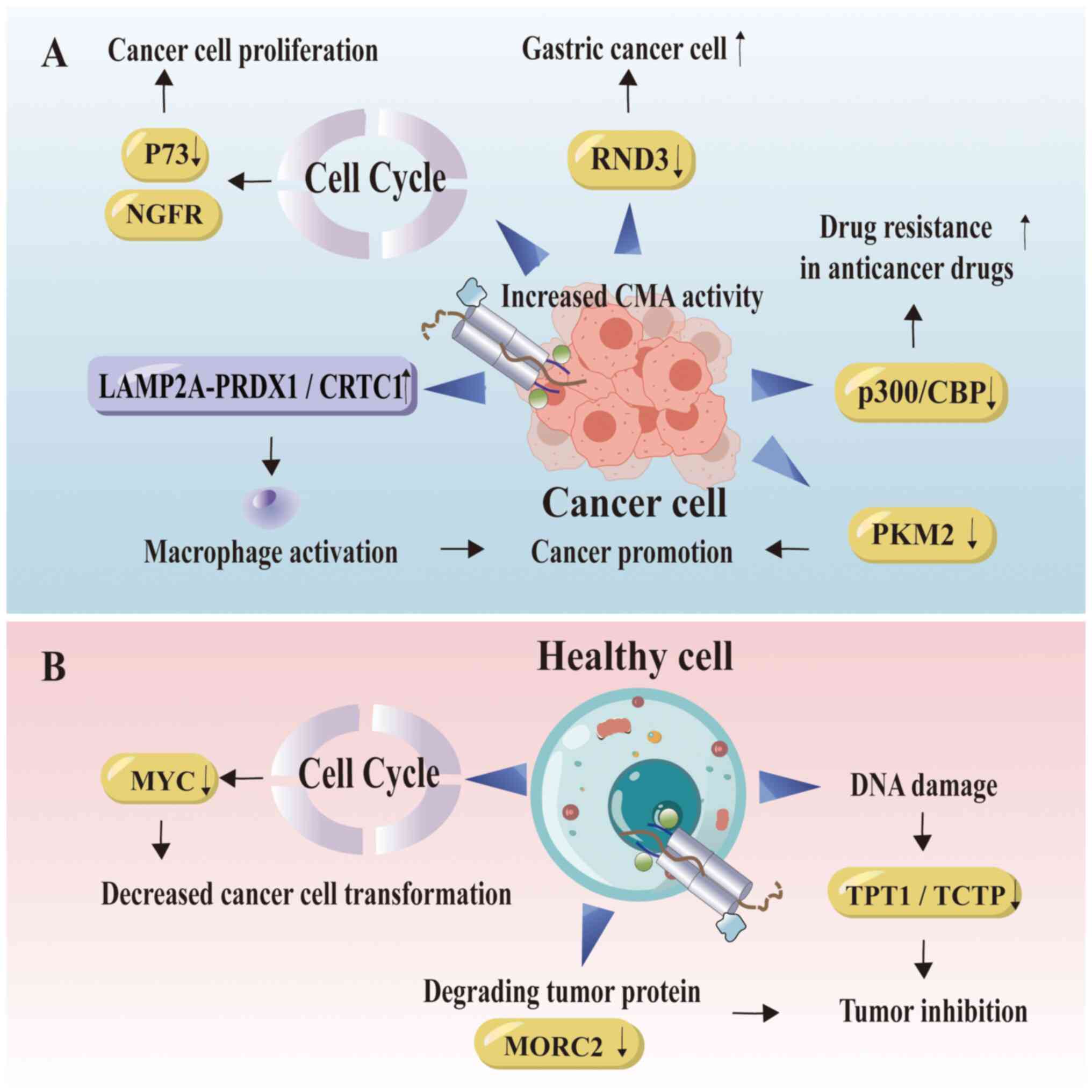 | Figure 4.CMA is associated with cancer. (A)
Activation of CMA in tumor cells is implicated in the promotion of
cancer development. Increased CMA activity influences cell cycle
alterations, downregulates the tumor suppressor factor P73, leads
to the degradation of RND3 and facilitates the rapid proliferation
of cancer cells. Tumor cells promote their own proliferation by
regulating the LAMP2A-PRDX1/CRTC1 axis. Proteins related to
degradation promote tumor development, which is manifested as
increased resistance to drugs for treating colorectal cancer. (B)
In non-carcinogenic cells, CMA exhibits antitumor effect. In normal
cells, CMA regulates the cell cycle, contributes to DNA damage
prevention to inhibit malignant tumor transformation and degrades
pro-oncogenic proteins. The figure was generated using Adobe
Illustrator 2021, version 25.0, by Adobe Inc. NGFR, nerve growth
factor receptor; RND3, Rho family GTPase 3; CMA, chaperone-mediated
autophagy; LAMP2A, lysosome-associated membrane protein 2A;
PRDX1/CRTC1, peroxiredoxin 1/CREB regulated transcription
coactivator 1; p300/CBP, p300/CREB-binding protein; PKM2, pyruvate
kinase M2; MYC, MYC proto-oncogene, BHLH transcription factor;
MORC2, microrchidia family cw-type zinc finger 2; TPT1/TCTP,
translationally controlled tumor protein. |
Neurodegenerative disorders
Post-mitotic neurons encounter challenges with
misfolded protein aggregates, which, undiluted by cell division,
necessitate clearance via the proteolytic system (166). Within the central nervous system,
multifactorial injuries result in neuronal dysfunction. CMA
controls the selective degradation of cytosolic proteins,
potentially protecting neurons from degeneration. Inhibition of CMA
leads to destabilization of proteins within neurons (166). Numerous proteins related to
neurodegeneration possess CMA targeting sequences, including
leucine-rich repeat kinase 2 (LRRK2) (167), ubiquitin C-terminal hydrolase
isozyme L1 (UCH-L1) (168),
α-synuclein and microtubule-associated protein t (TAU). These
proteins directly interact with LAMP2A and are degraded via CMA
(169). However,
post-translational modifications of α-synuclein affect its
degradation by CMA, since phosphorylated and dopamine-modified
forms of α-synuclein are resistant to CMA degradation (170). The persistent association of
α-synuclein with the lysosomal membrane further impairs CMA,
resulting in a vicious cycle. Vascular protein sorting 35 (VPS35),
crucial for endosome-to-Golgi retrieval, exhibits defects that
alter lysosomal morphology, reduce LAMP2A vesicles and contribute
to the elimination of α-synuclein accumulation (171).
When pathogenic proteins directly compromise CMA
components, thereby hindering their degradation capability, the
toxic effects of CMA dysregulation may act as a primary etiological
factor or as a secondary consequence when protein homeostasis is
indirectly affected (172).
Factors such as genetics, aging and the microenvironment contribute
to protein aggregation and neurodegenerative changes that result
from CMA dysregulation (Fig.
5).
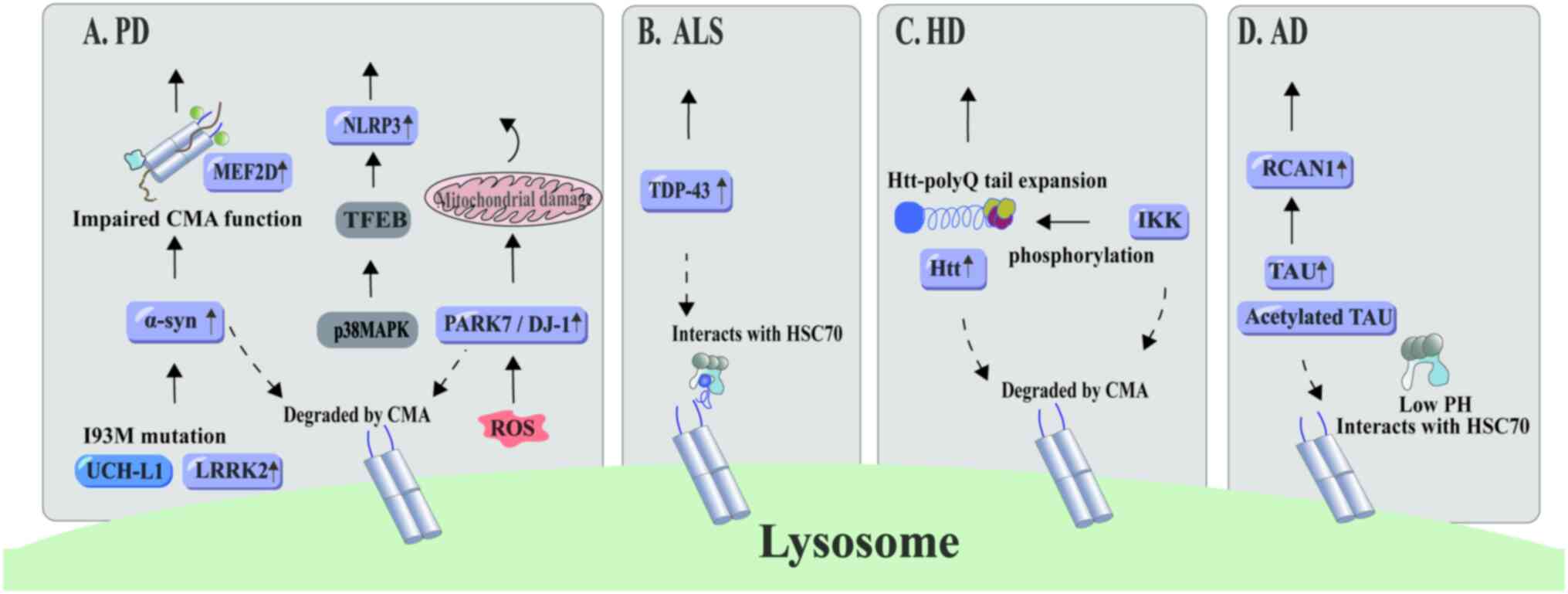 | Figure 5.CMA and neurodegenerative diseases.
(A) CMA involves proteins associated with PD. The primary
pathogenic proteins in PD, α-synuclein and LRRK2, are degraded via
CMA. Mutations in ubiquitin C-terminal hydrolase-L1 impair CMA
functionality, leading to α-synuclein accumulation. Upon ROS
activation, neuronal cells exposed to mitochondrial toxins show a
CMA-driven inclination to degrade PARK7/DJ-1. This may exacerbate
PD progression if CMA functionality declines. Additionally, the
P38-TFEB signaling pathway limits CMA-mediated NLRP3 inflammasome
degradation, thereby stimulating glial cell activity. (B) TDP-43, a
key protein in amyotrophic lateral sclerosis, undergoes degradation
through CMA. (C) After phosphorylation by inflammatory kinase IKK,
Huntington's protein becomes a target for CMA-mediated degradation.
(D) In an acidic environment, acetylated intraneuronal
microtubule-binding protein TAU forms a stable complex with HSC70,
which facilitates its CMA-mediated degradation. Concurrently,
calmodulin phosphatase 1 regulator undergoes degradation through
the CMA lysosomal pathway. The figure was generated using Adobe
Illustrator 2021, version 25.0, by Adobe Inc. PD, Parkinson's
disease; NLRP3, NLR family pyrin domain containing 3; MEF2D,
myocyte enhancer factor 2D; CMA, chaperone-mediated autophagy;
TFEB, transcription factor EB; PARK7/DJ-1, Parkinsonism associated
deglycase; UCH-L1, ubiquitin C-terminal hydrolase L1; LRRK2,
leucine-rich repeat kinase 2; ROS, reactive oxygen species; ALS,
amyotrophic lateral sclerosis; TDP-43, TAR DNA-binding protein 43;
HSC70, heat shock cognate 70; HD, Huntington's disease; IKK, IκΒ
kinase; Htt, huntingtin; AD, Alzheimer's disease; RCAN1, regulator
of calcineurin 1. |
Alzheimer's disease (AD) is the most prevalent form
of dementia in the elderly (173), primarily characterized by
extracellular amyloid-β (Aβ) deposition and intracellular
neurofibrillary tangles of TAU. CHIP, a ubiquitin ligase that
interacts directly with Hsp70/90, exhibits neuroprotective
properties (174,175), and can inhibit toxicities linked
to abnormal protein accumulation in Drosophila and mouse
disease models (176). Under
normal circumstances, TAU is eliminated by macroautophagy, and
acetylated TAU is degraded via CMA. Pathogenic TAU mutants that are
resistant to CMA associate with lysosomal membranes, thereby
interfering with the degradation of other proteins (177). Effective lysosomal translocation
of TAU requires its binding with HSC70 in acidic environments.
