Schizophrenia, a multifaceted psychiatric condition,
affects ~287 per 100,000 individuals globally (1), presenting important challenges due to
its diverse symptoms and effects on brain functionality. This
disorder is characterized by three primary categories of symptoms:
i) Positive symptoms, including hallucinations (predominantly
auditory) and delusions (2,3); ii)
negative symptoms, which comprise emotional numbness, apathy and
social isolation (4,5); and iii) cognitive symptoms, which
disrupt daily functioning by impairing memory, attention and
executive skills (6). A total of
>50% of patients with chronic schizophrenia experience at least
one negative symptom (7). These
symptoms do not tend to improve spontaneously throughout the
progression of the disease and about one-third of patients
demonstrate a poor response to the antipsychotics commonly used
today, such as Chlorpromazine (8,9). The
typical onset of schizophrenia varies by sex, with men usually
developing the disorder between the ages of 18 and 25 years,
whereas women tend to have a later onset, typically between 25 and
35 years (10). Schizophrenia
evolves through various stages: i) The prodromal phase, marked by
initial symptoms and a decline in brain function (11); ii) the acute phase, characterized
by intense psychotic symptoms and significant functional
degradation (12); and iii) the
residual phase, during which some symptoms may abate but
substantial functional difficulties persist (13,14).
Treatment of schizophrenia involves both medications and
psychosocial interventions (15).
Medication includes primarily antipsychotic drugs that target
dopamine receptors in the brain, which can alleviate positive
symptoms, such as hallucination and delusions (16). Alongside medication, therapies like
cognitive behavioral therapy aid the change of harmful thought
patterns (17), while
family-oriented interventions, such as psychoeducation and mutual
support among families of schizophrenia patients, can notably
enhance the patients' treatment satisfaction and adherence
(18). Additional options such as
electroconvulsive therapy may be used for severe cases (19). A comprehensive care approach,
integrating these treatments, is crucial for the effective
management of schizophrenia.
Ferroptosis is a distinct form of programmed cell
death dependent on iron and characterized by the lethal
accumulation of lipid peroxides (20). In contrast to other cell death
mechanisms, such as apoptosis or necrosis, ferroptosis is
predominantly caused by failure of the cellular antioxidant
systems, particularly the enzyme glutathione peroxidase 4 (GPX4)
(21,22). This enzyme normally detoxifies
lipid peroxides by catalyzing the transformation of lipid
hydroperoxides into harmless lipid alcohols, using glutathione
(GSH) as a reducing agent. This action is crucial in maintaining
the integrity of cellular membranes and preventing oxidative damage
leading to ferroptosis (23).
Researchers have also identified mechanisms of ferroptosis that do
not depend on GPX4, and a major component of this GPX4-independent
pathway is ferroptosis suppressor protein 1 (FSP1) (24). FSP1 uses NADPH to catalyze the
reduction of coenzyme Q10 (CoQ10), a potent lipophilic antioxidant.
This reduction is vital to prevent the accumulation of lipid
peroxides (25). From a
morphological perspective, cells undergoing ferroptosis demonstrate
several distinctive alterations (26). Electron microscopy reveals that
these cells typically have smaller mitochondria with denser
membranes. Furthermore, the mitochondria often exhibit a loss of
cristae and may show ruptures in their outer membranes (20,27,28).
Changes in the cellular membrane, including increased density and
notable damage, also occur, culminating in the disruption of
cellular integrity and eventual death (29,30).
This pathway of cell death is intricately connected to various
metabolic functions within the cell, such as lipid metabolism, iron
regulation and the management of reactive oxygen species (ROS),
underscoring its important impact on cellular health and disease
pathogenesis (31–33). Ferroptosis has been implicated in a
range of diseases, such as cancer (34) and neurodegenerative conditions
(35), where iron dysregulation
and oxidative stress are prominent.
The interconnection between ferroptosis and
schizophrenia is attracting considerable attention, triggered by
the pervasive role of oxidative stress and anomalies in iron
metabolism observed in schizophrenia (36,37).
Oxidative stress, a well-documented aspect of schizophrenia, is
linked to both the pathogenesis of the disorder and the
degeneration of neural circuits (38). Ferroptosis, with its fundamental
role in managing iron levels and oxidative responses (39), offers a compelling framework to
explore how disruptions in these cellular mechanisms may contribute
to the developmental and progressive phases of schizophrenia.
Examining the biochemical processes underlying ferroptosis, such as
iron accumulation, lipid peroxide formation and antioxidant system
failures, in alignment with the pathophysiological traits of
schizophrenia, could reveal potential contributions of ferroptosis
to the onset and progression of this disorder. Additionally,
insights into ferroptosis could lead to innovative therapeutic
strategies, potentially addressing the limitations of current
treatments by targeting the underlying cellular disturbances in
schizophrenia.
