Heart disease (HD) is a general term for various
diseases affecting the heart, such as heart failure (HF), coronary
heart disease (CHD) and cardiomyopathy. The heart is a highly
energy-efficient organ. Mitochondria play an important role in the
maintenance of myocardial cell bioenergetics via adenosine
triphosphate (ATP) production. Mitochondrial dysfunction can
promote oxidative stress, calcium imbalance, metabolic
reprogramming, abnormal intracellular signal transduction and
apoptosis in cardiomyocytes (1).
Therefore, mitochondrial-dependent pathways may represent
attractive therapeutic targets for human HD.
Peroxisome proliferator-activated receptor (PPAR) γ
coactivator-1α (PGC-1α) is encoded by the PPAGC1A gene
situated on chromosome 4p15.2, which is expressed in most cells and
is recognized as a coactivator transcription factor for maintaining
the transcriptional activation of target genes related to
mitochondrial biosynthesis, energy metabolism and oxidative stress
(2). The heart is a very efficient
tissue where ATP and PGC-1α are highly expressed. HDs caused by
changing PGC-1α comprise genetic or pathological stimuli factors
(such as hyperglycemia or hyperlipidemia). Gly482Ser (rs8192678)
polymorphism is the most frequently studied PPARGC1A
polymorphism. Genetic evidence suggests that the PGC-1α Gly482Ser
mutant variant increases the risk of type II diabetes, coronary
artery disease and hypertension induced-left ventricular
hypertrophy and diastolic dysfunction (3–5).
PGC-1α can also serve as an oxidative stress regulator or energy
receptor to respond to stimuli in the heart. For example, PGC-1α
mediates the elimination of reactive oxygen species (ROS) by
binding to and co-activating the nuclear factor erythroid 2 like-2
(Nrf2) and its downstream antioxidant genes (6). A heart-specific PGC-1α deficiency can
result in HF and is considered a model of energy-related HF, which
leads to the compromised utilization of both glucose and fatty
acids as well as reduced mitochondrial function (7). This suggests that mutations or
changes in PGC-1α may contribute to the pathogenesis of HD.
The present review elucidated recent research on the
roles of PGC-1α signaling pathways in various HDs, including
cardiac hypertrophy and HF, CHD, myocardial infarct (MI),
infarct/reperfusion (I/R), diabetic cardiomyopathy (DCM),
drug-induced cardiotoxicity (DIC) and arrhythmia. It focused on the
interaction between PGC-1α and HD, specifically examining the
upstream of PGC-1α and its post-translational modifications in HD
pathogenesis. In addition, it discussed the therapeutic potential
of PGC-1α in HD and its role as a diagnostic biomarker.
PGC-1α is highly conserved and located on the
reverse strand of human chromosome 4 (mouse chromosome 5). Although
variants have been reported, the most well-studied PGC-1α is
expressed from a proximal promoter and encodes a protein containing
797 amino acids. PGC-1α contains an amino terminal activation
domain with LXXLL/LXXLL/LLXXL motifs that mediate binding and
coactivation of several nuclear receptors and transcription
factors, such as PPARs or NRF1/2. This complex can serve as a
docking scaffold for histone-modifying enzymes, mediator complex
and RNA splicing machinery (2,8).
AMPK acts as a cellular energy receptor and
regulates lipid metabolism and glucose metabolism. Under stress,
such as hypoglycemia or hypoxia, the AMPK signaling pathway in
cells is activated in response to changes in the AMP/ATP ratio,
while the catabolic process of ATP production is promoted to
restore the energy balance (9).
The role of AMPK in HD is currently controversial. Certain studies
have found that AMPK is activated as an adaptive and protective
response in a number of models of cardiac injury such as pressure
overload-induced cardiac hypertrophy or ischemia (10,11).
However, mice with HF with preserved ejection fraction (HFpEF) show
a significant reduction in AMPK activity (12). Cardiomyocyte-specific AMPK knockout
mice can also develop left atrium (LA) remodeling and atrial
fibrillation (12). Regardless of
whether AMPK is activated or inhibited in HD, activating AMPK is
considered a beneficial effect (13).
The NF-κB signaling pathway is a central regulator
of immunity and inflammation (22), which has recently emerged as
important factors in a wide variety of HDs including
atherosclerosis, cardiac remodeling and HF (23). Recent research has revealed that
NF-κB and PGC-1α exert mutual regulatory effects. During
inflammation, NF-κB signaling is activated and p65 binding to the
PGC-1α promoter reduces PGC-1α expression and activity in a
dose-dependent manner (24). This
ultimately leads to downregulation of antioxidant target genes and
the oxidative stress response. Simultaneously, oxidative stress
will promote inhibitor kappa B alpha (IκBα) phosphorylation and
subsequently increase p65 nuclear translocation, thereby
exacerbating inflammatory factor release (25). Therefore, cross interaction between
NF-κB and PGC-1α regulates the inflammatory response and HD.
PGC-1α is a key regulatory factor for the
development and maturation of myocardial cells with functions in
energy metabolism, inflammation, oxidative stress and contraction
reaction. Therefore, the regulation of PGC-1α is crucial for
cardiac homeostasis and PGC-1α signal transduction disorders are
associated with various HDs. Specifically, PGC-1α deactivation will
lead to the occurrence and development of cardiac hypertrophy, HF,
CHD, DCM, DIC and arrhythmia.