However, continuous accumulation of acetylated TAU affects the pH
sensitivity of HSC70 binding, subsequently slowing down CMA
(178). While TAU's affinity for
isolated lysosomes may not be perfectly tailored for CMA substrate
interaction, truncated TAU mutants do not completely translocate
into the lysosomal lumen for degradation, despite the existence of
two CMA targeting motifs in TAU C-terminal region (179). Acetylation of soluble TAU
represents an early event in neurodegeneration, and blockade of CMA
exacerbates the proliferation of pathogenic TAU in mice, worsening
the disease state (178). RCAN1
plays a vital role in the pathogenesis of AD, with its expression
being significantly increased in AD brains. In addition to
degradation through the ubiquitin-proteasome pathway, RCAN1, which
possesses two CMA recognition motifs, undergoes lysosomal
degradation via CMA (180).
Inhibiting the intracellular degradation of RCAN1 further
diminishes calcineurin-NFAT activity.
PD ranks as the second most prevalent
neurodegenerative disorder worldwide (181). In the substantia nigra of
individuals affected by PD, LAMP2A and HSC70 levels are
significantly reduced (182),
highlighting CMA's role in the disease's etiology. Both α-synuclein
and LRRK2 have been identified as major pathogenic proteins in PD
and as substrates for CMA (167,183). Mutations in LRRK2 are among the
leading causes of hereditary neurodegenerative disorders. The most
common pathogenic mutation in LRRK2, G2019S, exhibits slowed
degradation and can inhibit CMA by disrupting the formation of
translocation complexes at the lysosomal membrane, leading to
impaired degradation of LRRK2 within lysosomes (167). Furthermore, other proteins
associated with PD also engage in CMA, reinforcing the relevance of
CMA dysregulation in the progression of PD. Myocyte enhancer factor
2D (MEF2D), critical for neuronal survival, relocates from the
nucleus to the cytosol for degradation in lysosomes. Elevated
levels of MEF2D in α-synuclein transgenic mice and the brains of
patients with PD indicate cytosolic degradation of MEF2D, which
interacts with HSC70 via CMA. However, this degradation process is
hindered by α-synuclein, resulting in its cytosolic accumulation
(184,185). Mutations in ubiquitin
carboxy-terminal hydrolase L1 (UCH-L1) have been linked to familial
PD, with observed changes in dopaminergic neurons in mice carrying
UCH-L1 mutations (186). UCH-L1,
functioning in the CMA pathway, interacts with LAMP2A and HSC70.
The I93M mutation causes structural instability, leading to
abnormally enhanced interactions, thus impairing CMA function and
facilitating the accumulation of α-synuclein proteins (155,187).
Amyotrophic lateral sclerosis (ALS) is a motor
neuron disorder characterized by the accumulation of misfolded
proteins within motor neurons, resulting in progressive paralysis
and eventual mortality. TAR DNA-binding protein 43 (TDP-43; also
known as TARDBP) is a pathological hallmark of neurodegenerative
diseases, with the majority of mutation-driven amyotrophic lateral
sclerosis cases leading to increased intrinsic aggregation of
TDP-43. Previously, it has been shown that TDP-43 is associated
with human HSC70 (HSPA8) in mammalian cells (188). Under chronic stress, the
Drosophila adult HSC70-4 protein forms complexes with TDP-43
in motor neurons (189). Both
naturally occurring and overexpressed TDP-43 are degraded through
macroautophagy and the proteasomal system, whereas ubiquitinated
TDP-43 undergoes degradation via the CMA pathway, interacting with
HSC70 (190). Ormeño et al
(191) demonstrated that TDP-43
could be degraded both in vivo and in vitro via CMA,
with the inhibition of CMA significantly inducing TDP-43
aggregation. However, in later stages of aggregation, TDP-43
affects LAMP2A peri-nuclear localization, leading to CMA-related
lysosomal dysfunction (191). In
peripheral blood mononuclear cells of patients with ALS, reduced
HSC70 mRNA and protein levels, decreased LAMP2A, and stable TDP-43
mRNA were observed, suggesting an elevation in TDP-43 protein
levels attributed to diminished protein clearance rates (192).
Huntington's disease is an autosomal dominant
disorder resulting from polyQ tail expansion due to mutations in
the Huntingtin (Htt) gene. Despite Htt not directly containing the
‘KFERQ’ HSC70 binding motif within its sequence, it can be
phosphorylated by the inflammatory IκB kinase (IKK). Following
phosphorylation, the N-terminal fragment of Htt may acquire a
KFERQ-like motif, affecting the levels of phosphorylated Htt
through CMA degradation (193).
In conditions where LAMP2A expression is high, mutated Htt can be
degraded prior to inducing toxicity; however, with advancing age
and reduced LAMP2A, mutated Htt accumulates, exacerbating the onset
of neurodegenerative diseases (193). One mechanism to mitigate the
toxicity of mutated Htt protein involves HSP40 and HSP70
chaperones, which capture it through hydrophobic interactions and
redirect it to lysosomes for degradation (194). Increased expression of HSP70
significantly improves motor function in transgenic motor neuron
disease mice by offering protection against polyQ (195). Hsp70s prevent the oligomerization
of Htt protein, and the expression of Hsp70s progressively inhibits
the development of Huntington's disease in mouse models by
counteracting Htt conformational effects (196). Consequently, upregulation of
Hsp70 can mitigate the toxicity of mutated Htt and restore various
physiological protein functions. In addition to the macroautophagy
degradation pathway for Htt, CMA-related proteins interact with Htt
fragments (Htt-552). Htt-552 can be recognized by HSC70 and
transported to lysosomes through LAMP2A, and alterations in
CMA-related proteins also influence Htt accumulation (197).
Cardiovascular diseases (CVDs)
CVDs continue to be the primary cause of global
mortality, representing 1/3 of all mortalities (198). Atherosclerosis is the main risk
factor. Areas abundant in lipids, necrosis and macrophages, along
with increased cytokine secretion and production of nitric oxide
(NO) and ROS, establish a pro-inflammatory and oxidative
environment. This environment induces the dedifferentiation of
vascular smooth muscle cells (VSMCs) from a contractile phenotype
to an activated, secretory and migratory phenotype. Activated VSMCs
migrate to the intima, further exacerbating inflammation, oxidative
stress, and the deposition of collagen and elastin proteins in the
fibrous cap (198). In mouse
models, aortic plaque associated with necrosis significantly
enlarged following the knockout of LAMP2A; however, increased CMA
activity markedly ameliorated these symptoms (199). Similarly, defects in CMA led to
reduced lipid metabolism, impaired clotting function, decreased
VSMC resistance to lipid toxicity and increased dedifferentiation
processes in mice, thereby further elevating the risk of
atherosclerosis (199).
As atherosclerosis progresses, CMA activity becomes
compromised. Macrophages are the primary cell type expressing CMA
biomarkers, with observed co-localization of LAMP2A and macrophages
in murine atherosclerotic lesions and colitis models (138). The NLRP3 inflammasome maintains
balance through CMA-mediated degradation. Inactivation of CMA in
macrophages leads to NLRP3 inflammasome overactivation, promoting
interleukin 1 beta (IL-1β) secretion, vascular inflammation and the
progression of atherosclerosis (138). Enhancing CMA activity may improve
systemic metabolic parameters and vascular function, and inhibit
pathological changes in VSMCs and macrophages (199). These strategies show promise as
innovative therapeutic approaches for preventing CVDs (200). NO, produced by endothelial NO
synthase (eNOS), helps prevent myocardial ischemia-reperfusion
injury. Mutations in NOS residues inhibit CMA, leading to further
damage. Under hypoxia-reoxygenation conditions, glutathionylated
eNOS, targeted by the HSC90 chaperone, reveals specific sequences,
leading to its transport to LAMP2A vesicles for degradation. Prompt
deglutathionylation can prevent CMA, thus ameliorating myocardial
ischemia (201). Hypoxia is a
major factor in myocardial infarction injuries. In samples from
patients with ischemic heart failure, LAMP2A expression levels were
found to be elevated. Overexpression of LAMP2A has been shown to
reduce hypoxia-induced cardiomyocyte apoptosis by 50%, indicating
the potential cardioprotective effects of CMA activation in
ischemic heart diseases (202)
(Fig. 6).
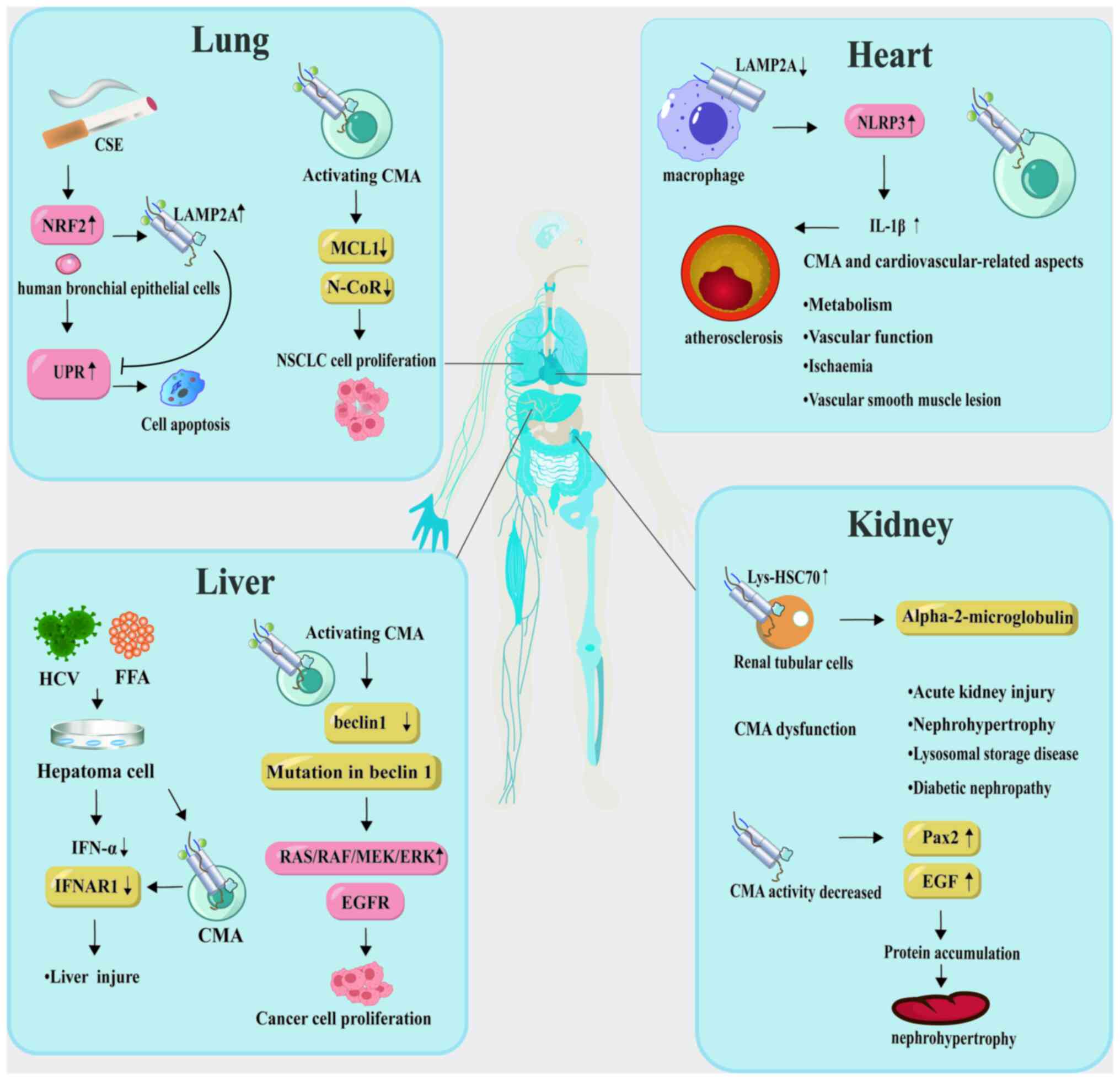 | Figure 6.CMA dysfunction contributes to
multiple diseases. In lung diseases, exposure to cigarette smoke
extract is known to induce an increase in the UPR in human
bronchial epithelial cells, thereby promoting apoptosis and
ultimately contributing to the development of chronic obstructive
pulmonary disease. Overexpression of LAMP2A has been shown to
mitigate this UPR and can potentially reverse apoptosis. CMA
enables the degradation of N-CoR, a factor that is associated with
MCL1 errors, thereby promoting the survival of NSCLC cells.