Iron is essential for various biological processes,
including oxygen transport, DNA synthesis and electron transfer,
due to its ability to exist in multiple oxidation states (40). The cellular uptake of iron is
primarily facilitated through the binding of transferrin-bound iron
to transferrin receptors, which internalize the iron into cells
(41). Once inside the cells, iron
is released from transferrin in acidic endosomes and then
distributed into the labile iron pool (LIP), readily participating
in critical cellular functions or being stored in ferritin
(42,43). Most of the intracellular iron is
sequestered in ferritin, where it is stored in a less reactive
ferric iron state, helping to stabilize the balance with the
volatile LIP (44). This storage
mechanism is key, especially in the brain, where specific cells,
such as oligodendrocytes, microglia and neurons, express ferritin
(45–48), highlighting its role in preserving
neural integrity and guarding against oxidative damage. Under
conditions of stress or specific cellular signals, ferritin can
undergo degradation via a process called ferritinophagy, typically
facilitated by nuclear receptor coactivator 4 (NCOA4) (49). This degradation liberates iron back
into the LIP, enhancing the availability of ferrous iron that can
initiate the production of harmful lipid peroxides, thereby
triggering the pathway of ferroptosis (50,51).
The export of iron from cells is controlled by ferroportin, the
sole known cellular iron exporter (52). The activity of ferroportin is
stringently regulated by hepcidin, a hormone produced in the liver
(53). Hepcidin binds to
ferroportin, inducing its internalization and degradation, which
diminishes iron export, and can result in an accumulation of
intracellular iron if overexpressed or when mutations impede the
function of ferroportin (50,54).
This chain reaction progresses until two lipid
peroxyl radicals react, thus terminating the process. However,
several reactive aldehydes, such as 4-hydroxynonenal (4-HNE) and
malondialdehyde, are produced during this phase (63,64).
These compounds can form adducts with DNA, proteins and other vital
macromolecules, further impairing cellular functions (65–67).
For instance, in the brain tissue of patients with Alzheimer's
disease, the peroxidation product 4-HNE can bind covalently to
critical neuronal mitochondrial, membrane and cytosolic proteins
via Michael addition reactions. This binding results in functional
impairments of important neuronal proteins, such as
glyceraldehyde-3-phosphate dehydrogenase and α-enolase, leading to
neuronal death and consequent cognitive decline (68). Moreover, lipid peroxidation
involves feedback mechanisms that exacerbate damage. Released iron
from damaged proteins or mitochondria can catalyze additional
Fenton reactions, perpetuating the cycle of ROS production and
lipid peroxidation (69). This
feedback loop is particularly harmful in environments with elevated
iron levels or compromised antioxidant defenses.
In the neurological context of schizophrenia, the
susceptibility of the brain to lipid peroxidation is important due
to its high lipid content and metabolic activity, coupled with
relatively low antioxidant protection (36,70,71).
Schizophrenia may be exacerbated by anomalies in iron metabolism
and antioxidant pathways, thereby increasing susceptibility to
oxidative damage, which is linked to the cognitive and behavioral
manifestations observed in schizophrenia (72,73).
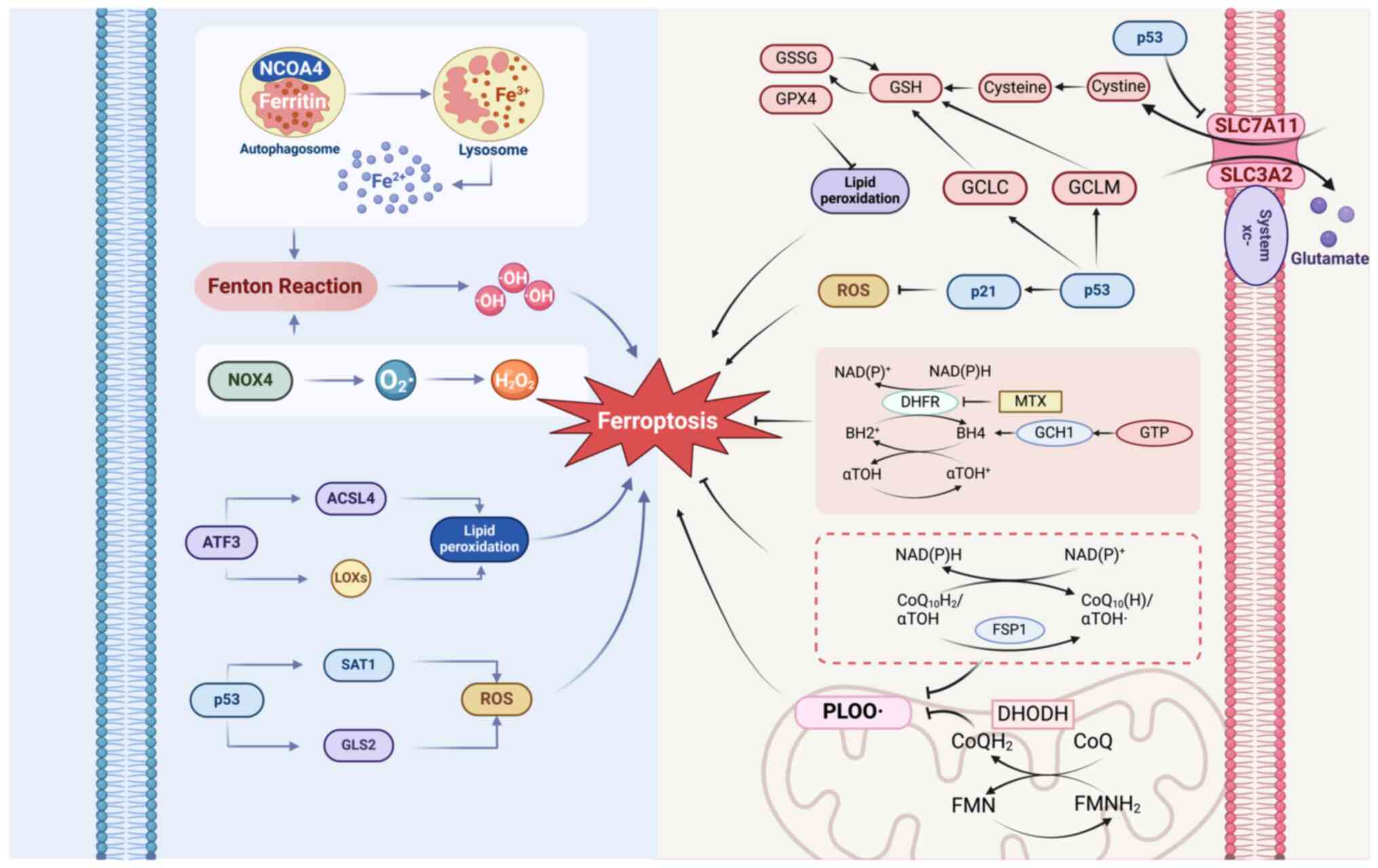
The intricate interplay of various cellular
mechanisms contributes to the initiation and amplification of
ferroptosis, particularly within the neural contexts relevant to
schizophrenia. Mainly the NCOA4-, NADPH oxidase 4 (NOX4)-,
activating transcription factor 3 (ATF3)-and p53-mediated
mechanisms are crucial in modulating iron metabolism, lipid
peroxidation and antioxidant defenses, thereby affecting the
vulnerability to ferroptosis under physiological and pathological
conditions (Fig. 1).