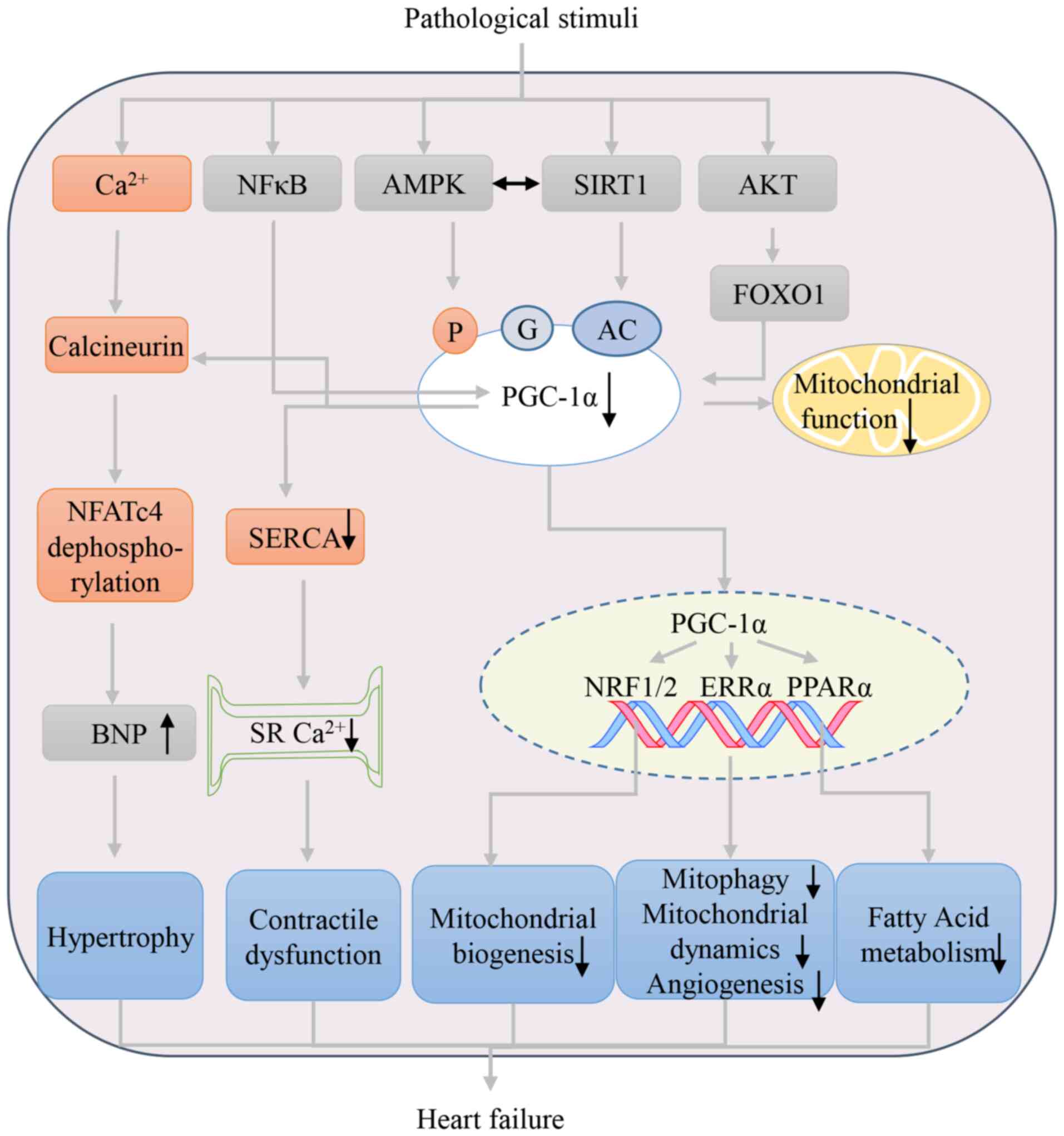
Myocardial hypertrophy is widely defined as an
increase in heart mass and volume to cope with various factors,
such as a continuous increase in blood pressure and blood volume,
including hypertrophy of myocardial cells, proliferation of
myocardial interstitial cells and changes in the extra-myocardial
matrix. Myocardial hypertrophy is divided into physiological and
pathological myocardial hypertrophy (31). Previous studies have demonstrated
that cardiac hypertrophy and HF are associated with the suppression
of PGC-1α (32,33). The inhibition of PGC-1α is
regulated by multiple factors. On one hand, the inactivation of
AMPK or AKT/Forkhead box protein O1 (FOXO1) and the activation of
STAT3 or NF-κB inhibit the promoter activity of PGC-1α, reducing
its mRNA and protein expression (33–36).
On other hand, post-translational modifications of PGC-1a affect
its ability. For example, SIRT1 repression led to PGC-1α
acetylation in a phenylephrine (PE)-induced cardiomyocyte
hypertrophy model (37).
Meanwhile, PE-induced cardiomyocyte hypertrophy also suppresses
PGC-1α expression by enhanced O-glycosylation (31). PGC-1α downregulation, or its
activity reduction, inhibits mitochondrial biogenesis, fatty acid
metabolism, mitochondrial oxidative phosphorylation, angiogenesis
and nuclear factor of activated T cell 4 dephosphorylation
(promoting the transcription of hypertrophic genes, in particular,
BNP), which are involved in the process of myocardial hypertrophy
(2,38,39).
Pathological myocardial hypertrophy is the main
predictive factor of the progression and poor prognosis of HD,
usually related to HF. As expected, a heart-specific PGC-1α
deficiency can result in HF and is considered a model of
energy-related HF, which leads to the compromised utilization of
both glucose and fatty acids as well as reduced mitochondrial
function (7). PGC-1α dysregulation
can also inhibit the recruitment of RNA polymerase II to metabolic
gene promoters in HF, which might be another mechanism underlying a
metabolic imbalance (7,40). Further research has shown that
PGC-1α is associated with dilated HF, including changes in
dyssynchronous local calcium release resulting from the disruption
of t-tubular structures of cardiomyocytes, depending on energy
metabolism (41). In addition,
PGC-1α can mediate the control of mitochondrial quality and,
thereby, the occurrence and development of HF by modulating
mitochondrial dynamics, mitochondrial biogenesis and mitophagy
(42). Fig. 1 shows a schematic diagram of the
involvement of PGC-1α in pathological hypertrophy and HF.
CHD, also called ischemic heart disease, is one of
the most common HDs. Its pathogenesis is mainly coronary artery
stenosis or blockage caused by atherosclerosis (AS), which leads to
long ischemic hypoxia or MI (43).
Studies have demonstrated that PGC-1α plays a key role in
endothelial damage, macrophage function and smooth muscle cell
proliferation and migration by affecting oxidative stress, energy
metabolism and inflammation (44–47).
In addition, PGC-1α regulates MI or I/R injury via effects on ROS
production, mitochondrial biogenesis, mitophagy and energy
metabolism (48,49). Given the complex role of PGC-1α in
CHD, fully understanding its function in different cells will
provide a basis for the future application of PGC-1α agonists. A
promising advantage of PGC-1α agonists is the ability to improve
multiple pathological pathways in CHD.