Macrophages are presumed to be the primary cell type expressing CMA
markers in cardiovascular diseases. A reduction in CMA activity
leads to the activation of the NLRP3 inflammasome in macrophages,
subsequently promoting IL-1β secretion, vascular inflammation and
atherosclerosis progression. Furthermore, CMA is instrumental in
the functionality and metabolism of new blood vessels. Chronic HCV
infection markedly increases the risk of hepatic steatosis.
Co-culturing HCV with free fatty acids leads to downregulation of
IFNAR1, subsequently inhibiting the antiviral activity of IFN-α and
inducing hepatic steatosis. IFNAR1 is capable of colocalizing with
LAMP2A for degradation through CMA. Activation of CMA results in
the degradation of beclin 1, thereby inhibiting the endocytosis of
EGFR and subsequently activating EGFR signaling. This causes
autophagosome-lysosome damage and the activation of downstream
carcinogenic signaling pathways such as RAS/RAF/MEK/ERK, eventually
leading to the growth of cirrhotic hepatocellular carcinoma.
Various forms of nephropathy are associated with abnormally high
levels of lys-HSC70, and CMA upregulates toxic modifications in
renal tubular cells in response to α2-microglobulin. Decreased
activity of LAMP2A leads to increased expression of the nuclear
transcription factor Pax2, thereby upregulating the proliferation
of renal tubular cells and promoting hypertrophy. Macrophages are
presumed to be the primary cell type expressing CMA markers in
cardiovascular diseases. A reduction in CMA activity leads to the
activation of the NLRP3 inflammasome in macrophages, subsequently
promoting IL-1β secretion, vascular inflammation and
atherosclerosis progression. Furthermore, CMA is instrumental in
the functionality and metabolism of new blood vessels. Chronic HCV
infection markedly increases the risk of hepatic steatosis.
Co-culturing HCV with free fatty acids leads to downregulation of
IFNAR1, subsequently inhibiting the antiviral activity of IFN-α and
inducing hepatic steatosis. IFNAR1 is capable of colocalizing with
LAMP2A for degradation through CMA. Activation of CMA results in
the degradation of beclin 1, thereby inhibiting the endocytosis of
EGFR and subsequently activating EGFR signaling. This causes
autophagosome-lysosome damage and the activation of downstream
carcinogenic signaling pathways such as RAS/RAF/MEK/ERK, eventually
leading to the growth of cirrhotic hepatocellular carcinoma.
Various forms of nephropathy are associated with abnormally high
levels of lys-HSC70, and CMA upregulates toxic modifications in
renal tubular cells in response to α2-microglobulin. Decreased
activity of LAMP2A leads to increased expression of the nuclear
transcription factor Pax2, thereby upregulating the proliferation
of renal tubular cells and promoting hypertrophy. The figure was
generated using Adobe Illustrator 2021, version 25.0, by Adobe Inc.
CSE, cigarette smoke extract; CMA, chaperone-mediated autophagy;
NRF2, nuclear factor, erythroid 2; LAMP2A, lysosome-associated
membrane protein 2A; UPR, unfolded protein response; MCL1, myeloid
cell leukemia 1; N-CoR, nuclear receptor co-repressor; NSCLC,
non-small cell lung cancer; NLRP3, NLR family pyrin domain
containing 3; IL-1β, interleukin 1 beta; HCV, hepatitis C virus;
FFA, free fatty acid; IFN-α, interferon alpha; IFNAR1, interferon
alpha and beta receptor subunit 1; EGFR, epidermal growth factor
receptor; HSC70, heat shock cognate 70; Pax2, paired box 2. |
Liver diseases
HCV is a major cause of chronic hepatitis, liver
cirrhosis and HCC. Characterized as an enveloped, positive-sense,
single-stranded RNA virus, HCV belongs to the Flaviviridae
family (203). In total, ~20% of
infected adults spontaneously clear the HCV infection, while the
remaining 80% develop chronic hepatitis, with increased risks of
cirrhosis, HCC and end-stage liver disease (204). The HCV genome consists of 3,010
kb RNA, encoding a polyprotein of 96 amino acids. This polyprotein
undergoes cleavage by viral proteases and host signal peptidases,
resulting in three structural proteins essential for viral particle
formation (205), and seven
non-structural proteins involved in viral replication (206). Chronic HCV infection
significantly increases the risk of hepatic steatosis. In
vitro studies demonstrated a considerably higher accumulation
of lipid droplets in HCV-infected hepatoma cells compared with
uninfected cells. Co-culturing HCV with free fatty acids (FFAs)
induces macrovesicular steatosis by downregulating the expression
of interferon (IFN)-α receptors, thereby inhibiting the antiviral
activity of IFN-α. The CMA-targeting amino acid sequence is found
in IFN alpha and beta receptor subunit 1 (IFNAR1) on the IFN-λ
receptor, and autophagy responses induced by HCV and FFA result in
decreased IFNAR1 expression and its selective degradation through
CMA. Activation induced by HCV and FFA leads to increased LAMP2A
protein expression, resulting in the formation of a protein complex
that includes HSC70, LAMP2A and IFNAR1 (207). HCV infection leads to the
downregulation of hepatocyte nuclear factor-1α (HNF-1α) expression,
both post-transcriptionally and translationally, and enhances its
interaction with HSC70. Importantly, the HNF-1α protein includes a
CMA targeting sequence within its POU domain. The HCV NS5A protein
aids in recruiting HSC70 to HNF-1α, directing the lysosomal
membrane-cytosolic LAMP2A recognition protein complex to the
lysosome for degradation (208).
It has been confirmed that HCV-induced degradation of HNF-1α via
CMA suppresses glucose transporter type 2 (GLUT2) gene expression,
leading to reduced surface GLUT2 expression and disrupted glucose
uptake into cells, culminating in glucose metabolic disorders
(209). During cellular responses
induced by microbial stress, autophagy regulation under
pathological conditions becomes essential, and the continual
replication of HCV in hepatocytes promotes autophagy regulation
through CMA activation, thus enhancing cell survival. Sorting nexin
10 (SNX-10), which plays a role in endo-lysosomal transport and
stabilization, regulates CMA activity by enhancing LAMP2A
stability. It does this by inhibiting the interaction with tissue
plasminogen activator in SNX-10 knockout mice, thereby mitigating
alcoholic liver injury and steatosis (210) (Fig.
6).
Renal disease
Elevated levels of lys-HSC70 have been reported in
various nephropathies, yet the significance of these alterations
remains to be elucidated. A previous study showed an upregulation
of CMA in renal tubular epithelial cells in response to toxic
modifications of lipid-binding proteins prevalent in the kidney
such as α-2-microglobulin. Enhanced CMA activity facilitates the
removal of these toxic protein forms prior to intracellular
accumulation (89).
Renal hypertrophy is a potential pathological
condition in the kidney where altered selective protein degradation
by CMA may play a role. This hypertrophic cell growth may originate
from an imbalance between protein synthesis and degradation.
Hypertrophy is a characteristic feature of diabetic kidneys,
characterized by reduced levels of LAMP2A. The nuclear
transcription factor paired box 2 (Pax2), crucial for renal cell
proliferation regulation, is overexpressed due to decreased CMA
activity, promoting tubular epithelial cell growth and maintaining
the hypertrophic state (211). In
conditions such as diabetes or acidosis, protein degradation in the
kidney is inhibited. The growth factor in diabetic nephropathy,
epidermal growth factor, does not affect the proteasomal
degradation pathway, implying that 30% of kidney proteins possess
the CMA targeting sequence KFERQ (212). In acute rat diabetic models,
streptozotocin-induced reductions in renal cortical protein
degradation were observed, along with increases in the quantity of
KFERQ-containing proteins. In vivo, CMA protein degradation
levels were inhibited (211),
suggesting that a reduction in CMA may primarily underlie diabetic
renal hypertrophy.
In lysosomal storage disorders (LSDs), the
accumulation of metabolites leads to cellular dysfunction.
Cystinosis, an LSD, is caused by defects in the cystine transporter
protein cystinosin (CTNS), primarily affecting the kidney. In
cystinosis, Rab11 and RILP are significantly downregulated,
resulting in decreased stability of LAMP2A and its mislocalization,
which disrupts CMA autophagic balance and directly impacts organ
function in patients. The application of CMA activators in
CTNS-deficient mice led to an increase in Rab11 expression and
transport, suggesting that upregulating CMA could be a potential
therapeutic strategy for cystinosis (213). Oxidative stress, production of
ROS and accumulation of lipid peroxidation are key factors in acute
kidney injury. CMA activation serves as a primary response
mechanism to oxidative stress and is closely linked to ferroptosis.
However, additional research is required to elucidate the interplay
between CMA and renal diseases (Fig.
6).
Pulmonary diseases
Chronic obstructive pulmonary disease (COPD) is
primarily caused by smoke exposure, which amplifies both
macroautophagy and CMA in mice (214). Exposure to smoke leads to
oxidative modifications in pulmonary proteins, resulting in the
accumulation of misfolded proteins and intensified unfolded protein
response (UPR). This process is closely associated with the
development of COPD and pulmonary inflammation (215). Therefore, CMA may play a crucial
role in the pathogenesis of COPD. Cigarette smoke extract (CSE)
induces LAMP2A expression and CMA activation in human bronchial
epithelial cells via an Nrf2-dependent mechanism. Knockdown of
LAMP2A intensifies the UPR due to CMA inhibition and is coupled
with enhanced cell apoptosis during CSE exposure. Overexpressing
LAMP2A reverses this apoptosis. This suggests a potential causal
association between Nrf2-regulated impairment of CMA and lung
epithelial cell apoptosis mediated by an enhanced UPR, contributing
to the development of COPD. Consequently, activating CMA may
represent a therapeutic approach for COPD (216). The etiological mechanisms of COPD
are diverse. Reduced autophagic function leads to bronchial
epithelial cell senescence. Both macroautophagy and CMA are
reported to have compensatory functions. Following CSE stimulation,
macroautophagy activity peaks at 24 h, whereas LAMP2A activity
continues for 72 h, suggesting a potential role for CMA in cell
autophagy during disease progression (217). CMA is involved in the mediation
of the onset of ferroptosis. The triterpenoid compound CDDO and
HSP90 obstruct a key complex involved in necrotic apoptosis, thus
blocking ferroptosis via CMA inhibition (82). However, the exact role of CMA in
regulating ferroptosis in relation to COPD pathogenesis is yet to
be fully understood (Fig. 6).
Therapeutic strategies targeting CMA
Increasing evidence has linked CMA with the
pathogenesis of various organ systems, making CMA modulation a
potential therapeutic target. Therapies based on the mechanism of
CMA may have therapeutic value for age-related diseases through CMA
intervention. In neurodegenerative diseases, the decline of CMA has
been closely associated with the abnormal aggregation of pathogenic
proteins in neurons. CMA can regulate amyloid proteins, potentially
serving as a therapeutic approach for AD. A previous study revealed
that resveratrol, a polyphenol found in red wine, can accelerate
the degradation of toxic Aβ through a CMA-dependent pathway. In a
transgenic Caenorhabditis elegans model expressing Aβ1-42,
resveratrol significantly inhibited paralysis by suppressing the
expression of LAMP2A (218).
Additionally, bioactive compounds derived from medicinal plants,
such as dihydromyricetin and salvianolic acid B, suppress the
accumulation of α-synuclein, enhance the CMA pathway, and exert
anti-inflammatory effects (219).
Chronic caffeine treatment counteracts pathologies induced by
mutant α-synuclein, such as dopaminergic neuronal cell death, while
preserving normal autophagic processes in the striatum, thereby
correcting defects in macroautophagy and CMA of α-synuclein
(220). MA activators such as
CA77.1 and QX77 have demonstrated significant therapeutic potential
by enhancing LAMP2A expression, aiding in the degradation of
pathogenic proteins, and showing efficacy in models of
neurodegenerative diseases (55).