NCOA4 is important in various cellular processes,
surpassing its originally identified role as a coactivator for
nuclear receptors, such as androgen and thyroid hormone receptors
(74,75). By interacting with these receptors,
NCOA4 promotes their transcriptional activities, aided by its
ability to recruit additional transcriptional machinery (76). The functions of NCOA4 are
attributed to its structural design, comprising an N-terminal
domain that facilitates protein-protein interactions, a central
coactivator domain essential for its coactivation role and a
C-terminal domain crucial for the autophagic breakdown of ferritin
(76–79).
NCOA4 has an essential function in iron metabolism,
which is crucial for the proper maintenance of cellular iron
balance (49,80,81).
NCOA4 is highly expressed in critical organs involved in iron
regulation, such as the liver, bone marrow and spleen (82,83).
The involvement of NCOA4 in ferritinophagy underscores its
capability to regulate iron storage and mobilization effectively
(84). In conditions of iron
sufficiency, NCOA4 is degraded to prevent excessive ferritinophagy,
thereby avoiding iron overload that can exacerbate oxidative stress
and cellular damage (79,85,86).
Conversely, under conditions of iron deficiency or cellular stress,
NCOA4 levels increase to promote ferritin degradation and maintain
essential iron supply for metabolic processes (81,87,88).
This regulatory mechanism ensures that ferritinophagy mediated by
NCOA4 adapts to the cellular iron status and environmental cues,
maintaining iron homeostasis and shielding cells from oxidative
stress due to uncontrolled iron release.
NCOA4 is primarily regulated at the
post-translational level, involving dual degradation pathways:
Autophagy, particularly through its role in ferritinophagy
(79,84), and proteasomal degradation, which
is facilitated by the E3 ubiquitin-protein ligase HERC2 under
iron-replete conditions (79).
This degradation is further fine-tuned by ferritin levels. Knockout
of ferritin heavy chain 1 (FTH1) has been demonstrated to reduce
NCOA4 levels by increasing free iron, which activates the
HERC2-mediated degradation of NCOA4 (79,89).
Conversely, overexpression of FTH1 can lead to NCOA4 accumulation,
indicating a protective sequestration from degradation pathways
(89). This regulation can alter
the interaction of NCOA4 with ferritin, potentially leading to its
degradation and influencing the efficacy of ferritinophagy.
Additionally, NCOA4 engages with autophagy components, such as
microtubule-associated protein 1A/1B-light chain 3, which are
integral to autophagosome assembly and the selective autophagic
degradation of ferritin (49,90,91).
This complex regulatory framework ensures that the activity of
NCOA4 is finely adjusted according to the internal iron conditions,
effectively responding to changes and protecting against
iron-related oxidative stress.
The NOX enzyme family, encompassing seven isoforms
(NOX1 to NOX5, DUOX1 and DUOX2), is pivotal for generating ROS,
which are integral to cellular signaling, host defense and redox
balance maintenance (92,93). Each isoform has specific tissue
distributions, regulatory mechanisms and functional roles, thus
contributing uniquely to cellular functions (94).
NOX4 is particularly notable within the NOX family
for its intrinsic activity without the need for cytosolic subunits
required by other isoforms, such as NOX1, NOX2 and NOX3 (95,96).
This unique attribute of NOX4 allows it to maintain a consistent
production of H2O2 rather than superoxide,
which is more commonly produced by its counterparts (97–99).
This ability to generate H2O2, which is
capable of diffusing across membranes and acting as a signaling
molecule, implies a role for NOX4 in cellular signaling and
regulation (100).