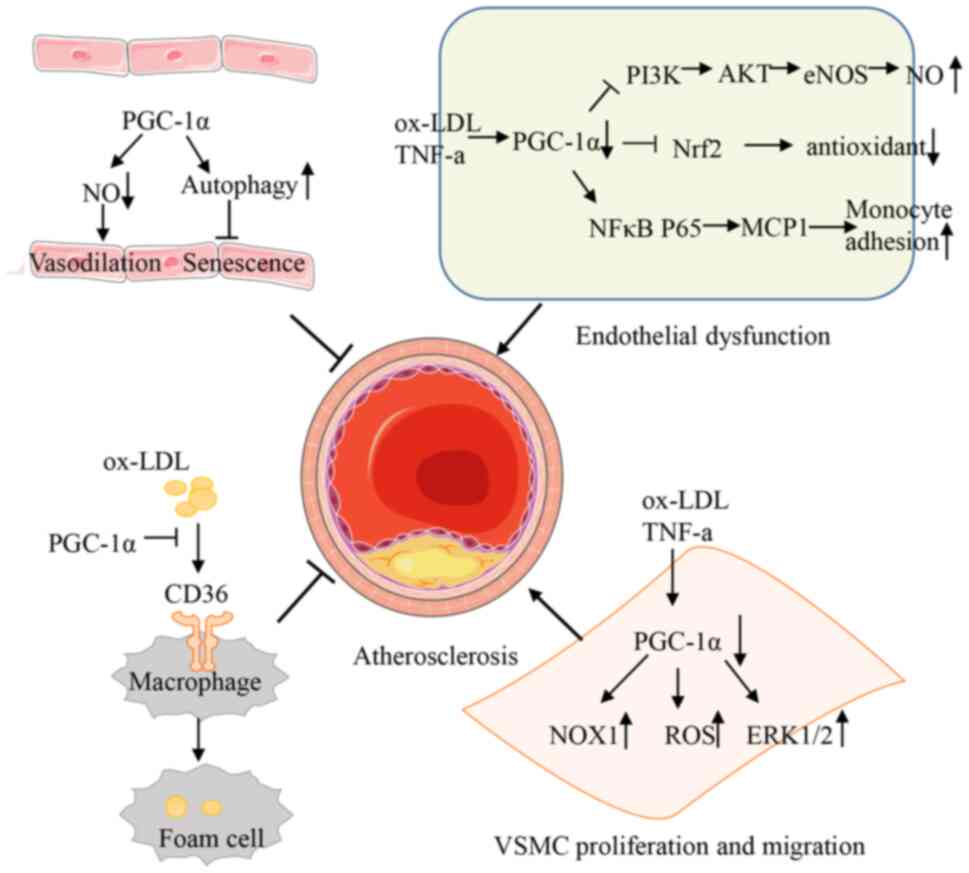
The pathological mechanism underlying AS relies on
an imbalance between blood flow and energy expenditure, leading to
the impairment of endothelial function, mononuclear macrophage
infiltration and vascular smooth muscle cell (VSMC) proliferation
and migration (50). Research
shows that the overexpression of PGC-1α in coronary artery disease
(CAD) vessels increases vascular intraluminal pressure and exerts a
therapeutic effect in patients with CAD via a shift from
mitochondria-derived hydrogen peroxide to nitric oxide
(NO)-mediated vasodilation (51).
These results indicate that PGC-1α is a promising target for
treating AS. Fig. 2 shows a
summary of the aforementioned data.
Endothelial dysfunction is considered a gatekeeper
of vascular diseases and one of the signs of AS (52). PGC-1α participates in the
regulation of endothelial function by maintaining vascular tension
and via antioxidant and anti-inflammatory factors. For example,
PGC-1α can activate the phosphatidylinositol 3-kinase/AKT signaling
pathway, leading to the decrease of endothelial nitric oxide
synthase (eNOS) serine 1177 phosphorylation and NO production; this
maintains vascular tension (53).
Additionally, PGC-1α can combine with Nrf2 to form a complex that
exerts antioxidant effects and inhibits endothelial dysfunction
caused by high glucose/oxidation low lipoprotein (oxLDL) (45). PGC-1α can inhibit NF-κB signaling
and reduce monocyte chemoattractant protein 1 and vascular cellular
adhesion molecule-1 (VCAM-1) expression in endothelial cells, which
can lead to a decrease in monocyte aggregation and slow the
progression of AS. Meanwhile, PGC-1α inhibits ROS production by
regulating the NF-κB and VEGFA signaling pathways and alleviating
oxidative stress and inflammatory responses (46,54).
The presence of macrophages is an obvious sign of
atherosclerotic plaque. Increased levels of inflammatory factors,
such as vascular VCAM-1 and intercellular adhesion molecules, can
mediate the adhesion between the surface of monocytes and the
endothelium, leading to the recruitment of monocyte-derived cells
under the endothelium and differentiation into macrophages.
Macrophages engulf excessive oxidized lipoproteins under the
endothelium and eventually become foam cells, a sign of ‘fat
streaks’ and early atherosclerotic plaques (55). A study has shown that PGC-1α
inhibits adhesion molecule gene expression and cell adhesion
(56). Furthermore, the
overexpression of PGC-1α inhibits oxLDL uptake in macrophages. By
contrast, the macrophage-specific deletion of PGC-1α accelerates
atherosclerosis in LDLR−/− mice by promoting foam cell
formation (47).
In addition to endothelial cells and macrophages,
VSMC proliferation and migration play important roles in vascular
homeostasis. Reports show that PGC-1α inhibits VSMC proliferation
and migration by attenuating NOX1 or upregulating the antioxidant
enzyme superoxide dismutase (SOD)2 to mediate the generation of ROS
and prevent extracellular signal-regulated kinase 1/2
phosphorylation (57–59).
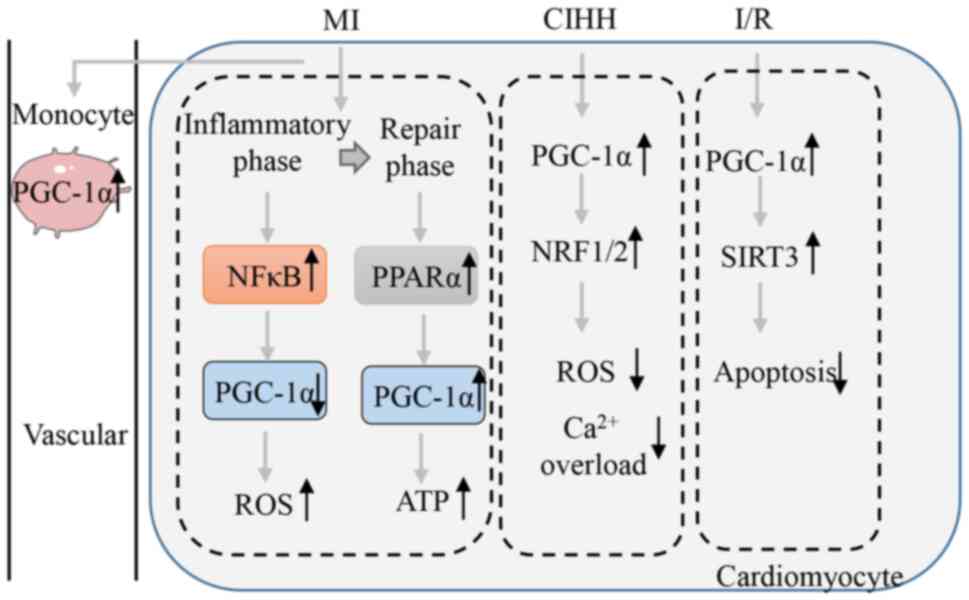
The damage caused to the myocardium during MI is the
result of two processes: Ischemia and subsequent reperfusion.
Cardiac tissue will go through two phases after MI: Inflammatory
phase (3 h to 7 days) and repair phase (7–21 days) (60). When subjected to hypoxic pressure,
the mitochondrial function of myocardial cells is impaired and
NF-κB p65 activation increases, thereby silencing PGC-1α promoter
activity (24). Transcriptomics
analysis has shown that the enrichment of PPAR/retinoid X receptor
binding sites is decreased and levels of the target gene PGC-1α are
lower in post-MI border zone tissues than they are in the healthy
left ventricle 7 days after infarction (61). In animal models of I/R, PGC-1α
expression is reduced at 3 days but partially recovers at 16 days
in the infarcted area, with no changes in remote areas (62). Lou et al (63) discovered that infarct-remodeled
hearts (6 weeks after infarction) show activation of fatty acid
β-oxidation and mobilization of fatty acids from the endogenous
triglycerides store via increased PPARα/PGC-1α signaling. These
results suggest that the activation of NF-κB may inhibit the
expression of PGC-1α during the inflammatory phase of MI. PGC-1α
expression gradually recovers after inflammation disappears. To
maintain the energy required by the heart, it is hypothesized that
PGC-1α expression may be elevated in infarct-remodeled hearts. More
evidence is needed to verify this hypothesis.