CMA exhibits a dual role in cancer, making it an
interesting therapeutic target. On one hand, overactivation of CMA
can promote tumor cell survival and metastasis. Inhibiting CMA, for
example by reducing LAMP2A levels, can suppress tumor growth and
spread. Conversely, activating CMA can lead to the degradation of
specific oncogenic mutants, thereby inhibiting tumor growth. For
instance, ManA promotes the interaction between CMA substrates and
LAMP2A, slowing tumor growth and intensifying the anti-tumorigenic
action of activated CMA under hypoxia. Additionally, compounds such
as chloroquine, which neutralize lysosomal pH, regulate lysosomal
activity and have been applied in clinical trials for cancer
treatment (221,222). However, CMA activation requires a
combination of nutritional stress and autophagy blockade, which may
lead to side effects.
The role of CMA in autoimmune diseases has also
garnered widespread attention. In lupus erythematosus mouse models,
increased levels of LAMP2A and HSC70 result in overactivation of
CMA. The immunomodulatory agent phosphorylated peptide P140 binds
to HSPA8, targeting CMA, and demonstrates potential as an inhibitor
for lupus treatment. P140 has shown promising results in phase II
and III clinical trials, indicating its efficacy and safety in
reducing lupus disease activity and maintaining remission (223,224).
CMA is crucial for the self-renewal and
differentiation of stem cells. Studies on embryonic and adult stem
cells may lay the foundation for potential new treatment methods
and applications in clinical cell therapy. In metabolic diseases,
the regulation of CMA also holds potential therapeutic value.
Metformin, a commonly used type 2 diabetes medication, is a novel
activator of CMA, promoting the phosphorylation of the key CMA
mediator HSC70 via the TAK1-IKKα/β signaling cascade. Enhancing CMA
activity can improve metabolic disorders, reduce fat accumulation,
and thereby benefit diabetes and related metabolic diseases
(225,226).
As the understanding of the CMA mechanism deepens,
more innovative CMA modulators are being developed. For example,
the natural compound trehalose modulates autophagy via transient
lysosomal enlargement and membrane permeabilization, enhancing
LAMP2A expression (225). These
modulators reveal potential not only in laboratory studies but also
for future clinical applications.
Overall, research on CMA as a therapeutic target
continues to deepen, and there is hope that precise regulation of
the CMA pathway will provide new therapeutic strategies for various
diseases. However, translating these research findings into
clinical treatments still presents significant challenges,
requiring more clinical trials and studies to verify their safety
and efficacy. By continuously exploring the biological functions of
CMA and its roles in different diseases, researchers can aim to
open new therapeutic avenues and improve patient outcomes.
Discussion
Backer and Dice (30) were the first to elucidate the
selective autophagy process of CMA based on the KFERQ motif.
Subsequently, the work of Cuervo and Dice (35) highlighted the crucial roles of the
chaperone-substrate complex and LAMP2A in the CMA pathway. Recent
advancements have shed light on the molecular mechanisms and
pathophysiological implications of CMA. Protein quality control is
fundamental to CMA, orchestrating various cellular functions by
regulating the quality and abundance of target proteins. At the
molecular level, CMA plays a crucial role in maintaining organismal
homeostasis, cellular energy balance and protein degradation
mechanisms. Alterations in CMA activity can predispose individuals
to disease pathogenesis. Although current therapeutic targets for
autophagy in human diseases are limited, methodologies for CMA
research are undergoing refinement. Kaushik and Cuervo (55) provided detailed assays for
assessing CMA functionality in cells and tissues. Assessing LAMP2A
levels in cultured cells or tissues is one of the most effective
and reliable methods for measuring CMA activity. Enhanced CMA
activity positively correlates with elevated levels of lysosomal
LAMP2A and HSC70. Another diagnostic technique involves measuring
the translocation and degradation of CMA substrates in purified
CMA-rich lysosomes from target cells or tissues (227). Substrate turnover can be detected
by incubating intact lysosomes with CMA substrates in the absence
of exogenous lysosomal protease inhibitors. Subsequent
immunoblotting and quantification enable the identification of
lysosomes specific to CMA. The use of fluorescent CMA reporter
genes aids in assessing CMA substrate delivery and degradation
(37). Dong et al (228), using a transgenic mouse model
expressing a CMA reporter, validated its effectiveness for
spatiotemporal monitoring of CMA at the cellular and tissue levels.
This image-based approach enables the differentiation of CMA
activity disparities, even in organs where hepatocytes predominate
by 80%, as shown in the KFERQ-Dendra mouse model.
CMA is a specialized type of autophagy, differing
significantly from other autophagic pathways in substrate
selectivity and mechanism. The past decade has significantly
enhanced the present understanding of the intricate molecular
underpinnings of CMA and its wide-ranging physiological and
pathophysiological implications. At the core of CMA is its unique
mechanism of substrate recognition. Understanding the precise
determinants of this specificity can inform therapeutic strategies
targeting CMA activity manipulation. The dynamic regulation of
LAMP2A, including its rapid turnover and factors affecting its
stability, has been a focus of intense study, shedding light on how
CMA activity is modulated under various physiological and stress
conditions. In conclusion, although the understanding of molecular
CMA has greatly expanded, several aspects remain unexplored. This
includes identifying the full range of CMA substrates,
understanding the detailed regulation of LAMP2A, and developing
targeted therapeutics that precisely modulate CMA activity. As
research in this field progresses, it promises to yield novel
insights into cellular homeostasis mechanisms and potential
therapeutic strategies for a diverse array of pathologies.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Scientific and
Technological Research Project of Henan (grant no. 232102311212)
and the China Postdoc Innovation Talent Support Program (grant no.
BX2021093).
Availability of data and materials
Not applicable.
Authors' contributions
JW conceptualized the study, conducted the project
administration and acquired the funding. HJ wrote the original
draft, reviewed and edited the manuscript, performed the
visualization and supervised the study. Both authors read and
approved the final manuscript. Data authentication is not
applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
CMA
|
chaperone-mediated autophagy
|
|
VTC
|
vacuolar transporter chaperone
|
|
ATG5
|
autophagy-related 5
|
|
ULK1
|
Unc-51 like autophagy activating
kinase
|
|
PAS
|
phagophore assembly site
|
|
FIP200
|
FAK family kinase-interacting protein
of 200 kDa
|
|
mTOR
|
mammalian target of rapamycin
|
|
ER
|
endoplasmic reticulum
|
|
LAMP2A
|
lysosome-associated membrane protein
2A
|
|
HSC70
|
heat shock cognate 70
|
|
HSP70
|
heat shock protein 70
|
|
cyt-HSC70
|
cytoplasmic HSC70
|
|
PKM2
|
pyruvate kinase M2
|
|
PLIN2
|
perilipin 2
|
|
Htt
|
huntingtin
|
|
RNA
|
ribonucleic acid
|
|
ROS
|
reactive oxygen species
|
|
Nrf2
|
nuclear factor, erythroid 2
|
|
GFAP
|
glial fibrillary acidic protein
|
|
Akt
|
protein kinase B
|
|
IKK
|
IκB kinase
|
|
ATRA
|
all-trans retinoic acid
|
|
AMPK
|
AMP-activated protein kinase
|
|
N-CoR
|
Nuclear receptor co-repressor
|
|
SIRT3
|
sirtuin 3
|
|
MAPK
|
mitogen-activated protein kinase
|
|
NLRP3
|
NLR family pyrin domain containing
3
|
|
TFEB
|
transcription factor EB
|
|
TCR
|
T cell receptor
|
|
NFAT
|
nuclear factor of activated T
cells
|
|
PI3K/Akt
|
phosphoinositide 3-kinases/protein
kinase B
|
|
MYC
|
MYC proto-oncogene, BHLH
transcription factor
|
|
RND3
|
Rho family GTPase 3
|
|
CDKS
|
cyclin-dependent kinases
|
|
EF2
|
elongation factor 2
|
|
CHK1
|
checkpoint kinase 1
|
|
HIF-1α
|
hypoxia-inducible factor 1 alpha
|
|
IFITM3
|
interferon-induced transmembrane
protein 3
|
|
C11orf54
|
chromosome 11 open reading frame
54
|
|
TGFβ
|
transforming growth factor beta;
|
References
|
1
|
De Duve C and Wattiaux R: Functions of
lysosomes. Annu Rev Physiol. 28:435–492. 1966. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Etlinger JD and Goldberg AL: A soluble
ATP-dependent proteolytic system responsible for the degradation of
abnormal proteins in reticulocytes. Proc Natl Acad Sci USA.
74:54–58. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bright NA, Davis LJ and Luzio JP:
Endolysosomes are the principal intracellular sites of acid
hydrolase activity. Curr Biol. 26:2233–2245. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mizushima N: Autophagy: Process and
function. Genes Dev. 21:2861–2873. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Levine B and Kroemer G: Biological
functions of autophagy genes: A disease perspective. Cell.
176:11–42. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mizushima N and Komatsu M: Autophagy:
Renovation of cells and tissues. Cell. 147:728–741. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Klionsky DJ: Autophagy: From phenomenology
to molecular understanding in less than a decade. Nat Rev Mol Cell
Biol. 8:931–937. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nakatogawa H: Mechanisms governing
autophagosome biogenesis. Nat Rev Mol Cell Biol. 21:439–458. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tsukada M and Ohsumi Y: Isolation and
characterization of autophagy-defective mutants of Saccharomyces
cerevisiae. FEBS Lett. 333:169–174. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Klionsky DJ, Cregg JM, Dunn WA Jr, Emr SD,
Sakai Y, Sandoval IV, Sibirny A, Subramani S, Thumm M, Veenhuis M
and Ohsumi Y: A unified nomenclature for yeast autophagy-related
genes. Dev Cell. 5:539–545. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Feng Y, He D, Yao Z and Klionsky DJ: The
machinery of macroautophagy. Cell Res. 24:24–41. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chan EYW, Longatti A, McKnight NC and
Tooze SA: Kinase-inactivated ULK proteins inhibit autophagy via
their conserved C-terminal domains using an Atg13-independent
mechanism. Mol Cell Biol. 29:157–171. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Suzuki K, Kirisako T, Kamada Y, Mizushima
N, Noda T and Ohsumi Y: The pre-autophagosomal structure organized
by concerted functions of APG genes is essential for autophagosome
formation. EMBO J. 20:5971–5981. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Griffey CJ and Yamamoto A: Macroautophagy
in CNS health and disease. Nat Rev Neurosci. 23:411–427. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mari M, Griffith J, Rieter E, Krishnappa
L, Klionsky DJ and Reggiori F: An Atg9-containing compartment that
functions in the early steps of autophagosome biogenesis. J Cell
Biol. 190:1005–1022. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hara T, Takamura A, Kishi C, Iemura S,
Natsume T, Guan JL and Mizushima N: FIP200, a ULK-interacting
protein, is required for autophagosome formation in mammalian
cells. J Cell Biol. 181:497–510. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hosokawa N, Hara T, Kaizuka T, Kishi C,
Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, et
al: Nutrient-dependent mTORC1 association with the
ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell.
20:1981–1991. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Weidberg H, Shvets E and Elazar Z:
Biogenesis and cargo selectivity of autophagosomes. Annu Rev
Biochem. 80:125–156. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yokota S and Dariush Fahimi H: Degradation
of excess peroxisomes in mammalian liver cells by autophagy and
other mechanisms. Histochem Cell Biol. 131:455–458. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kundu M, Lindsten T, Yang CY, Wu J, Zhao
F, Zhang J, Selak MA, Ney PA and Thompson CB: Ulk1 plays a critical
role in the autophagic clearance of mitochondria and ribosomes
during reticulocyte maturation. Blood. 112:1493–1502. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Oku M and Sakai Y: Three distinct types of
microautophagy based on membrane dynamics and molecular
machineries. Bioessays. 40:e18000082018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Uttenweiler A, Schwarz H, Neumann H and
Mayer A: The vacuolar transporter chaperone (VTC) complex is
required for microautophagy. Mol Biol Cell. 18:166–175. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Uttenweiler A, Schwarz H and Mayer A:
Microautophagic vacuole invagination requires calmodulin in a
Ca2+-independent function. J Biol Chem. 280:33289–33297. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Müller O, Sattler T, Flötenmeyer M,
Schwarz H, Plattner H and Mayer A: Autophagic tubes: Vacuolar
invaginations involved in lateral membrane sorting and inverse
vesicle budding. J Cell Biol. 151:519–528. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Epple UD, Suriapranata I, Eskelinen EL and
Thumm M: Aut5/Cvt17p, a putative lipase essential for
disintegration of autophagic bodies inside the vacuole. J
Bacteriol. 183:5942–5955. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yang Z and Klionsky DJ: Permeases recycle
amino acids resulting from autophagy. Autophagy. 3:149–150. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Roberts P, Moshitch-Moshkovitz S, Kvam E,
O'Toole E, Winey M and Goldfarb DS: Piecemeal microautophagy of
nucleus in Saccharomyces cerevisiae. Mol Biol Cell. 14:129–141.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kawamura N, Sun-Wada GH, Aoyama M, Harada
A, Takasuga S, Sasaki T and Wada Y: Delivery of endosomes to
lysosomes via microautophagy in the visceral endoderm of mouse
embryos. Nat Commun. 3:10712012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kaushik S and Cuervo AM:
Chaperone-mediated autophagy: A unique way to enter the lysosome
world. Trends Cell Biol. 22:407–417. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dice JF, Walker CD, Byrne B and Cardiel A:
General characteristics of protein degradation in diabetes and
starvation. Proc Natl Acad Sci USA. 75:2093–2097. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Backer JM and Dice JF: Covalent linkage of
ribonuclease S-peptide to microinjected proteins causes their
intracellular degradation to be enhanced during serum withdrawal.