In the context of ferroptosis, the involvement of
NOX4 in the continuous production of H2O2 is
critical. In the presence of free iron, H2O2
is converted into highly reactive •OH via the Fenton reaction
(101–103). These radicals are potent inducers
of lipid peroxidation, leading to the oxidative breakdown of PUFAs
within cellular membranes, which is a hallmark of ferroptosis
(104,105). Moreover, the activity of NOX4
further intersects with iron metabolism, thereby modulating the
ferroptotic process (106). For
example, NOX4 affects the expression of ferritin, thus controlling
the availability of free iron necessary for lipid peroxidation, and
influences the formation of iron-sulfur clusters essential for
numerous cellular enzymes, including aconitase and mitochondrial
respiratory chain complexes (107,108).
Pharmacological interventions through the use of NOX
inhibitors, including diphenyleneiodonium and GKT137831, have
demonstrated that inhibiting NOX4 can substantially reduce ROS
levels and lipid peroxidation, and thus protect against ferroptosis
(109,110). These findings are further
corroborated by experimental models that show increased NOX4
activity under oxidative stress conditions, including treatment
with RAS-selective lethal small molecule 3 (RSL3) or erastin, which
have been shown to enhance lipid peroxidation and subsequent cell
death (111,112). This highlights the crucial role
of NOX4 not only in inducing cellular oxidative stress but also in
regulating iron-dependent cell death, indicating its importance in
both normal physiological and pathological conditions.
ATF3, part of the ATF/cAMP-response element-binding
protein family, is recognized as a stress-induced transcription
factor critically affecting ferroptosis (113). ATF3 participates in various
aspects of ferroptosis, influencing iron metabolism and lipid
processing, and modifying cellular antioxidant defense mechanisms
(114–116).
Directly, ATF3 facilitates ferroptosis by regulating
genes that play vital roles in iron management and the initiation
of lipid peroxidation (115,117). ATF3 upregulates heme oxygenase 1
that is integral in controlling internal iron levels conducive to
lipid peroxidation (118).
Additionally, ATF3 regulates genes such as acyl-CoA synthetase long
chain family member 4 and lipoxygenases, essential for producing
and incorporating PUFAs into cellular membranes, which is a
critical step in lipid peroxidation (119–121).
Indirectly, ATF3 undermines cellular antioxidant
defense mechanisms and promotes ferroptosis by downregulating
components of the GSH pathway and system xc−, which
safeguard cells against oxidative damage (113). Specifically, ATF3 reduces the
expression of GPX4, which is crucial for converting lipid
hydroperoxides into non-toxic compounds, thus promoting lipid
peroxidation (115). Furthermore,
the interactions of ATF3 with other stress-responsive transcription
factors, such as NF-κB and p53, alter their regulatory activities,
enhancing conditions that favor oxidative stress and ferroptosis
(122–126). These interactions, especially
with p53, may intensify pro-oxidant functions, thereby increasing
the likelihood of ferroptosis under stress conditions (113,123).
The tumor suppressor protein p53 intricately
regulates cell fate through its dual role in promoting and
inhibiting ferroptosis (127,128). p53 facilitates ferroptosis by
diminishing antioxidant defenses and elevating oxidative stress.
p53 suppresses the expression of solute carrier family 7 member 11
(SLC7A11), a critical component of the system xc−
cystine/glutamate antiporter necessary for cystine importation into
cells (129). Cystine is
transformed into cysteine, a precursor of GSH, which is crucial for
detoxifying lipid peroxides (130). By lowering GSH levels, p53
increases the cellular vulnerability to oxidative stress and
ferroptosis (129). Additionally,
p53 increases the expression of spermidine/spermine
N1-acetyltransferase 1 (SAT1), which leads to the acetylation of
polyamines, thus reducing their antioxidant activity and elevating
acetyl polyamine levels, which contribute to ROS production
(128,131). Furthermore, p53 stimulates
glutaminase 2 (GLS2) expression, enhancing the conversion of
glutamine to glutamate, which increases ROS levels through the
tricarboxylic acid cycle, thereby creating conditions conducive to
ferroptosis (132,133).
Conversely, p53 also activates pathways that enhance
the cellular antioxidant capabilities to counteract ferroptosis.
p53 promotes CDKN1A gene transcription (coding for p21), which is
known for regulating the cell cycle (134,135). However, p21 also affects the
cellular antioxidant response, possibly by altering the stability
and function of nuclear factor erythroid 2-related factor 2 (NRF2),
which is a key regulator of antioxidant genes (136,137). This action enhances the cell
ability to counteract oxidative stress and avert lipid peroxidation
(138). p53 also promotes the
expression of genes essential for synthesizing and recycling GSH,
including glutamate-cysteine ligase modifier subunit (GCLM) and
glutamate-cysteine ligase catalytic subunit (GCLC), as well as
those involved in restoring oxidized GSH to its reduced state
(139–141). These mechanisms demonstrate the
comprehensive regulatory abilities of p53, enabling precise control
over cell responses to stress signals, and switching between
promoting and preventing ferroptosis depending on the cellular
conditions.
The disruption of cellular defense mechanisms
against ferroptosis underscores the pathological processes observed
in schizophrenia. Specifically, the GPX4-, FSP1-, dihydroorotate
dehydrogenase (DHODH)- and GCH1-mediated mechanisms are crucial in
safeguarding neural cells from oxidative stress and lipid
peroxidation. The impairment of these defense mechanisms increases
the susceptibility to ferroptosis, highlighting potential
therapeutic targets (Fig. 1).