PGC-1α participates in the regulation of MI risk
factors, including low adiponectin (APN) levels and an increased
risk of type 2 diabetes in patients with MI (68). APN activates AMPK-PGC-1α signaling
in cardiomyocytes and reduces apoptosis to protect against post-MI
remodeling and dysfunction (69).
PGC-1α expression is increased in blood mononuclear cells of
patients with st-elevated myocardial infarction and the expression
level was correlated with the infarct size (70). These results indicate that PGC-1α
plays an important role in MI or I/R and may serve as a blood
marker for MI. Fig. 3. shows a
schematic diagram of the involvement of PGC-1α in the process of MI
and I/R.
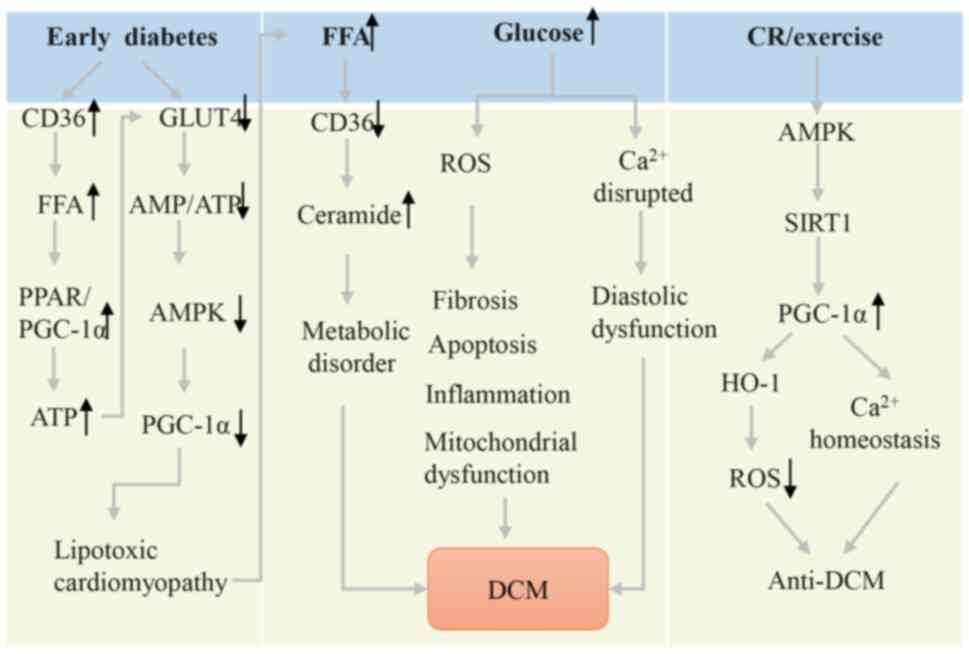
DCM refers to myocardial disease that occurs in
patients with diabetes and cannot be explained by hypertensive
heart disease, coronary atherosclerotic heart disease, heart valve
disease and other HDs. The PPARα/PGC-1α pathway plays an important
role in the occurrence and development of DCM by promoting
metabolic inflexibility. In the early diabetic heart, PPARα and
PGC-1α are activated to increase fatty acid oxidation and lipid
uptake rates, instead of glucose oxidation (71). This leads to increased ATP
generation and a decreased AMP/ATP ratio, which leads to AMPK
inactivation and subsequent PGC-1α inhibition, ultimately resulting
in an excessive supply of fatty acids and lipid accumulation in the
DCM heart (9,72). Cardiac lipotoxicity will lead to
increased total ceramide levels. The accumulation of ceramides in
the heart leads to oxidative stress and mitochondrial dysfunction
by inhibiting PGC-1α, PPARα and CD36 expression (72,73).
PGC-1α also participates in the pathological
mechanism of DCM by regulating ROS production, inflammatory
response and Ca2+ homeostasis. On one hand, PGC-1α can
interact with heme-oxygenase-1 to improve antioxidant defense by
ROS clearance (74). The anti-DCM
effect has been validated in caloric restriction and exercise
models by activating the AMPK/SIRT1/PGC-1α signaling pathway
(75,76). Moreover, moderate overexpression of
PGC-1α maintains Ca2+ homeostasis by increasing the
expression of sarcoplasmic/endoplasmic reticulum Ca2+
transporting ATPase 2 (77).
Fig. 4 shows a schematic diagram
of the involvement of PGC-1α in DCM.
Cardiotoxicity caused by drugs is essentially a
harmful reaction in the heart that occurs during drug use. For
example, the cardiotoxicity produced by the anti-cancer agent
doxorubicin limits its wide clinical applications. Energy
homeostasis, oxidative stress, apoptosis and mitophagy disorders
are considered the main factors associated with cardiotoxicity
(78,79). PGC-1α mainly functions in the
myocardium by participating in energy metabolism and mitochondrial
oxidative stress. PGC-1α can also affect the production of ROS,
mitochondrial biogenesis, mitochondrial autophagy and ultimately
cell apoptosis through NRF2 (80).
A recently discovered function of PGC-1α is the ability to promote
autophagy and inhibit apoptosis by binding to nucleolin (81). At present, certain drugs have shown
protective effects in DIC by upregulating PGC-1α, such as
hydropersulfides and the hydroethanolic extract of Cirsium
(80,82). In the future, the aim will be to
identify drugs for treating DIC by upregulating PGC-1α.
Cardiac rhythm is controlled by various ion channels
and electrogenic ion transporters. Intracellular sarcoplasmic
reticulum and mitochondria regulate these channel and transporter
changes (83). Recent research has
shown that PGC-1α participates in the occurrence and development of
arrhythmia. A transcription profiling analysis of
PGC-1α−/− mouse atrial tissues showed that genes related
to Na+-K+-ATPase activity, hyperpolarized
activation of cyclic nucleotide gated ion channels, Na+
channel-dependent action potential activation and propagation,
Ca2+ current generation and Ca2+ homeostasis
were downregulated. Compared with the levels in wild-type mice,
NaV1.5 channel protein expression is reduced, while the
gap junction protein expression remains unchanged (84). In PGC-1α−/− mouse
ventricular tissues, genes related to
Na+-/K+- ATPase activity, Ca2+
influx, action potential repolarization, autonomous function and
morphological characteristics are also downregulated. The
expression of NaV1.5 decreases and tissue fibrosis
increases (85). Naumenko et
al (41) specifically knocked
out myocardial PGC-1α and found that mice exhibit dilated HF and
myocardial electrophysiological remodeling related to energy
metabolism inhibition, with abnormal SR absorption and release of
Ca2+. The findings of previous research confirm that
PGC-1α participates in cardiac electrophysiology, provides
substrates for the occurrence of arrhythmia and may be related to
Na+/Ca2+ homeostasis (84–86).