Proc Natl Acad Sci USA. 83:5830–5834. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chiang HL and Dice JF: Peptide sequences
that target proteins for enhanced degradation during serum
withdrawal. J Biol Chem. 263:6797–6805. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chiang HL, Terlecky SR, Plant CP and Dice
JF: A role for a 70-kilodalton heat shock protein in lysosomal
degradation of intracellular proteins. Science. 246:382–385. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cuervo AM and Dice JF: A receptor for the
selective uptake and degradation of proteins by lysosomes. Science.
273:501–503. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cuervo AM and Dice JF: Age-related decline
in chaperone-mediated autophagy. J Biol Chem. 275:31505–31513.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Dice JF, Chiang HL, Spencer EP and Backer
JM: Regulation of catabolism of microinjected ribonuclease A.
Identification of residues 7–11 as the essential pentapeptide. J
Biol Chem. 261:6853–6859. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Koga H, Martinez-Vicente M, Macian F,
Verkhusha VV and Cuervo AM: A photoconvertible fluorescent reporter
to track chaperone-mediated autophagy. Nat Commun. 2:3862011.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Dice JF: Peptide sequences that target
cytosolic proteins for lysosomal proteolysis. Trends Biochem Sci.
15:305–309. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kirchner P, Bourdenx M, Madrigal-Matute J,
Tiano S, Diaz A, Bartholdy BA, Will B and Cuervo AM: Proteome-wide
analysis of chaperone-mediated autophagy targeting motifs. PLoS
Biol. 17:e30003012019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Glick BS: Can Hsp70 proteins act as
force-generating motors? Cell. 80:11–14. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bandyopadhyay U, Kaushik S, Varticovski L
and Cuervo AM: The chaperone-mediated autophagy receptor organizes
in dynamic protein complexes at the lysosomal membrane. Mol Cell
Biol. 28:5747–5763. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Gong Z, Tasset I, Diaz A, Anguiano J, Tas
E, Cui L, Kuliawat R, Liu H, Kühn B, Cuervo AM and Muzumdar R:
Humanin is an endogenous activator of chaperone-mediated autophagy.
J Cell Biol. 217:635–647. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Massey AC, Kaushik S, Sovak G, Kiffin R
and Cuervo AM: Consequences of the selective blockage of
chaperone-mediated autophagy. Proc Natl Acad Sci USA.
103:5805–5810. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Majeski AE and Dice JF: Mechanisms of
chaperone-mediated autophagy. Int J Biochem Cell Biol.
36:2435–2444. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Bimston D, Song J, Winchester D, Takayama
S, Reed JC and Morimoto RI: BAG-1, a negative regulator of Hsp70
chaperone activity, uncouples nucleotide hydrolysis from substrate
release. EMBO J. 17:6871–6878. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Gough NR, Hatem CL and Fambrough DM: The
family of LAMP-2 proteins arises by alternative splicing from a
single gene: Characterization of the avian LAMP-2 gene and
identification of mammalian homologs of LAMP-2b and LAMP-2c. DNA
Cell Biol. 14:863–867. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chi C, Leonard A, Knight WE, Beussman KM,
Zhao Y, Cao Y, Londono P, Aune E, Trembley MA, Small EM, et al:
LAMP-2B regulates human cardiomyocyte function by mediating
autophagosome-lysosome fusion. Proc Natl Acad Sci USA. 116:556–565.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Fujiwara Y, Furuta A, Kikuchi H, Aizawa S,
Hatanaka Y, Konya C, Uchida K, Yoshimura A, Tamai Y, Wada K and
Kabuta T: Discovery of a novel type of autophagy targeting RNA.
Autophagy. 9:403–409. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Schnebert S, Goguet M, Vélez EJ, Depincé
A, Beaumatin F, Herpin A and Seiliez I: Diving into the
evolutionary history of HSC70-linked selective autophagy pathways:
Endosomal microautophagy and chaperone-mediated autophagy. Cells.
11:19452022. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Cuervo AM and Dice JF: Regulation of
lamp2a levels in the lysosomal membrane. Traffic. 1:570–583. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Cuervo AM, Mann L, Bonten EJ, d'Azzo A and
Dice JF: Cathepsin A regulates chaperone-mediated autophagy through
cleavage of the lysosomal receptor. EMBO J. 22:47–59. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Tanaka Y, Guhde G, Suter A, Eskelinen EL,
Hartmann D, Lüllmann-Rauch R, Janssen PM, Blanz J, von Figura K and
Saftig P: Accumulation of autophagic vacuoles and cardiomyopathy in
LAMP-2-deficient mice. Nature. 406:902–906. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Ferreira JV, da Rosa Soares A, Ramalho J,
Máximo Carvalho C, Cardoso MH, Pintado P, Carvalho AS, Beck HC,
Matthiesen R, Zuzarte M, et al: LAMP2A regulates the loading of
proteins into exosomes. Sci Adv. 8:eabm11402022. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Kaushik S, Massey AC and Cuervo AM:
Lysosome membrane lipid microdomains: Novel regulators of
chaperone-mediated autophagy. EMBO J. 25:3921–3933. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Kaushik S and Cuervo AM: The coming of age
of chaperone-mediated autophagy. Nat Rev Mol Cell Biol. 19:365–381.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Duong V and Rochette-Egly C: The molecular
physiology of nuclear retinoic acid receptors. From health to
disease. Biochim Biophys Acta. 1812:1023–1031. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Anguiano J, Garner TP, Mahalingam M, Das
BC, Gavathiotis E and Cuervo AM: Chemical modulation of
chaperone-mediated autophagy by retinoic acid derivatives. Nat Chem
Biol. 9:374–382. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Gomez-Sintes R, Xin Q, Jimenez-Loygorri
JI, McCabe M, Diaz A, Garner TP, Cotto-Rios XM, Wu Y, Dong S,
Reynolds CA, et al: Targeting retinoic acid receptor
alpha-corepressor interaction activates chaperone-mediated
autophagy and protects against retinal degeneration. Nat Commun.
13:42202022. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Tian Y, Liu B, Li Y, Zhang Y, Shao J, Wu
P, Xu C, Chen G and Shi H: Activation of RARα receptor attenuates
neuroinflammation after SAH via promoting M1-to-M2 phenotypic
polarization of microglia and regulating Mafb/Msr1/PI3K-Akt/NF-κB
pathway. Front Immunol. 13:8397962022. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Alers S, Löffler AS, Wesselborg S and
Stork B: Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy:
Cross talk, shortcuts, and feedbacks. Mol Cell Biol. 32:2–11. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Carling D: AMPK signalling in health and
disease. Curr Opin Cell Biol. 45:31–37. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Zhang K, Zhu S, Li J, Jiang T, Feng L, Pei
J, Wang G, Ouyang L and Liu B: Targeting autophagy using
small-molecule compounds to improve potential therapy of
Parkinson's disease. Acta Pharm Sin B. 11:3015–3034. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Kaushik S and Cuervo AM: Degradation of
lipid droplet-associated proteins by chaperone-mediated autophagy
facilitates lipolysis. Nat Cell Biol. 17:759–770. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Schweiger M and Zechner R: Breaking the
barrier-chaperone-mediated autophagy of perilipins regulates the
lipolytic degradation of fat. Cell Metab. 22:60–61. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Zhang T, Liu J, Shen S, Tong Q, Ma X and
Lin L: SIRT3 promotes lipophagy and chaperon-mediated autophagy to
protect hepatocytes against lipotoxicity. Cell Death Differ.
27:329–344. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Zhang T, Liu J, Tong Q and Lin L: SIRT3
Acts as a positive autophagy regulator to promote lipid
mobilization in adipocytes via activating AMPK. Int J Mol Sci.
21:3722020. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Li W, Zhu J, Dou J, She H, Tao K, Xu H,
Yang Q and Mao Z: Phosphorylation of LAMP2A by p38 MAPK couples ER
stress to chaperone-mediated autophagy. Nat Commun. 8:17632017.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Wang L, Cai J, Zhao X, Ma L, Zeng P, Zhou
L, Liu Y, Yang S, Cai Z, Zhang S, et al: Palmitoylation prevents
sustained inflammation by limiting NLRP3 inflammasome activation
through chaperone-mediated autophagy. Mol Cell. 83:281–297.e10.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Chen J, Mao K, Yu H, Wen Y, She H, Zhang
H, Liu L, Li M, Li W and Zou F: p38-TFEB pathways promote microglia
activation through inhibiting CMA-mediated NLRP3 degradation in
Parkinson's disease. J Neuroinflammation. 18:2952021. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Song JX, Liu J, Jiang Y, Wang ZY and Li M:
Transcription factor EB: An emerging drug target for
neurodegenerative disorders. Drug Discov Today. 26:164–172. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Valdor R, Mocholi E, Botbol Y,
Guerrero-Ros I, Chandra D, Koga H, Gravekamp C, Cuervo AM and
Macian F: Chaperone-mediated autophagy regulates T cell responses
through targeted degradation of negative regulators of T cell
activation. Nat Immunol. 15:1046–1054. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Bader AG, Kang S, Zhao L and Vogt PK:
Oncogenic PI3K deregulates transcription and translation. Nat Rev
Cancer. 5:921–929. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Sarbassov DD, Guertin DA, Ali SM and
Sabatini DM: Phosphorylation and regulation of Akt/PKB by the
rictor-mTOR complex. Science. 307:1098–1101. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Li Y, Lu L, Luo N, Wang YQ and Gao HM:
Inhibition of PI3K/AKt/mTOR signaling pathway protects against
d-galactosamine/lipopolysaccharide-induced acute liver failure by
chaperone-mediated autophagy in rats. Biomed Pharmacother.
92:544–553. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Endicott SJ, Ziemba ZJ, Beckmann LJ,
Boynton DN and Miller RA: Inhibition of class I PI3K enhances
chaperone-mediated autophagy. J Cell Biol. 219:e2020010312020.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
He Q, Qin R, Glowacki J, Zhou S, Shi J,
Wang S, Gao Y and Cheng L: Synergistic stimulation of osteoblast
differentiation of rat mesenchymal stem cells by leptin and
25(OH)D3 is mediated by inhibition of chaperone-mediated
autophagy. Stem Cell Res Ther. 12:5572021. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Bourdenx M, Gavathiotis E and Cuervo AM:
Chaperone-mediated autophagy: A gatekeeper of neuronal
proteostasis. Autophagy. 17:2040–2042. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Andrade-Tomaz M, de Souza I, Rocha CRR and
Gomes LR: The role of chaperone-mediated autophagy in cell cycle
control and its implications in cancer. Cells. 9:21402020.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Arias E and Cuervo AM: Chaperone-mediated
autophagy in protein quality control. Curr Opin Cell Biol.
23:184–189. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Li W, Nie T, Xu H, Yang J, Yang Q and Mao
Z: Chaperone-mediated autophagy: Advances from bench to bedside.
Neurobiol Dis. 122:41–48. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao
N, Sun B and Wang G: Ferroptosis: Past, present and future. Cell
Death Dis. 11:882020. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Wu Z, Geng Y, Lu X, Shi Y, Wu G, Zhang M,
Shan B, Pan H and Yuan J: Chaperone-mediated autophagy is involved
in the execution of ferroptosis. Proc Natl Acad Sci USA.