GPX4 is a critical enzyme in mitigating oxidative
stress by specifically targeting lipid peroxidation, which is a
pivotal trigger of ferroptosis (22). GPX4 uses GSH as a reducing
substrate to transform lipid hydroperoxides into lipid alcohols
(142). This crucial reaction
occurs at the active site of GPX4, distinguished by the presence of
a selenocysteine residue that is vital for its activity (143). By limiting the accumulation of
lipid peroxides, GPX4 contributes to maintaining the structural
integrity of cellular membranes and preventing cell death (144).
GPX4 engages directly with lipid hydroperoxides
found in cellular membranes or lipoproteins (144,145). By reducing the levels of these
hydroperoxides, GPX4 inhibits the formation of more reactive and
damaging lipid radicals, particularly protecting cells with
membranes rich in PUFAs, which are highly susceptible to
peroxidation (143,146). Furthermore, GPX4 extends its
influence to regulate various signaling pathways that control cell
proliferation and apoptosis, highlighting its broad functional
implications beyond its antioxidant capabilities (144,147). GPX4 primarily regulates cell
proliferation by inhibiting ferroptosis. Additionally, it acts as
an anti-apoptotic factor by preventing the release of cytochrome C,
inactivating caspase-3 and reducing hydroperoxide production, thus
preventing mitochondrial apoptosis (144,147).
Ferroptosis is tightly linked to the disruption of
cellular antioxidant defenses, notably through mechanisms impacting
GPX4 (22). Central to this
process are two primary factors: i) The depletion of GSH, which is
critical for the antioxidant function of GPX4 (111); and ii) direct effects on GPX4,
including oxidative damage to its structure, genetic alterations
affecting its expression and functionality and the use of specific
pharmacological inhibitors (148–150). Depletion of GSH due to
intensified oxidative stress or the action of inhibitors
significantly reduces the ability of GPX4 to counteract lipid
peroxidation, leading to increased lipid peroxides and subsequent
ferroptosis (151). Similarly,
any direct disablement of GPX4 removes the critical barrier against
lipid peroxidation, thereby inducing cell death via ferroptosis
(149,150).
Originally identified as apoptosis-inducing factor
(AIF) mitochondria-associated 2 due to its resemblance to AIF, FSP1
has since been characterized through detailed studies as playing a
crucial role in preventing ferroptosis rather than facilitating
apoptosis (24,152). The FSP1 gene, located on human
chromosome 10, encodes a protein comprising ~373 amino acids,
including a flavin adenine dinucleotide (FAD)-binding motif
critical for its enzymatic activity (153–155).
The enzymatic function of FSP1 is uniquely
characterized by its ability to reduce CoQ10 to ubiquinol within
the lipid bilayers of cell membranes via its FAD-binding domain
(24,25). CoQ10, a lipid-soluble component of
the electron transport chain that is prevalent in cellular
membranes, serves as an electron carrier (156). The transformation of CoQ10 to
ubiquinol by FSP1 markedly reinforces the cellular antioxidant
capacity by providing ubiquinol (24,25),
a powerful lipophilic antioxidant that captures lipid peroxyl
radicals and prevents the peroxidation of PUFAs within the cell
membranes (157).
Functioning independently from the traditional
GSH-dependent antioxidant pathways, which are central to most
cellular defense mechanisms against oxidative stress, FSP1 is
instrumental in the inhibition of ferroptosis by directly limiting
lipid peroxidation (24,25). The reduction of CoQ10 to ubiquinol
not only impedes the onset but also the propagation of lipid
peroxidation processes in the membranes (157,158). By neutralizing lipid peroxyl
radicals produced when ROS interact with PUFAs, ubiquinol
interrupts the lipid peroxidation chain reaction, thereby
maintaining cellular integrity and function (159–161). This distinct role positions FSP1
as an essential, non-traditional regulator of ferroptosis.
DHODH is an essential mitochondrial enzyme
strategically positioned on the outer surface of the inner
mitochondrial membrane (162).
DHODH comprises two distinct domains: An α/β barrel domain that
extends into the mitochondrial matrix and a larger domain anchored
to the membrane, which houses the binding sites for the substrate
dihydroorotate and the coenzyme flavin mononucleotide (163–166). This structural arrangement is
crucial as it enables the direct transfer of electrons from
dihydroorotate to the mitochondrial respiratory chain, specifically
to CoQ10 (167). This connection
bridges the synthesis of pyrimidines with mitochondrial electron
transport.
Beyond its primary biochemical functions, DHODH
impacts cellular redox homeostasis and the process of ferroptosis
(171). The inhibition of DHODH
by brequinar disrupts mitochondrial electron flow, resulting in an
increase in ROS. This increase in ROS enhances lipid peroxidation,
which is a critical marker of ferroptosis (171,172). Furthermore, the inhibition of
DHODH also reduces the levels of vital antioxidants, such as GSH,
which are necessary for counteracting lipid peroxides, thus
rendering cells more prone to oxidative stress and lipid damage
(173,174). Therefore, DHODH has emerged as a
potential regulatory hub, where its inhibition may enhance
ferroptotic cell death, offering a novel angle for therapeutic
intervention, particularly in cancers such as pancreatic
adenocarcinoma (175) and breast
cancer (176), where certain
cells show resistance to other forms of cell death (171,177).
GTP cyclohydrolase-1 (GCH1) is the rate-limiting
enzyme in the biosynthesis of tetrahydrobiopterin (BH4), a critical
cofactor necessary for various biochemical processes, including the
synthesis of monoamine neurotransmitters and the regulation of
nitric oxide levels (178,179).