The present review systematically analyzed the role
of PGC-1α in the development of HD. Various HDs are closely related
to energy metabolism, calcium signaling and antioxidant capacity.
PGC-1α is involved in these processes. Summarizing the pathogenesis
of different types of HDs clearly reveals that impaired heart
function can lead to the downregulation of PGC-1α at different
times and to varying degrees, leading to oxidative stress
reactions. This result provides a basis for PGC-1α as a therapeutic
target. However, the mechanisms underlying the effects of PGC-1α in
HD are extremely complex and remain to be elucidated. Based on this
review, future studies should focus on the following.
First, the mechanism by which PGC-1α genetic
variations lead to HD is unclear. Although studies have been
published proving PGC-1α genetic variation is closely related to
diabetes and CAD disease, most existing studies are focused on the
effect of PGC-1α genetic variation on diabetes in different ethnic
groups (3–5). More detailed analysis, such as
considering the differences between diabetes cardiomyopathy or
coronary heart disease caused by diabetes, is lacking. However,
these studies are helpful for using PGC-1α genetic variations as
biomarkers.
Second, the role of PGC-1α in HDs has not been fully
explored. The post transcriptional translation and promoter of
PGC-1α are regulated by multiple factors. The effects of multiple
signaling pathways on PGC-1α when myocardial cells are stimulated
need to be simultaneously studied. Concurrently, different reasons
for the decrease in PGC-1α were discovered among different stimuli.
Similarly, PGC-1α plays a dual role in the formation of lipotoxic
cardiomyopathy in early diabetes. Although the role of PGC-1α in
DCM has been clarified, studying it in human DCM is difficult. As
there are no clear indicators for categorizing diabetes, the use of
PGC-1α agonists or inhibitors is challenging.
Third, controlling the amount of PGC-1α
overexpression is difficult. Previous studies show that
overexpression of PGC-1α can cause cardiomyopathy (87). Whitehead et al (77) found that moderate overexpression of
PGC-1α improves cardiac function and fibrosis in aged mice hearts.
These results indicate that the dosage of PGC-1α is critical.
Moreover, they are inconsistent with our expectations, thereby
limiting PGC-1α as a therapeutic target for HDs.
Overall, the present study showed that PGC-1α plays
a crucial role in HDs and is one of the key targets for treating
HDs. Clarifying the mechanism of PGC-1α in HDs will promote the
precision of HD treatment.
Not applicable.
This study was supported by the Henan Provincial Department of
Science and Technology Research Project (grant no. 232102311128)
and The First Affiliated Hospital of Xinxiang Medical University
Youth Foundation (grant nos. QN-2022-B10, QN-2021-B11 and
QN-2021-B02).
Not applicable.
SS was responsible for conceptualization, literature
research, writing the original draft and funding acquisition. HG
was responsible for writing, review and editing. GC and ZZ were
responsible for acquisition, analysis and interpretation of data.
HZ drafted the manuscript and created the figures. DL, XL and XW
were responsible for the acquisition of funding and revised the
manuscript for critically for important intellectual content. GZ
and FL were responsible for project administration,
conceptualization and the designing the method for writing the
review. Data authentication is not applicable. All authors have
read and approved the final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Harrington JS, Ryter SW, Plataki M, Price
DR and Choi AMK: Mitochondria in health, disease and aging. Physiol
Rev. 103:2349–2422. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jannig PR, Dumesic PA, Spiegelman BM and
Ruas JL: SnapShot: Regulation and biology of PGC-1α. Cell.
185:1444.e12022. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Andrulionyte L, Peltola P, Chiasson JL and
Laakso M; STOP-NIDDM Study Group, : Single nucleotide polymorphisms
of PPARD in combination with the Gly482Ser substitution of PGC-1A
and the Pro12Ala substitution of PPARG2 predict the conversion from
impaired glucose tolerance to type 2 diabetes: The STOP-NIDDM
trial. Diabetes. 55:2148–2152. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yongsakulchai P, Settasatian C,
Settasatian N, Komanasin N, Kukongwiriyapan U, Cote ML,
Intharapetch P and Senthong V: Association of combined genetic
variations in PPARγ, PGC-1α and LXRα with coronary artery disease
and severity in Thai population. Atherosclerosis. 248:140–148.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rojek A, Cielecka-Prynda M,
Przewlocka-Kosmala M, Laczmanski L, Mysiak A and Kosmala W: Impact
of the PPARGC1A Gly482Ser polymorphism on left ventricular
structural and functional abnormalities in patients with
hypertension. J Hum Hypertens. 28:557–563. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yao Y, Chen T, Wu H, Yang N and Xu S:
Melatonin attenuates bisphenol A-induced colon injury by dual
targeting mitochondrial dynamics and Nrf2 antioxidant system via
activation of SIRT1/PGC-1α signaling pathway. Free Radic Biol Med.
195:13–22. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kärkkäinen O, Tuomainen T, Mutikainen M,
Lehtonen M, Ruas JL, Hanhineva K and Tavi P: Heart specific PGC-1α
deletion identifies metabolome of cardiac restricted metabolic
heart failure. Cardiovasc Res. 115:107–118. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rowe GC, Jiang A and Arany Z: PGC-1
coactivators in cardiac development and disease. Circ Res.
107:825–838. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Garcia D and Shaw RJ: AMPK: Mechanisms of
cellular energy sensing and restoration of metabolic balance. Mol
Cell. 66:789–800. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tian R, Musi N, D'Agostino J, Hirshman MF
and Goodyear LJ: Increased adenosine monophosphate-activated
protein kinase activity in rat hearts with pressure-overload
hypertrophy. Circulation. 104:1664–1669. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nishino Y, Miura T, Miki T, Sakamoto J,
Nakamura Y, Ikeda Y, Kobayashi H and Shimamoto K: Ischemic
preconditioning activates AMPK in a PKC-dependent manner and
induces GLUT4 up-regulation in the late phase of cardioprotection.
Cardiovasc Res. 61:610–619. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tong D, Schiattarella GG, Jiang N, Daou D,
Luo Y, Link MS, Lavandero S, Gillette TG and Hill JA: Impaired
AMP-Activated protein kinase signaling in heart failure with
preserved ejection Fraction-associated atrial fibrillation.