116:2996–3005. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Wu K, Yan M, Liu T, Wang Z, Duan Y, Xia Y,
Ji G, Shen Y, Wang L, Li L, et al: Creatine kinase B suppresses
ferroptosis by phosphorylating GPX4 through a moonlighting
function. Nat Cell Biol. 25:714–725. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Yu S, Li Z, Zhang Q, Wang R, Zhao Z, Ding
W, Wang F, Sun C, Tang J, Wang X, et al: GPX4 degradation via
chaperone-mediated autophagy contributes to antimony-triggered
neuronal ferroptosis. Ecotoxicol Environ Saf. 234:1134132022.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Chen C, Wang D, Yu Y, Zhao T, Min N, Wu Y,
Kang L, Zhao Y, Du L, Zhang M, et al: Legumain promotes tubular
ferroptosis by facilitating chaperone-mediated autophagy of GPX4 in
AKI. Cell Death Dis. 12:652021. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Liu C, Sun W, Zhu T, Shi S, Zhang J, Wang
J, Gao F, Ou Q, Jin C, Li J, et al: Glia maturation factor-β
induces ferroptosis by impairing chaperone-mediated autophagic
degradation of ACSL4 in early diabetic retinopathy. Redox Biol.
52:1022922022. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Xie W, Zhang L, Jiao H, Guan L, Zha J, Li
X, Wu M, Wang Z, Han J and You H: Chaperone-mediated autophagy
prevents apoptosis by degrading BBC3/PUMA. Autophagy. 11:1623–1635.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Kiffin R, Christian C, Knecht E and Cuervo
AM: Activation of chaperone-mediated autophagy during oxidative
stress. Mol Biol Cell. 15:4829–4840. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Cuervo AM, Hildebrand H, Bomhard EM and
Dice JF: Direct lysosomal uptake of alpha 2-microglobulin
contributes to chemically induced nephropathy. Kidney Int.
55:529–545. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Schneider JL, Villarroya J, Diaz-Carretero
A, Patel B, Urbanska AM, Thi MM, Villarroya F, Santambrogio L and
Cuervo AM: Loss of hepatic chaperone-mediated autophagy accelerates
proteostasis failure in aging. Aging Cell. 14:249–264. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Kaushik S, Juste YR and Cuervo AM:
Circadian remodeling of the proteome by chaperone-mediated
autophagy. Autophagy. 18:1205–1207. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Hao Y, Kacal M, Ouchida AT, Zhang B,
Norberg E and Vakifahmetoglu-Norberg H: Targetome analysis of
chaperone-mediated autophagy in cancer cells. Autophagy.
15:1558–1571. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Dong S, Wang Q, Kao YR, Diaz A, Tasset I,
Kaushik S, Thiruthuvanathan V, Zintiridou A, Nieves E,
Dzieciatkowska M, et al: Chaperone-mediated autophagy sustains
haematopoietic stem-cell function. Nature. 591:117–123. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Lv L, Li D, Zhao D, Lin R, Chu Y, Zhang H,
Zha Z, Liu Y, Li Z, Xu Y, et al: Acetylation targets the M2 isoform
of pyruvate kinase for degradation through chaperone-mediated
autophagy and promotes tumor growth. Mol Cell. 42:719–730. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Susan PP and Dunn WA Jr:
Starvation-induced lysosomal degradation of aldolase B requires
glutamine 111 in a signal sequence for chaperone-mediated
transport. J Cell Physiol. 187:48–58. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Schneider JL, Suh Y and Cuervo AM:
Deficient chaperone-mediated autophagy in liver leads to metabolic
dysregulation. Cell Metab. 20:417–432. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Xu Y, Zhang Y, García-Cañaveras JC, Guo L,
Kan M, Yu S, Blair IA, Rabinowitz JD and Yang X: Chaperone-mediated
autophagy regulates the pluripotency of embryonic stem cells.
Science. 369:397–403. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
TeSlaa T, Chaikovsky AC, Lipchina I,
Escobar SL, Hochedlinger K, Huang J, Graeber TG, Braas D and
Teitell MA: α-Ketoglutarate accelerates the initial differentiation
of primed human pluripotent stem cells. Cell Metab. 24:485–493.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Carey BW, Finley LW, Cross JR, Allis CD
and Thompson CB: Intracellular α-ketoglutarate maintains the
pluripotency of embryonic stem cells. Nature. 518:413–416. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Tasset I and Cuervo AM: Role of
chaperone-mediated autophagy in metabolism. FEBS J. 283:2403–2413.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Kaushik S, Juste YR, Lindenau K, Dong S,
Macho-González A, Santiago-Fernández O, McCabe M, Singh R,
Gavathiotis E and Cuervo AM: Chaperone-mediated autophagy regulates
adipocyte differentiation. Sci Adv. 8:eabq27332022. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Schroeder B, Schulze RJ, Weller SG,
Sletten AC, Casey CA and McNiven MA: The small GTPase Rab7 as a
central regulator of hepatocellular lipophagy. Hepatology.
61:1896–1907. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Rodriguez-Navarro JA, Kaushik S, Koga H,
Dall'Armi C, Shui G, Wenk MR, Di Paolo G and Cuervo AM: Inhibitory
effect of dietary lipids on chaperone-mediated autophagy. Proc Natl
Acad Sci USA. 109:E705–E714. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
García-Gutiérrez L, Delgado MD and León J:
MYC oncogene contributions to release of cell cycle brakes. Genes
(Basel). 10:2442019. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Gomes LR, Menck CFM and Cuervo AM:
Chaperone-mediated autophagy prevents cellular transformation by
regulating MYC proteasomal degradation. Autophagy. 13:928–940.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Zhou J, Yang J, Fan X, Hu S, Zhou F, Dong
J, Zhang S, Shang Y, Jiang X, Guo H, et al: Chaperone-mediated
autophagy regulates proliferation by targeting RND3 in gastric
cancer. Autophagy. 12:515–528. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Patil M, Pabla N and Dong Z: Checkpoint
kinase 1 in DNA damage response and cell cycle regulation. Cell Mol
Life Sci. 70:4009–4021. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Park C, Suh Y and Cuervo AM: Regulated
degradation of Chk1 by chaperone-mediated autophagy in response to
DNA damage. Nat Commun. 6:68232015. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Hubbi ME, Gilkes DM, Hu H, Kshitiz Ahmed I
and Semenza GL: Cyclin-dependent kinases regulate lysosomal
degradation of hypoxia-inducible factor 1α to promote cell-cycle
progression. Proc Natl Acad Sci USA. 111:E3325–E3334. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Spence J, Sadis S, Haas AL and Finley D: A
ubiquitin mutant with specific defects in DNA repair and
multiubiquitination. Mol Cell Biol. 15:1265–1273. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Ferreira JV, Soares AR, Ramalho JS,
Pereira P and Girao H: K63 linked ubiquitin chain formation is a
signal for HIF1A degradation by chaperone-mediated autophagy. Sci
Rep. 5:102102015. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Ferreira JV, Fôfo H, Bejarano E, Bento CF,
Ramalho JS, Girão H and Pereira P: STUB1/CHIP is required for HIF1A
degradation by chaperone-mediated autophagy. Autophagy.
9:1349–1366. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Jin C, Ou Q, Chen J, Wang T, Zhang J, Wang
Z, Wang Y, Tian H, Xu JY, Gao F, et al: Chaperone-mediated
autophagy plays an important role in regulating retinal progenitor
cell homeostasis. Stem Cell Res Ther. 13:1362022. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Tan J, Wang W, Liu X, Xu J, Che Y, Liu Y,
Hu J, Hu L, Li J and Zhou Q: C11orf54 promotes DNA repair via
blocking CMA-mediated degradation of HIF1A. Commun Biol. 6:6062023.
View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Gong Y, Li Z, Zou S, Deng D, Lai P, Hu H,
Yao Y, Hu L, Zhang S, Li K, et al: Vangl2 limits chaperone-mediated
autophagy to balance osteogenic differentiation in mesenchymal stem
cells. Dev Cell. 56:2103–2120.e9. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Zhu L, He S, Huang L, Ren D, Nie T, Tao K,
Xia L, Lu F, Mao Z and Yang Q: Chaperone-mediated autophagy
degrades Keap1 and promotes Nrf2-mediated antioxidative response.
Aging Cell. 21:e136162022. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Zhang L, Sun Y, Fei M, Tan C, Wu J, Zheng
J, Tang J, Sun W, Lv Z, Bao J, et al: Disruption of
chaperone-mediated autophagy-dependent degradation of MEF2A by
oxidative stress-induced lysosome destabilization. Autophagy.
10:1015–1035. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Kahle PJ, Waak J and Gasser T: DJ-1 and
prevention of oxidative stress in Parkinson's disease and other
age-related disorders. Free Radic Biol Med. 47:1354–1361. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Wang B, Cai Z, Tao K, Zeng W, Lu F, Yang
R, Feng D, Gao G and Yang Q: Essential control of mitochondrial
morphology and function by chaperone-mediated autophagy through
degradation of PARK7. Autophagy. 12:1215–1228. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Nie T, Tao K, Zhu L, Huang L, Hu S, Yang
R, Xu P, Mao Z and Yang Q: Chaperone-mediated autophagy controls
the turnover of E3 ubiquitin ligase MARCHF5 and regulates
mitochondrial dynamics. Autophagy. 17:2923–2938. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Kiffin R, Kaushik S, Zeng M, Bandyopadhyay
U, Zhang C, Massey AC, Martinez-Vicente M and Cuervo AM: Altered
dynamics of the lysosomal receptor for chaperone-mediated autophagy
with age. J Cell Sci. 120:782–791. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Zhang C and Cuervo AM: Restoration of
chaperone-mediated autophagy in aging liver improves cellular
maintenance and hepatic function. Nat Med. 14:959–965. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Unno K, Asakura H, Shibuya Y, Kaiho M,
Okada S and Oku N: Increase in basal level of Hsp70, consisting
chiefly of constitutively expressed Hsp70 (Hsc70) in aged rat
brain. J Gerontol A Biol Sci Med Sci. 55:B329–B335. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Calabrese V, Scapagnini G, Ravagna A,
Colombrita C, Spadaro F, Butterfield DA and Giuffrida Stella AM:
Increased expression of heat shock proteins in rat brain during
aging: Relationship with mitochondrial function and glutathione
redox state. Mech Ageing Dev. 125:325–335. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Gleixner AM, Pulugulla SH, Pant DB, Posimo
JM, Crum TS and Leak RK: Impact of aging on heat shock protein
expression in the substantia nigra and striatum of the female rat.
Cell Tissue Res. 357:43–54. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Loeffler DA: Influence of normal aging on
brain autophagy: A complex scenario. Front Aging Neurosci.
11:492019. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Huang J, Xu J, Pang S, Bai B and Yan B:
Age-related decrease of the LAMP-2 gene expression in human
leukocytes. Clin Biochem. 45:1229–1232. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Zhou J, Chong SY, Lim A, Singh BK, Sinha
RA, Salmon AB and Yen PM: Changes in macroautophagy,
chaperone-mediated autophagy, and mitochondrial metabolism in
murine skeletal and cardiac muscle during aging. Aging (Albany NY).
9:583–599. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Muñoz-Espín D and Serrano M: Cellular
senescence: From physiology to pathology. Nat Rev Mol Cell Biol.
15:482–496. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Rovira M, Sereda R, Pladevall-Morera D,
Ramponi V, Marin I, Maus M, Madrigal-Matute J, Díaz A, García F,
Muñoz J, et al: The lysosomal proteome of senescent cells
contributes to the senescence secretome. Aging Cell. 21:e137072022.
View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Ye W, Xu K, Huang D, Liang A, Peng Y, Zhu
W and Li C: Age-related increases of macroautophagy and
chaperone-mediated autophagy in rat nucleus pulposus. Connect
Tissue Res. 52:472–478. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Kang HT, Lee KB, Kim SY, Choi HR and Park
SC: Autophagy impairment induces premature senescence in primary
human fibroblasts. PLoS One. 6:e233672011. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Hubbi ME and Semenza GL: An essential role
for chaperone-mediated autophagy in cell cycle progression.