GCH1 catalyzes the conversion of GTP to formic acid and
dihydroneopterin triphosphate, the initial and rate-limiting step
in BH4 synthesis (180,181). Following this, BH4 can be
recycled back from its oxidized form, dihydrobiopterin, with the
help of dihydrofolate reductase using NAD(P)H (182,183). Structurally, GCH1 is organized
into a homodecameric formation, comprising two pentameric rings
aligned into a toroidal configuration, with each monomer possessing
a catalytic domain essential for the activity of GCH1 (184).
Previous studies have identified a significant role
for GCH1 in regulating oxidative stress and its potential influence
on ferroptosis (185,186). As an antioxidant, BH4, the
enzymatic product of GCH1, effectively neutralizes ROS and reactive
nitrogen species (187,188). This function is pivotal in
preventing the uncoupling of nitric oxide synthase, which can
elevate oxidative stress by shifting the production from nitric
oxide to superoxide (189). BH4,
along with α-tocopherol, which exhibits similar activity, enhances
antioxidant capacities when used together. This combination shows a
synergistic effect, significantly improving protection against
lipid peroxidation compared to their individual use, and
effectively blocking ferroptosis (190). The ability of GCH1 to influence
cellular resistance to oxidative stress conditions and ferroptosis
has profound implications. Research indicates that elevating GCH1
expression or supplementing with BH4 can protect various cell types
against ferroptosis by diminishing lipid peroxidation and oxidative
stress (185,190). Conversely, a lack or inhibition
of GCH1 increases the vulnerability to this type of cell death
(185,191).
Studies using animal models have provided compelling
evidence linking alterations in iron metabolism to
schizophrenia-associated behaviors. In one investigation, male
Sprague-Dawley rats subjected to social isolation, an environmental
stressor known to affect mental health, demonstrated notable iron
level discrepancies in the brain. Specifically, isolated rats
showed increased iron levels in the prefrontal cortex and decreased
levels in the hippocampus compared with their group-housed
counterparts. These iron imbalances were associated with behavioral
changes characteristic of schizophrenia, including increased
anxiety, altered locomotor activity and impaired cognitive
functions, suggesting that the altered distribution of iron in
these brain regions may contribute to the development of
schizophrenia-associated symptoms (192).
Further research indicates that iron imbalances at
different life stages can have profound effects. Rats experiencing
iron deficiency during the perinatal period exhibited enduring
neurochemical and behavioral abnormalities in adulthood, which
persisted even after rectifying the iron deficiency through
supplementation (193). This
points to the lasting impact of early-life iron deficiency on brain
function and its potential to contribute to psychiatric disorders.
Additionally, experiments that induced iron overload in the
hippocampus and prefrontal cortex of male rats led to the
development of schizophrenia-associated behaviors (37), suggesting that both an excess and a
deficiency of iron could foster changes in the brain associated
with schizophrenia.
Another innovative study involved transferring gut
microbiota from patients with schizophrenia into germ-free mice.
This transfer caused the mice to exhibit schizophrenia-associated
behaviors, such as decreased social interaction and hyperactivity
(increased total distance traveled and higher average speed in the
Open-field test). Furthermore, a bioinformatics analysis comparing
transcriptional changes between the brains of mice that received
schizophrenia-associated fecal microbiota and those given a control
(phosphate-buffered saline) highlighted notable overlaps in
differentially expressed genes associated with critical brain areas
and pathways, including those involved in ferroptosis (194).
Advances in genomic research have shed light on the
involvement of ferroptosis-related genes in schizophrenia. A recent
study specifically explored the differential expression of
ferroptosis-related genes in schizophrenia by employing
bioinformatics analysis to compare patient and control groups. Key
among the identified genes are TP53, VEGFA and PTGS2 (195), which have been highlighted as
potential markers for ferroptosis within the context of
schizophrenia. These genes are implicated in the regulation of
oxidative stress responses and cell survival (196–198), and their altered expression in
patients with schizophrenia indicates a potential disruption in
cellular mechanisms that regulate oxidative stress and iron
metabolism.
The impact of ancient viral elements on
schizophrenia also presents a compelling avenue of research,
particularly the role of the endogenous retrovirus group W member 1
(ERVW-1) retroviral element. This element has been shown to promote
ferroptosis in neuronal cells by targeting and degrading key
regulators, such as GPX4 and solute carrier family 3 member 2
(201). This contributes to
increased iron levels and oxidative stress markers, decreased GSH
levels and disrupted mitochondrial membrane potential, all of which
are hallmarks of ferroptosis (202,203). Furthermore, the ERVW-1-induced
effects could be reversed by the ferroptosis inhibitor
ferrostatin-1 (201).
In a case study of a patient initially diagnosed
with Sydenham's chorea who later developed schizophrenia, autopsies
revealed mineral accumulations, including predominantly iron, in
the basal ganglia. These mineral deposits have been associated with
disturbances in dopaminergic signaling, symptoms typical of
schizophrenia and disorders characterized by irregular movements
(204). In patients with
schizophrenia, there was notably intense iron staining in the
caudate nucleus compared with that in normal controls; however, it
remains uncertain whether antipsychotic treatments induced this
increase in iron staining (205).