Circulation. 146:73–76. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang T, Xu L, Guo X, Tao H, Liu Y, Liu X,
Zhang Y and Meng X: The potential of herbal drugs to treat heart
failure: The roles of Sirt1/AMPK. J Pharm Anal. 14:157–176. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jäger S, Handschin C, St-Pierre J and
Spiegelman BM: AMP-activated protein kinase (AMPK) action in
skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl
Acad Sci USA. 104:12017–12022. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang Y, Li X, Guo Y, Chan L and Guan X:
Alpha-Lipoic acid increases energy expenditure by enhancing
adenosine monophosphate-activated protein kinase-peroxisome
proliferator-activated receptor-gamma coactivator-1alpha signaling
in the skeletal muscle of aged mice. Metabolism. 59:967–976. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Malik N, Ferreira BI, Hollstein PE, Curtis
SD, Trefts E, Weiser Novak S, Yu J, Gilson R, Hellberg K, Fang L,
et al: Induction of lysosomal and mitochondrial biogenesis by AMPK
phosphorylation of FNIP1. Science. 380:eabj55592023. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xu CQ, Li J, Liang ZQ, Zhong YL, Zhang ZH,
Hu XQ, Cao YB and Chen J: Sirtuins in macrophage immune metabolism:
A novel target for cardiovascular disorders. Int J Biol Macromol.
256:1282702024. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Komen JC and Thorburn DR: Turn up the
power-pharmacological activation of mitochondrial biogenesis in
mouse models. Br J Pharmacol. 171:1818–1836. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang L, Quan N, Sun W, Chen X, Cates C,
Rousselle T, Zhou X, Zhao X and Li J: Cardiomyocyte-specific
deletion of Sirt1 gene sensitizes myocardium to ischaemia and
reperfusion injury. Cardiovasc Res. 114:805–821. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen Q and Lesnefsky EJ: A new strategy to
decrease cardiac injury in aged heart following
Ischaemia-reperfusion: Enhancement of the interaction between AMPK
and SIRT1. Cardiovasc Res. 114:771–772. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bugga P, Alam MJ, Kumar R, Pal S,
Chattopadyay N and Banerjee SK: Sirt3 ameliorates mitochondrial
dysfunction and oxidative stress through regulating mitochondrial
bioge-nesis and dynamics in cardiomyoblast. Cell Signal.
94:1103092022. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Capece D, Verzella D, Flati I, Arboretto
P, Cornice J and Franzoso G: NF-κB: Blending metabolism, immunity
and inflammation. Trends Immunol. 43:757–775. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bertero E, Dudek J, Cochain C, Delgobo M,
Ramos G, Gerull B, Higuchi T, Vaeth M, Zernecke A, Frantz S, et al:
Immuno-metabolic interfaces in cardiac disease and failure.
Cardiovasc Res. 118:37–52. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rabinovich-Nikitin I, Blant A, Dhingra R,
Kirshenbaum LA and Czubryt MP: NF-κB p65 attenuates cardiomyocyte
PGC-1α expression in hypoxia. Cells. 11:21932022. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhao MM, Xu MJ, Cai Y, Zhao G, Guan Y,
Kong W, Tang C and Wang X: Mitochondrial reactive oxygen species
promote p65 nuclear translocation mediating high-phosphate-induced
vascular calcification in vitro and in vivo. Kidney Int.
79:1071–1079. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang P, Xu S, Xu J, Xin Y, Lu Y, Zhang H,
Zhou B, Xu H, Sheu SS, Tian R and Wang W: Elevated MCU expression
by CaMKIIδB limits pathological cardiac remodeling. Circulation.
145:1067–1083. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wright DC, Geiger PC, Han DH, Jones TE and
Holloszy JO: Calcium induces increases in peroxisome
proliferator-activated receptor gamma coactivator-1alpha and
mitochondrial biogenesis by a pathway leading to p38
mitogen-activated protein kinase activation. J Biol Chem.
282:18793–18799. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kim HK, Ko TH, Song IS, Jeong YJ, Heo HJ,
Jeong SH, Kim M, Park NM, Seo DY, Kha PT, et al: BH4 activates
CaMKK2 and rescues the cardiomyopathic phenotype in rodent models
of diabetes. Life Sci Alliance. 3:e2019006192020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Watanabe S, Horie T, Nagao K, Kuwabara Y,
Baba O, Nishi H, Sowa N, Narazaki M, Matsuda T, Takemura G, et al:
Cardiac-specific inhibition of kinase activity in
calcium/calmodulin-dependent protein kinase kinase-β leads to
accelerated left ventricular remodeling and heart failure after
transverse aortic constriction in mice. PLoS One. 9:e1082012014.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gill JF, Delezie J, Santos G, McGuirk S,
Schnyder S, Frank S, Rausch M, St-Pierre J and Handschin C:
Peroxisome proliferator-activated receptor γ coactivator 1α
regulates mitochondrial calcium homeostasis, sarcoplasmic reticulum
stress and cell death to mitigate skeletal muscle aging. Aging
Cell. 18:e129932019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Oldfield CJ, Duhamel TA and Dhalla NS:
Mechanisms for the transition from physiological to pathological
cardiac hypertrophy. Can J Physiol Pharmacol. 98:74–84. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Brainard RE and Facundo HT: Cardiac
hypertrophy drives PGC-1α suppression associated with enhanced
O-glycosylation. Biochim Biophys Acta Mol Basis Dis.
1867:1660802021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Xu Z, Li M, Lyu D, Xiao H, Li S, Li Z, Li
M, Xiao J and Huang H: Cinnamaldehyde activates AMPK/PGC-1α pathway
via targeting GRK2 to ameliorate heart failure. Phytomedicine.
133:1558942024. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hu X, Xu X, Huang Y, Fassett J, Flagg TP,
Zhang Y, Nichols CG, Bache RJ and Chen Y: Disruption of sarcolemmal
ATP-sensitive potassium channel activity impairs the cardiac
response to systolic overload. Circ Res. 103:1009–1017. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhuang L, Jia K, Chen C, Li Z, Zhao J, Hu
J, Zhang H, Fan Q, Huang C, Xie H, et al: DYRK1B-STAT3 drives
cardiac hypertrophy and heart failure by impairing mitochondrial
bioenergetics. Circulation. 145:829–846. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang S, Tang F, Yang Y, Lu M, Luan A,
Zhang J, Yang J and Wang H: Astragaloside IV protects against
isoproterenol-induced cardiac hypertrophy by regulating
NF-κB/PGC-1α signaling mediated energy biosynthesis. PLoS One.
10:e01187592015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Planavila A, Iglesias R, Giralt M and
Villarroya F: Sirt1 acts in association with PPARα to protect the
heart from hypertrophy, metabolic dysregulation and inflammation.
Cardiovasc Res. 90:276–284. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Liu XP, Gao H, Huang XY, Chen YF, Feng XJ,
He YH, Li ZM and Liu PQ: Peroxisome proliferator-activated receptor
gamma coactivator 1 alpha protects cardiomyocytes from hypertrophy
by suppressing calcineurin-nuclear factor of activated T cells c4
signaling pathway. Transl Res. 166:459–473.e3. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Pereira RO, Wende AR, Crum A, Hunter D,
Olsen CD, Rawlings T, Riehle C, Ward WF and Abel ED: Maintaining
PGC-1α expression following pressure overload-induced cardiac
hypertrophy preserves angiogenesis but not contractile or
mitochondrial function. FASEB J. 28:3691–3702. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bhat S, Chin A, Shirakabe A, Ikeda Y,
Ikeda S, Zhai P, Hsu CP, Sayed D, Abdellatif M, Byun J, et al:
Recruitment of RNA polymerase II to metabolic gene promoters is
inhibited in the failing heart possibly through PGC-1α (Peroxisome
proliferator-activated Receptor-γ coactivator-1α) Dysregulation.