Autophagy. 11:850–851. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Koshiji M, Kageyama Y, Pete EA, Horikawa
I, Barrett JC and Huang LE: HIF-1alpha induces cell cycle arrest by
functionally counteracting Myc. EMBO J. 23:1949–1956. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Liu J, Wang L, He H, Liu Y, Jiang Y and
Yang J: The complex role of chaperone-mediated autophagy in cancer
diseases. Biomedicines. 11:20502023. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Koga H and Cuervo AM: Chaperone-mediated
autophagy dysfunction in the pathogenesis of neurodegeneration.
Neurobiol Dis. 43:29–37. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Hosaka Y, Araya J, Fujita Y and Kuwano K:
Role of chaperone-mediated autophagy in the pathophysiology
including pulmonary disorders. Inflamm Regen. 41:292021. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Qiao L, Ma J, Zhang Z, Sui W, Zhai C, Xu
D, Wang Z, Lu H, Zhang M, Zhang C, et al: Deficient
chaperone-mediated autophagy promotes inflammation and
atherosclerosis. Circ Res. 129:1141–1157. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Mastoridou EM, Goussia AC, Kanavaros P and
Charchanti AV: Involvement of lipophagy and chaperone-mediated
autophagy in the pathogenesis of non-alcoholic fatty liver disease
by regulation of lipid droplets. Int J Mol Sci. 24:158912023.
View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Franch HA: Chaperone-mediated autophagy in
the kidney: The road more traveled. Semin Nephrol. 34:72–83. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Kon M, Kiffin R, Koga H, Chapochnick J,
Macian F, Varticovski L and Cuervo AM: Chaperone-mediated autophagy
is required for tumor growth. Sci Transl Med. 3:109ra1172011.
View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Nguyen D, Yang K, Chiao L, Deng Y, Zhou X,
Zhang Z, Zeng SX and Lu H: Inhibition of tumor suppressor p73 by
nerve growth factor receptor via chaperone-mediated autophagy. J
Mol Cell Biol. 12:700–712. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Wang R, Liu Y, Liu L, Chen M, Wang X, Yang
J, Gong Y, Ding BS, Wei Y and Wei X: Tumor cells induce LAMP2a
expression in tumor-associated macrophage for cancer progression.
EBioMedicine. 40:118–134. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Ding ZB, Fu XT, Shi YH, Zhou J, Peng YF,
Liu WR, Shi GM, Gao Q, Wang XY, Song K, et al: Lamp2a is required
for tumor growth and promotes tumor recurrence of hepatocellular
carcinoma. Int J Oncol. 49:2367–2376. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Desideri E, Castelli S, Dorard C, Toifl S,
Grazi GL, Ciriolo MR and Baccarini M: Impaired degradation of YAP1
and IL6ST by chaperone-mediated autophagy promotes proliferation
and migration of normal and hepatocellular carcinoma cells.
Autophagy. 19:152–162. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Shi YH, Ding ZB, Zhou J, Qiu SJ and Fan J:
Prognostic significance of beclin 1-dependent apoptotic activity in
hepatocellular carcinoma. Autophagy. 5:380–382. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Al-Shenawy HAS: Expression of beclin-1, an
autophagy-related marker, in chronic hepatitis and hepatocellular
carcinoma and its relation with apoptotic markers. APMIS.
124:229–237. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh
H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, et al:
Promotion of tumorigenesis by heterozygous disruption of the beclin
1 autophagy gene. J Clin Invest. 112:1809–1820. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Aydin Y, Stephens CM, Chava S, Heidari Z,
Panigrahi R, Williams DD, Wiltz K, Bell A, Wilson W, Reiss K and
Dash S: Chaperone-mediated autophagy promotes beclin1 degradation
in persistently infected hepatitis C virus cell culture. Am J
Pathol. 188:2339–2355. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Ichikawa A, Fujita Y, Hosaka Y, Kadota T,
Ito A, Yagishita S, Watanabe N, Fujimoto S, Kawamoto H, Saito N, et
al: Chaperone-mediated autophagy receptor modulates tumor growth
and chemoresistance in non-small cell lung cancer. Cancer Sci.
111:4154–4165. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Suzuki J, Nakajima W, Suzuki H, Asano Y
and Tanaka N: Chaperone-mediated autophagy promotes lung cancer
cell survival through selective stabilization of the pro-survival
protein, MCL1. Biochem Biophys Res Commun. 482:1334–1340. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Ali AB, Nin DS, Tam J and Khan M: Role of
chaperone mediated autophagy (CMA) in the degradation of misfolded
N-CoR protein in non-small cell lung cancer (NSCLC) cells. PLoS
One. 6:e252682011. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Du C, Huang D, Peng Y, Yao Y, Zhao Y, Yang
Y, Wang H, Cao L, Zhu WG and Gu J: 5-Fluorouracil targets histone
acetyltransferases p300/CBP in the treatment of colorectal cancer.
Cancer Lett. 400:183–193. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Li P, Ji M, Lu F, Zhang J, Li H, Cui T, Li
Wang X, Tang D and Ji C: Degradation of AF1Q by chaperone-mediated
autophagy. Exp Cell Res. 327:48–56. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Fan Y, Hou T, Gao Y, Dan W, Liu T, Liu B,
Chen Y, Xie H, Yang Z, Chen J, et al: Acetylation-dependent
regulation of TPD52 isoform 1 modulates chaperone-mediated
autophagy in prostate cancer. Autophagy. 17:4386–4400. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Losmanova T, Zens P, Scherz A, Schmid RA,
Tschan MP and Berezowska S: Chaperone-mediated autophagy markers
LAMP2A and HSPA8 in advanced non-small cell lung cancer after
neoadjuvant therapy. Cells. 10:27312021. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Hubbi ME, Hu H, Kshitiz Ahmed I, Levchenko
A and Semenza GL: Chaperone-mediated autophagy targets
hypoxia-inducible factor-1α (HIF-1α) for lysosomal degradation. J
Biol Chem. 288:10703–10714. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Dice JF: Altered degradation of proteins
microinjected into senescent human fibroblasts. J Biol Chem.
257:14624–14627. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Valdor R, García-Bernal D, Riquelme D,
Martinez CM, Moraleda JM, Cuervo AM, Macian F and Martinez S:
Glioblastoma ablates pericytes antitumor immune function through
aberrant up-regulation of chaperone-mediated autophagy. Proc Natl
Acad Sci USA. 116:20655–20665. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Arias E and Cuervo AM: Pros and cons of
chaperone-mediated autophagy in cancer biology. Trends Endocrinol
Metab. 31:53–66. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Lu TL, Huang GJ, Wang HJ, Chen JL, Hsu HP
and Lu TJ: Hispolon promotes MDM2 downregulation through
chaperone-mediated autophagy. Biochem Biophys Res Commun.
398:26–31. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Bonhoure A, Vallentin A, Martin M,
Senff-Ribeiro A, Amson R, Telerman A and Vidal M: Acetylation of
translationally controlled tumor protein promotes its degradation
through chaperone-mediated autophagy. Eur J Cell Biol. 96:83–98.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Yang F, Xie HY, Yang LF, Zhang L, Zhang
FL, Liu HY, Li DQ and Shao ZM: Stabilization of MORC2 by estrogen
and antiestrogens through GPER1-PRKACA-CMA pathway contributes to
estrogen-induced proliferation and endocrine resistance of breast
cancer cells. Autophagy. 16:1061–1076. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Sohn EJ, Kim JH, Oh SO and Kim JY:
Regulation of self-renewal in ovarian cancer stem cells by fructose
via chaperone-mediated autophagy. Biochim Biophys Acta Mol Basis
Dis. 1869:1667232023. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Tang J, Zhan MN, Yin QQ, Zhou CX, Wang CL,
Wo LL, He M, Chen GQ and Zhao Q: Impaired p65 degradation by
decreased chaperone-mediated autophagy activity facilitates
epithelial-to-mesenchymal transition. Oncogenesis. 6:e3872017.
View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Cuervo AM and Wong E: Chaperone-mediated
autophagy: Roles in disease and aging. Cell Res. 24:92–104. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Orenstein SJ, Kuo SH, Tasset I, Arias E,
Koga H, Fernandez-Carasa I, Cortes E, Honig LS, Dauer W, Consiglio
A, et al: Interplay of LRRK2 with chaperone-mediated autophagy. Nat
Neurosci. 16:394–406. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Andersson FI, Werrell EF, McMorran L,
Crone WJ, Das C, Hsu ST and Jackson SE: The effect of
Parkinson's-disease-associated mutations on the deubiquitinating
enzyme UCH-L1. J Mol Biol. 407:261–272. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Wang YT and Lu JH: Chaperone-mediated
autophagy in neurodegenerative diseases: Molecular mechanisms and
pharmacological opportunities. Cells. 11:22502022. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Martinez-Vicente M, Talloczy Z, Kaushik S,
Massey AC, Mazzulli J, Mosharov EV, Hodara R, Fredenburg R, Wu DC,
Follenzi A, et al: Dopamine-modified alpha-synuclein blocks
chaperone-mediated autophagy. J Clin Invest. 118:777–788.
2008.PubMed/NCBI
|
|
171
|
Tang FL, Erion JR, Tian Y, Liu W, Yin DM,
Ye J, Tang B, Mei L and Xiong WC: VPS35 in dopamine neurons is
required for endosome-to-golgi retrieval of Lamp2a, a receptor of
chaperone-mediated autophagy that is critical for α-synuclein
degradation and prevention of pathogenesis of Parkinson's disease.
J Neurosci. 35:10613–10628. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Scrivo A, Bourdenx M, Pampliega O and
Cuervo AM: Selective autophagy as a potential therapeutic target
for neurodegenerative disorders. Lancet Neurol. 17:802–815. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Scheltens P, De Strooper B, Kivipelto M,
Holstege H, Chételat G, Teunissen CE, Cummings J and van der Flier
WM: Alzheimer's disease. Lancet. 397:1577–1590. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Yang Y, Nishimura I, Imai Y, Takahashi R
and Lu B: Parkin suppresses dopaminergic neuron-selective
neurotoxicity induced by Pael-R in Drosophila. Neuron.
37:911–924. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Dou F, Netzer WJ, Tanemura K, Li F, Hartl
FU, Takashima A, Gouras GK, Greengard P and Xu H: Chaperones
increase association of tau protein with microtubules. Proc Natl
Acad Sci USA. 100:721–726. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Auluck PK, Chan HYE, Trojanowski JQ, Lee
VMY and Bonini NM: Chaperone suppression of alpha-synuclein
toxicity in a Drosophila model for Parkinson's disease.
Science. 295:865–868. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Caballero B, Wang Y, Diaz A, Tasset I,
Juste YR, Stiller B, Mandelkow EM, Mandelkow E and Cuervo AM:
Interplay of pathogenic forms of human tau with different
autophagic pathways. Aging Cell. 17:e126922018. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Caballero B, Bourdenx M, Luengo E, Diaz A,
Sohn PD, Chen X, Wang C, Juste YR, Wegmann S, Patel B, et al:
Acetylated tau inhibits chaperone-mediated autophagy and promotes
tau pathology propagation in mice. Nat Commun. 12:22382021.
View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Wang Y, Martinez-Vicente M, Krüger U,
Kaushik S, Wong E, Mandelkow EM, Cuervo AM and Mandelkow E: Tau
fragmentation, aggregation and clearance: The dual role of
lysosomal processing. Hum Mol Genet. 18:4153–4170. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Liu H, Wang P, Song W and Sun X:
Degradation of regulator of calcineurin 1 (RCAN1) is mediated by
both chaperone-mediated autophagy and ubiquitin proteasome
pathways. FASEB J. 23:3383–3392. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Kalia LV and Lang AE: Parkinson's disease.
Lancet. 386:896–912. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Alvarez-Erviti L, Rodriguez-Oroz MC,
Cooper JM, Caballero C, Ferrer I, Obeso JA and Schapira AH:
Chaperone-mediated autophagy markers in Parkinson disease brains.
Arch Neurol. 67:1464–1472. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Cuervo AM, Stefanis L, Fredenburg R,
Lansbury PT and Sulzer D: Impaired degradation of mutant
alpha-synuclein by chaperone-mediated autophagy. Science.
305:1292–1295. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Yang Q, She H, Gearing M, Colla E, Lee M,
Shacka JJ and Mao Z: Regulation of neuronal survival factor MEF2D
by chaperone-mediated autophagy. Science. 323:124–127. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Yang Q and Mao Z: The complexity in
regulation of MEF2D by chaperone-mediated autophagy. Autophagy.