Further comprehensive autopsy analyses have indicated elevated iron
concentrations in the prefrontal cortex, which is a critical area
for cognitive processes, in patients with schizophrenia compared to
matched controls (16.4 vs. 12.7 µmol/g). These iron levels were not
associated with increased ferritin, which sequesters iron in a
biologically inert form, thus suggesting the accumulation of
potentially harmful free iron (37). Additionally, unlike in healthy
individuals where iron accumulation increases with age, in patients
with schizophrenia elevated iron levels were observed at a much
younger age and remained stable across age, despite both groups
having a similar age range at death (17–85 years for controls and
17–84 years for schizophrenia). And the difference in
covariate-adjusted iron was substantial in the younger subcohort
(age <35; 1.53 µmol/g) but marginal in the older subcohort (age
≥35 years; 0.46 µmol/g). This suggests that disruptions in iron
regulation may be linked with developmental anomalies occurring
early in the course of schizophrenia (37).
In the field of imaging studies, advanced
techniques such as magnetic resonance spectroscopy and quantitative
susceptibility mapping have provided a non-invasive method to
examine the levels of brain iron in living patients with
schizophrenia (206). Studies
using these methods have shown that individuals with schizophrenia
have distinct patterns of iron deposition. Particularly, increased
iron levels have been documented in subcortical structures such as
the thalamus and putamen, which are integral to dopaminergic
pathways and are often implicated in schizophrenia (207,208). These imaging findings not only
corroborate the autopsy studies regarding iron accumulation but
also suggest that iron may modulate neurotransmitter systems
directly involved in the symptomatology of schizophrenia (207).
Serological findings complement the
neuropathological and imaging evidence of iron dysregulation in
schizophrenia. Various studies have noted alterations in serum iron
levels among patients with schizophrenia compared with healthy
controls (209–211). Notably, lower serum
log10ferritin:AST ratio levels and higher total serum
iron have been observed in patients with schizophrenia (211), which may reflect an underlying
imbalance in iron metabolism. These serological abnormalities are
not isolated findings but are connected to broader physiological
disruptions, including liver function, which is crucial in iron
metabolism (209). Understanding
these systemic changes is vital for developing targeted
interventions that address the complex interplay between iron
dysregulation and schizophrenia, potentially leading to more
effective treatments for this disorder.
Decreased levels of antioxidant GSH in patients
with schizophrenia have been well-documented and are considered a
target for therapeutic intervention (212,213). NAC, as a precursor of GSH, plays
a crucial role in attenuating oxidative stress and mitochondrial
dysfunction (214), which are key
contributors to the pathophysiology of schizophrenia. NAC has been
shown to modulate neuroinflammatory pathways, protect against
apoptosis and improve mitochondrial function (215). In GSH-deficient mouse models of
schizophrenia, neurochemical changes were observed in the cortex,
including elevated levels of glutamine, glutamate,
N-acetylaspartate, myo-inositol, lactate, and alanine. NAC
supplementation can effectively normalize these neurochemical
changes during the development of the model mice, suggesting that
NAC may play a therapeutic role in halting or mitigating the
progression of schizophrenia (216).
Clinical trials have shown that NAC can markedly
improve the symptoms of schizophrenia, particularly negative
symptoms that are often less responsive to conventional
antipsychotics (Table I) (217–226). For example, Berk et al
(217) revealed notable
improvements in negative symptoms as measured using the Positive
and Negative Syndrome Scale in patients treated with NAC as an
adjunct to their ongoing antipsychotic regimen. These clinical
improvements were noted alongside enhancements in overall
psychopathology scores, suggesting a broad therapeutic impact of
NAC. In terms of cognitive improvements, NAC has been shown to
exhibit a positive effect on cognitive functions in patients with
schizophrenia. This includes enhancements in areas such as working
memory and executive function (227). These cognitive improvements are
vital, as they address core schizophrenia deficits that affect
daily life and overall quality of life (228).
The therapeutic potential of NAC supplements is not
limited to mitigating oxidative stress. For instance, NAC has been
shown to exhibit neuroprotective effects by reducing
pro-inflammatory cytokines, such as interleukin (IL)-6 and tumor
necrosis factor (TNF)-α, linked to schizophrenia. NAC also
regulates glutamate levels through the cysteine-glutamate
antiporter, influencing neurotransmitter pathways. These effects
suggest that NAC may help prevent the progression of schizophrenia
symptoms from prodromal stages to full-blown psychosis (229,230). Additionally, it may ameliorate
side effects related to antipsychotic medications, such as
akathisia and metabolic disturbances, thus improving patient
adherence and overall quality of life (227,231). The safety and tolerability
profile of NAC is also an advantage. It has been reported that NAC
is generally well-tolerated by patients with minimal side effects.
This aspect is crucial for long-term management strategies in
schizophrenia, where treatments often continue for 1 to 5 years
after the condition stabilizes (232).