Circ Heart Fail. 12:e0055292019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Naumenko N, Mutikainen M, Holappa L, Ruas
JL, Tuomainen T and Tavi P: PGC-1α deficiency reveals sex-specific
links between cardiac energy metabolism and EC-coupling during
development of heart failure in mice. Cardiovasc Res.
118:1520–1534. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chen L, Qin Y, Liu B, Gao M, Li A, Li X
and Gong G: PGC-1α-mediated mitochondrial quality control:
Molecular mechanisms and implications for heart failure. Front Cell
Dev Biol. 10:8713572022. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hausenloy DJ and Yellon DM: Ischaemic
conditioning and reperfusion injury. Nature reviews. Cardiology.
13:193–209. 2016.PubMed/NCBI
|
|
44
|
Kadlec AO, Chabowski DS, Ait-Aissa K and
Gutterman DD: Role of PGC-1α in Vascular Regulation: Implications
for Atherosclerosis. Arterioscler Thromb Vasc Biol. 36:1467–1474.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wang ZC, Niu KM, Wu YJ, Du KR, Qi LW, Zhou
YB and Sun HJ: A dual Keap1 and p47phox inhibitor Ginsenoside Rb1
ameliorates high glucose/ox-LDL-induced endothelial cell injury and
atherosclerosis. Cell Death Dis. 13:8242022. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kim HJ, Park KG, Yoo EK, Kim YH, Kim YN,
Kim HS, Kim HT, Park JY, Lee KU, Jang WG, et al: Effects of
PGC-1alpha on TNF-alpha-induced MCP-1 and VCAM-1 expression and
NF-kappaB activation in human aortic smooth muscle and endothelial
cells. Antioxid Redox Signal. 9:301–307. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
McCarthy C, Lieggi NT, Barry D, Mooney D,
de Gaetano M, James WG, McClelland S, Barry MC, Escoubet-Lozach L,
Li AC, et al: Macrophage PPAR gamma Co-activator-1 alpha
participates in repressing foam cell formation and atherosclerosis
in response to conjugated linoleic acid. EMBO Mol Med. 5:1443–1457.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Li YQ, Jiao Y, Liu YN, Fu JY, Sun LK and
Su J: PGC-1α protects from myocardial ischaemia-reperfusion injury
by regulating mitonuclear communication. J Cell Mol Med.
26:593–600. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Chen Y, Wang Y, Chen J, Chen X, Cao W,
Chen S, Xu S, Huang H and Liu P: Roles of transcriptional
corepressor RIP140 and coactivator PGC-1α in energy state of
chronically infarcted rat hearts and mitochondrial function of
cardiomyocytes. Mol Cell Endocrinol. 362:11–18. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Caligiuri G: Mechanotransduction,
immunoregulation and metabolic functions of CD31 in cardiovascular
pathophysiology. Cardiovasc Res. 115:1425–1434. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kadlec AO, Chabowski DS, Ait-Aissa K,
Hockenberry JC, Otterson MF, Durand MJ, Freed JK, Beyer AM and
Gutterman DD: PGC-1α (Peroxisome proliferator-activated receptor γ
coactivator 1-α) overexpression in coronary artery disease recruits
NO and hydrogen peroxide during flow-mediated dilation and protects
against increased intraluminal pressure. Hypertension. 70:166–173.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Xu S, Ilyas I, Little PJ, Li H, Kamato D,
Zheng X, Luo S, Li Z, Liu P, Han J, et al: Endothelial dysfunction
in atherosclerotic cardiovascular diseases and beyond: From
mechanism to pharmacotherapies. Pharmacol Rev. 73:924–967. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Li J, Geng XY and Cong XL: PGC-1α
ameliorates Angiotensin II-induced eNOS dysfunction in human aortic
endothelial cells. Vascul Pharmacol. 83:90–97. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
García-Quintans N, Prieto I, Sánchez-Ramos
C, Luque A, Arza E, Olmos Y and Monsalve M: Regulation of
endothelial dynamics by PGC-1α relies on ROS control of VEGF-A
signaling. Free Radical Biol Med. 93:41–51. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Moore KJ, Koplev S, Fisher EA, Tabas I,
Björkegren JLM, Doran AC and Kovacic JC: Macrophage trafficking,
inflammatory resolution and genomics in atherosclerosis: JACC
Macrophage in CVD Series (Part 2). J Am Coll Cardiol. 72:2181–2197.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Minsky N and Roeder RG: Inhibition of
adhesion molecule gene expression and cell adhesion by the
metabolic regulator PGC-1α. PLoS One. 11:e01655982016. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Qu A, Jiang C, Xu M, Zhang Y, Zhu Y, Xu Q,
Zhang C and Wang X: PGC-1alpha attenuates neointimal formation via
inhibition of vascular smooth muscle cell migration in the injured
rat carotid artery. Am J Physiol Cell Physiol. 297:C645–C653. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Zhu L, Sun G, Zhang H, Zhang Y, Chen X,
Jiang X, Jiang X, Krauss S, Zhang J, Xiang Y and Zhang CY:
PGC-1alpha is a key regulator of glucose-induced proliferation and
migration in vascular smooth muscle cells. PLoS One. 4:e41822009.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zhao Q, Zhang J and Wang H: PGC-1α limits
angiotensin II-induced rat vascular smooth muscle cells
proliferation via attenuating NOX1-mediated generation of reactive
oxygen species. Biosci Rep. 35:e002522015. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Nah DY and Rhee MY: The inflammatory
response and cardiac repair after myocardial infarction. Korean
Circ J. 39:393–398. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Günthel M, van Duijvenboden K, de Bakker
DEM, Hooijkaas IB, Bakkers J, Barnett P and Christoffels VM:
Epigenetic state changes underlie metabolic switch in mouse
post-infarction border zone cardiomyocytes. J Cardiovasc Dev Dis.
8:1342021.PubMed/NCBI
|
|
62
|
Oehler D, Spychala A, Gödecke A, Lang A,
Gerdes N, Ruas J, Kelm M, Szendroedi J and Westenfeld R:
Full-length transcriptomic analysis in murine and human heart
reveals diversity of PGC-1α promoters and isoforms regulated
distinctly in myocardial ischemia and obesity. BMC Biol.