5:1073–1074. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Setsuie R, Wang YL, Mochizuki H, Osaka H,
Hayakawa H, Ichihara N, Li H, Furuta A, Sano Y, Sun YJ, et al:
Dopaminergic neuronal loss in transgenic mice expressing the
Parkinson's disease-associated UCH-L1 I93M mutant. Neurochem Int.
50:119–129. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Kabuta T, Furuta A, Aoki S, Furuta K and
Wada K: Aberrant interaction between Parkinson disease-associated
mutant UCH-L1 and the lysosomal receptor for chaperone-mediated
autophagy. J Biol Chem. 283:23731–23738. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Freibaum BD, Chitta RK, High AA and Taylor
JP: Global analysis of TDP-43 interacting proteins reveals strong
association with RNA splicing and translation machinery. J Proteome
Res. 9:1104–1120. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Coyne AN, Lorenzini I, Chou CC, Torvund M,
Rogers RS, Starr A, Zaepfel BL, Levy J, Johannesmeyer J, Schwartz
JC, et al: Post-transcriptional inhibition of Hsc70-4/HSPA8
expression leads to synaptic vesicle cycling defects in multiple
models of ALS. Cell Rep. 21:110–125. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Huang CC, Bose JK, Majumder P, Lee KH,
Huang JT, Huang JK and Shen CK: Metabolism and mis-metabolism of
the neuropathological signature protein TDP-43. J Cell Sci.
127:3024–3038. 2014.PubMed/NCBI
|
|
191
|
Ormeño F, Hormazabal J, Moreno J, Riquelme
F, Rios J, Criollo A, Albornoz A, Alfaro IE and Budini M: Chaperone
mediated autophagy degrades TDP-43 protein and is affected by
TDP-43 aggregation. Front Mol Neurosci. 13:192020. View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Arosio A, Cristofani R, Pansarasa O,
Crippa V, Riva C, Sirtori R, Rodriguez-Menendez V, Riva N, Gerardi
F, Lunetta C, et al: HSC70 expression is reduced in lymphomonocytes
of sporadic ALS patients and contributes to TDP-43 accumulation.
Amyotroph Lateral Scler Frontotemporal Degener. 21:51–62. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
193
|
Thompson LM, Aiken CT, Kaltenbach LS,
Agrawal N, Illes K, Khoshnan A, Martinez-Vincente M, Arrasate M,
O'Rourke JG, Khashwji H, et al: IKK phosphorylates huntingtin and
targets it for degradation by the proteasome and lysosome. J Cell
Biol. 187:1083–1099. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
194
|
Jana NR, Tanaka M, Wang G and Nukina N:
Polyglutamine length-dependent interaction of Hsp40 and Hsp70
family chaperones with truncated N-terminal huntingtin: Their role
in suppression of aggregation and cellular toxicity. Hum Mol Genet.
9:2009–2018. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Adachi H, Katsuno M, Minamiyama M, Sang C,
Pagoulatos G, Angelidis C, Kusakabe M, Yoshiki A, Kobayashi Y, Doyu
M and Sobue G: Heat shock protein 70 chaperone overexpression
ameliorates phenotypes of the spinal and bulbar muscular atrophy
transgenic mouse model by reducing nuclear-localized mutant
androgen receptor protein. J Neurosci. 23:2203–2211. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Hay DG, Sathasivam K, Tobaben S, Stahl B,
Marber M, Mestril R, Mahal A, Smith DL, Woodman B and Bates GP:
Progressive decrease in chaperone protein levels in a mouse model
of Huntington's disease and induction of stress proteins as a
therapeutic approach. Hum Mol Genet. 13:1389–1405. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
197
|
Qi L and Zhang XD: Role of
chaperone-mediated autophagy in degrading Huntington's
disease-associated huntingtin protein. Acta Biochim Biophys Sin
(Shanghai). 46:83–91. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Virani SS, Alonso A, Aparicio HJ, Benjamin
EJ, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Cheng
S, Delling FN, et al: Heart disease and stroke statistics-2021
update: A report from the american heart association. Circulation.
143:e254–e743. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
199
|
Madrigal-Matute J, de Bruijn J, van Kuijk
K, Riascos-Bernal DF, Diaz A, Tasset I, Martín-Segura A, Gijbels
MJJ, Sander B, Kaushik S, et al: Protective role of
chaperone-mediated autophagy against atherosclerosis. Proc Natl
Acad Sci USA. 119:e21211331192022. View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Madrigal-Matute J, Cuervo AM and Sluimer
JC: Chaperone-mediated autophagy protects against atherosclerosis.
Autophagy. 18:2505–2507. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
201
|
Subramani J, Kundumani-Sridharan V and Das
KC: Chaperone-mediated autophagy of eNOS in myocardial
ischemia-reperfusion injury. Circ Res. 129:930–945. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
202
|
Ghosh R, Gillaspie JJ, Campbell KS, Symons
JD, Boudina S and Pattison JS: Chaperone-mediated autophagy
protects cardiomyocytes against hypoxic-cell death. Am J Physiol
Cell Physiol. 323:C1555–C1575. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
203
|
GBD 2017 Causes of Death Collaborators, .
Global, regional, and national age-sex-specific mortality for 282
causes of death in 195 countries and territories, 1980–2017: A
systematic analysis for the global burden of disease study 2017.
Lancet. 392:1736–1788. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
204
|
Sebastiani G, Gkouvatsos K and Pantopoulos
K: Chronic hepatitis C and liver fibrosis. World J Gastroenterol.
20:11033–11053. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
205
|
Blight KJ, Kolykhalov AA and Rice CM:
Efficient initiation of HCV RNA replication in cell culture.
Science. 290:1972–1974. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
206
|
Lohmann V, Körner F, Koch J, Herian U,
Theilmann L and Bartenschlager R: Replication of subgenomic
hepatitis C virus RNAs in a hepatoma cell line. Science.
285:110–113. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
207
|
Kurt R, Chandra PK, Aboulnasr F, Panigrahi
R, Ferraris P, Aydin Y, Reiss K, Wu T, Balart LA and Dash S:
Chaperone-mediated autophagy targets IFNAR1 for lysosomal
degradation in free fatty acid treated HCV cell culture. PLoS One.
10:e01259622015. View Article : Google Scholar : PubMed/NCBI
|
|
208
|
Matsui C, Deng L, Minami N, Abe T, Koike K
and Shoji I: Hepatitis C virus NS5A protein promotes the lysosomal
degradation of hepatocyte nuclear factor 1α via chaperone-mediated
autophagy. J Virol. 92:e00639–18. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
209
|
Matsui C, Shoji I, Kaneda S, Sianipar IR,
Deng L and Hotta H: Hepatitis C virus infection suppresses GLUT2
gene expression via downregulation of hepatocyte nuclear factor 1α.
J Virol. 86:12903–12911. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
210
|
You Y, Li WZ, Zhang S, Hu B, Li YX, Li HD,
Tang HH, Li QW, Guan YY, Liu LX, et al: SNX10 mediates
alcohol-induced liver injury and steatosis by regulating the
activation of chaperone-mediated autophagy. J Hepatol. 69:129–141.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
211
|
Sooparb S, Price SR, Shaoguang J and
Franch HA: Suppression of chaperone-mediated autophagy in the renal
cortex during acute diabetes mellitus. Kidney Int. 65:2135–2144.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
212
|
Wing SS, Chiang HL, Goldberg AL and Dice
JF: Proteins containing peptide sequences related to
Lys-Phe-Glu-Arg-Gln are selectively depleted in liver and heart,
but not skeletal muscle, of fasted rats. Biochem J. 275:165–169.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
213
|
Napolitano G, Johnson JL, He J, Rocca CJ,
Monfregola J, Pestonjamasp K, Cherqui S and Catz SD: Impairment of
chaperone-mediated autophagy leads to selective lysosomal
degradation defects in the lysosomal storage disease cystinosis.
EMBO Mol Med. 7:158–174. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
214
|
Lee CH, Lee KH, Jang AH and Yoo CG: The
impact of autophagy on the cigarette smoke extract-induced
apoptosis of bronchial epithelial cells. Tuberc Respir Dis (Seoul).
80:83–89. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
215
|
Kelsen SG: The unfolded protein response
in chronic obstructive pulmonary disease. Ann Am Thorac Soc. 13
(Suppl 2):S138–S145. 2016.PubMed/NCBI
|
|
216
|
Hosaka Y, Araya J, Fujita Y, Kadota T,
Tsubouchi K, Yoshida M, Minagawa S, Hara H, Kawamoto H, Watanabe N,
et al: Chaperone-mediated autophagy suppresses apoptosis via
regulation of the unfolded protein response during chronic
obstructive pulmonary disease pathogenesis. J Immunol.
205:1256–1267. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
217
|
Fujii S, Hara H, Araya J, Takasaka N,
Kojima J, Ito S, Minagawa S, Yumino Y, Ishikawa T, Numata T, et al:
Insufficient autophagy promotes bronchial epithelial cell
senescence in chronic obstructive pulmonary disease.
Oncoimmunology. 1:630–641. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
218
|
Regitz C, Fitzenberger E, Mahn FL, Dußling
LM and Wenzel U: Resveratrol reduces amyloid-beta
(Aβ1-42)-induced paralysis through targeting
proteostasis in an Alzheimer model of Caenorhabditis
elegans. Eur J Nutr. 55:741–747. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
219
|
Wu JZ, Ardah M, Haikal C, Svanbergsson A,
Diepenbroek M, Vaikath NN, Li W, Wang ZY, Outeiro TF, El-Agnaf OM
and Li JY: Dihydromyricetin and salvianolic acid B inhibit
alpha-synuclein aggregation and enhance chaperone-mediated
autophagy. Transl Neurodegener. 8:182019. View Article : Google Scholar : PubMed/NCBI
|
|
220
|
Luan Y, Ren X, Zheng W, Zeng Z, Guo Y, Hou
Z, Guo W, Chen X, Li F and Chen JF: Chronic caffeine treatment
protects against α-synucleinopathy by reestablishing autophagy
activity in the mouse striatum. Front Neurosci. 12:3012018.
View Article : Google Scholar : PubMed/NCBI
|
|
221
|
Sotelo J, Briceño E and López-González MA:
Adding chloroquine to conventional treatment for glioblastoma
multiforme: A randomized, double-blind, placebo-controlled trial.
Ann Intern Med. 144:337–343. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
222
|
Galan-Acosta L, Xia H, Yuan J and
Vakifahmetoglu-Norberg H: Activation of chaperone-mediated
autophagy as a potential anticancer therapy. Autophagy.
11:2370–2371. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
223
|
Wang F and Muller S: Manipulating
autophagic processes in autoimmune diseases: A special focus on
modulating chaperone-mediated autophagy, an emerging therapeutic
target. Front Immunol. 6:2522015. View Article : Google Scholar : PubMed/NCBI
|
|
224
|
Maueröder C, Schall N, Meyer F, Mahajan A,
Garnier B, Hahn J, Kienhöfer D, Hoffmann MH and Muller S:
Capability of neutrophils to form NETs is not directly influenced
by a CMA-targeting peptide. Front Immunol. 8:162017. View Article : Google Scholar : PubMed/NCBI
|
|
225
|
Rusmini P, Cortese K, Crippa V, Cristofani
R, Cicardi ME, Ferrari V, Vezzoli G, Tedesco B, Meroni M, Messi E,
et al: Trehalose induces autophagy via lysosomal-mediated TFEB
activation in models of motoneuron degeneration. Autophagy.
15:631–651. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
226
|
Xu X, Sun Y, Cen X, Shan B, Zhao Q, Xie T,
Wang Z, Hou T, Xue Y, Zhang M, et al: Metformin activates
chaperone-mediated autophagy and improves disease pathologies in an
Alzheimer disease mouse model. Protein Cell. 12:769–787. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
227
|
Cuervo AM, Dice JF and Knecht E: A
population of rat liver lysosomes responsible for the selective
uptake and degradation of cytosolic proteins. J Biol Chem.
272:5606–5615. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
228
|
Dong S, Aguirre-Hernandez C, Scrivo A,
Eliscovich C, Arias E, Bravo-Cordero JJ and Cuervo AM: Monitoring
spatiotemporal changes in chaperone-mediated autophagy in vivo. Nat
Commun. 11:6452020. View Article : Google Scholar : PubMed/NCBI
|