20:1692022. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Lou PH, Zhang L, Lucchinetti E, Heck M,
Affolter A, Gandhi M, Kienesberger PC, Hersberger M, Clanachan AS
and Zaugg M: Infarct-remodelled hearts with limited oxidative
capacity boost fatty acid oxidation after conditioning against
ischaemia/reperfusion injury. Cardiovasc Res. 97:251–261. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Bugger H and Pfeil K: Mitochondrial ROS in
myocardial ischemia reperfusion and remodeling. Biochim Biophys
Acta Mol Basis Dis. 1866:1657682020. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Gu S, Hua H, Guo X, Jia Z, Zhang Y, Maslov
LN, Zhang X and Ma H: PGC-1α participates in the protective effect
of chronic intermittent hypobaric hypoxia on cardiomyocytes. Cell
Physiol Biochem. 50:1891–1902. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Papatheodorou I, Makrecka-Kuka M, Kuka J,
Liepinsh E, Dambrova M and Lazou A: Pharmacological activation of
PPARβ/δ preserves mitochondrial respiratory function in
ischemia/reperfusion via stimulation of fatty acid oxidation-linked
respiration and PGC-1α/NRF-1 signaling. Front Endocrinol.
13:9418222022. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Yu LM, Dong X, Xue XD, Zhang J, Li Z, Wu
HJ, Yang ZL, Yang Y and Wang HS: Naringenin improves mitochondrial
function and reduces cardiac damage following ischemia-reperfusion
injury: The role of the AMPK-SIRT3 signaling pathway. Food
Function. 10:2752–2765. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Lindberg S, Jensen JS, Pedersen SH,
Galatius S, Frystyk J, Flyvbjerg A, Bjerre M and Mogelvang R: Low
adiponectin levels and increased risk of type 2 diabetes in
patients with myocardial infarction. Diabetes Care. 37:3003–3008.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Xia Y, Zhang F, Zhao S, Li Y, Chen X, Gao
E, Xu X, Xiong Z, Zhang X, Zhang J, et al: Adiponectin determines
farnesoid X receptor agonism-mediated cardioprotection against
post-infarction remodelling and dysfunction. Cardiovasc Res.
114:1335–1349. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Monsalve M: Induction of PGC-1α expression
can be detected in blood samples of patients with ST-segment
elevation acute myocardial infarction. PLoS One. 6:e269132011.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Duncan JG, Fong JL, Medeiros DM, Finck BN
and Kelly DP: Insulin-resistant heart exhibits a mitochondrial
biogenic response driven by the peroxisome proliferator-activated
receptor-alpha/PGC-1alpha gene regulatory pathway. Circulation.
115:909–917. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Kim Y, Lim JH, Kim EN, Hong YA, Park HJ,
Chung S, Choi BS, Kim YS, Park JY, Kim HW and Park CW: Adiponectin
receptor agonist ameliorates cardiac lipotoxicity via enhancing
ceramide metabolism in type 2 diabetic mice. Cell Death Disease.
13:2822022. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Bekhite M, González-Delgado A, Hübner S,
Haxhikadrija P, Kretzschmar T, Müller T, Wu JMF, Bekfani T, Franz
M, Wartenberg M, et al: The role of ceramide accumulation in human
induced pluripotent stem cell-derived cardiomyocytes on
mitochondrial oxidative stress and mitophagy. Free Radical Biol
Med. 167:66–80. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Waldman M, Arad M, Abraham NG and
Hochhauser E: The peroxisome Proliferator-activated receptor-gamma
coactivator-1α-heme oxygenase 1 axis a powerful antioxidative
pathway with potential to attenuate diabetic cardiomyopathy.
Antioxid Redox Signal. 32:1273–1290. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Wang SY, Zhu S, Wu J, Zhang M, Xu Y, Xu W,
Cui J, Yu B, Cao W and Liu J: Exercise enhances cardiac function by
improving mitochondrial dysfunction and maintaining energy
homoeostasis in the development of diabetic cardiomyopathy. J Mol
Med (Berl). 98:245–261. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Waldman M, Cohen K, Yadin D, Nudelman V,
Gorfil D, Laniado-Schwartzman M, Kornwoski R, Aravot D, Abraham NG,
Arad M and Hochhauser E: Regulation of diabetic cardiomyopathy by
caloric restriction is mediated by intracellular signaling pathways
involving ‘SIRT1 and PGC-1α’. Cardiovasc Diabetol. 17:1112018.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Whitehead N, Gill JF, Brink M and
Handschin C: Moderate modulation of cardiac PGC-1α expression
partially affects age-associated transcriptional remodeling of the
heart. Front Physiol. 9:2422018. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Mamoshina P, Rodriguez B and Bueno-Orovio
A: Toward a broader view of mechanisms of drug cardiotoxicity. Cell
Rep Med. 2:1002162021. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Zhao X, Tian Z, Sun M and Dong D: Nrf2: A
dark horse in doxorubicin-induced cardiotoxicity. Cell Death
Discov. 9:2612023. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Song JH, Kim MS, Lee SH, Hwang JT, Park
SH, Park SW, Jeon SB, Lee RR, Lee J and Choi HK: Hydroethanolic
extract of Cirsium setidens ameliorates doxorubicin-induced
cardiotoxicity by AMPK-PGC-1α-SOD-mediated mitochondrial
protection. Phytomedicine. 129:1556332024. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Yin L, Yuan L, Tang Y, Luo Z, Lin X, Wang
S, Liang P and Jiang B: Nucleolin promotes autophagy through PGC-1α
In LPS-induced myocardial injury. Shock. 60:227–237. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Pharoah BM, Zhang C, Khodade VS, Keceli G,
McGinity C, Paolocci N and Toscano JP: Hydropersulfides (RSSH)
attenuate doxorubicin-induced cardiotoxicity while boosting its
anticancer action. Redox Biol. 60:1026252023. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Terrar DA: Timing mechanisms to control
heart rhythm and initiate arrhythmias: Roles for intracellular
organelles, signalling pathways and subsarcolemmal Ca2.
Philos Trans R Soc Lond B Biol Sci. 378:202201702023. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Chadda KR, Edling CE, Valli H, Ahmad S,
Huang CL and Jeevaratnam K: Gene and protein expression profile of
selected molecular targets mediating electrophysiological function
in Pgc-1α deficient murine atria. Int J Mol Sci. 19:34502018.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Saadeh K, Chadda KR, Ahmad S, Valli H,
Nanthakumar N, Fazmin IT, Edling CE, Huang CL and Jeevaratnam K:
Molecular basis of ventricular arrhythmogenicity in a Pgc-1α
deficient murine model. Mol Genet Metab Rep. 27:1007532021.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Liu GZ, Hou TT, Yuan Y, Hang PZ, Zhao JJ,
Sun L, Zhao GQ, Zhao J, Dong JM, Wang XB, et al: Fenofibrate
inhibits atrial metabolic remodelling in atrial fibrillation
through PPAR-α/sirtuin 1/PGC-1α pathway. Br J Pharmacol.
173:1095–1109. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Lehman JJ, Barger PM, Kovacs A, Saffitz
JE, Medeiros DM and Kelly DP: Peroxisome proliferator-activated
receptor gamma coactivator-1 promotes cardiac mitochondrial
biogenesis. J Clin Invest. 106:847–856. 2000. View Article : Google Scholar : PubMed/NCBI
|