Given the role of oxidative stress in ferroptosis,
treatments that reinforce the antioxidant defenses of the body,
such as selenium, may help mitigate ferroptosis. Selenium, a vital
trace element known for its antioxidant properties (233), has attracted interest in
schizophrenia research due to the observed abnormalities in its
levels among patients (234,235). Studies have shown that patients
with schizophrenia typically have lower selenium concentrations in
their bloodstream compared with healthy individuals (235,236). Additionally, a connection has
been drawn between these depleted selenium levels and an increased
risk of schizophrenia. Extensive epidemiological data from the
European population have indicated an important association between
lower selenium levels and increased schizophrenia prevalence
(235). Selenium achieves its
antioxidant effects primarily through selenoproteins, such as GPx
and thioredoxin reductase, which reduce ROS and lipid
hydroperoxides, thus protecting against oxidative stress. Moreover,
these selenoproteins act as antioxidant enzymes that influence
eicosanoid synthesis (lipid mediators involved in inflammatory
responses), thereby affecting the balance between pro-inflammatory
and anti-inflammatory eicosanoids and exerting an anti-inflammatory
effect (237). This association
highlights the role of selenium in combating oxidative stress and
inflammation (238,239), factors that are believed to
contribute to the onset and exacerbation of schizophrenia.
Clinical trials exploring the benefits of selenium
supplementation for patients with schizophrenia have produced
promising results. For example, it has been revealed that restoring
selenium levels can lead to notable improvements in cognitive
functions, such as memory, executive function and attention, which
are frequently compromised in schizophrenia (240). Additionally, broader psychiatric
improvements, including reductions in both positive and negative
symptoms, have been observed, underscoring the wide-ranging
benefits of correcting selenium deficiency (241). These outcomes suggest that
selenium supplementation may be an effective adjunctive therapy in
schizophrenia, capable of not only enhancing symptom management but
also potentially addressing the underlying oxidative imbalances
that contribute to disease progression.
While PUFAs are substrates for lipid peroxidation,
their balanced intake can also modulate the composition and
function of the cell membrane, affecting ferroptosis indirectly.
Research into the effects of omega-3 fatty acids, particularly
docosahexaenoic acid and eicosapentaenoic acid (EPA), reveals their
potential to mitigate inflammatory processes and oxidative stress
(242,243), which are pivotal in managing
ferroptosis associated with schizophrenia.
Clinical studies and systematic reviews have
indicated that supplementation with omega-3 fatty acids can
ameliorate various psychopathological symptoms and metabolic
disorders in patients with schizophrenia (244–247). For instance, a 12-week trial
showed that omega-3 fatty acids supplementation significantly
improved cognitive function and increased brain-derived
neurotrophic factor (BDNF) levels, while reducing inflammatory
markers such as IL-6, TNF-α and C-reactive protein (CRP) (244). Furthermore, omega-3 fatty acids
have been reported to reduce the adverse effects of metabolic
syndrome, conditions often exacerbated by antipsychotic
medications, which typically promote weight gain and insulin
resistance (244). Additionally,
a review of 1,494 patients found significant improvements in
general psychopathology and positive symptoms, particularly in
severely ill patients receiving EPA at doses greater than 1 g/day,
along with favorable effects on metabolic parameters such as serum
triglycerides (247).
The incorporation of omega-3 fatty acids into cell
membranes alters lipid profiles, changing the cellular
vulnerability to lipid peroxidation that is central to ferroptosis
(248,249). These fatty acids can decrease the
production of pro-inflammatory cytokines such as IL-1β, IL-6 and
TNF-α, while simultaneously enhancing the production of
anti-inflammatory cytokines like IL-10 and IL-4 (250). The activation of inflammation can
lead to ferroptosis (251),
indicating that the anti-neuroinflammatory properties of omega-3
fatty acids may also contribute to reducing the levels of
ferroptosis in neuronal cells (250,252,253). These capabilities suggest that
omega-3 fatty acids could serve not only as supplementary
treatments for symptom relief in schizophrenia but also as
influential agents in modifying the fundamental mechanisms of the
disease through the modulation of ferroptosis.
Iron chelation therapy offers a promising avenue
for treating schizophrenia, given its efficacy in other
neurological disorders, such as Parkinson's disease and Alzheimer's
disease, where oxidative stress and iron dysregulation are
implicated (254). Iron
chelators, such as deferiprone, function by binding free iron in
the body, thus mitigating the oxidative stress caused by excess
iron (255). This is particularly
important in the brain, where elevated iron levels can lead to the
production of ROS, contributing to neuronal damage and
neurodegeneration (256,257). Reducing iron-induced oxidative
damage through chelation is an effective strategy in managing
diseases such as Alzheimer's disease, Parkinson's disease and
multiple sclerosis, where iron chelators have been shown to
effectively lower iron levels in brain regions associated with
these conditions, thereby potentially slowing disease progression
and improving neurological outcomes (254,258,259).
In schizophrenia, similar mechanisms of
iron-induced oxidative stress could play a role in the
pathophysiology of the disorder. The accumulation of iron in the
brain not only enhances oxidative stress but also promotes
ferroptosis. In a study involving seven psychiatric patients with
iron overload evidenced by abnormal serum ferritin, transferrin
saturation, or excessive urinary iron, treatment with iron
chelators led to significant clinical improvements (260). Iron chelation could, therefore,
offer dual benefits in schizophrenia treatment by reducing both
oxidative stress and ferroptosis. This therapeutic approach may be
particularly relevant for patients exhibiting elevated ferritin
levels or other indicators of disturbed iron metabolism. However,
further research is needed to systematically assess the efficacy of
iron chelation in schizophrenia and to validate this approach as a
viable treatment option.
Exploring ferroptosis in the context of
schizophrenia represents a promising frontier in psychiatric
research, offering new perspectives on the cellular and molecular
mechanisms underlying this complex disorder. The association
between iron dysregulation, oxidative stress and lipid peroxidation
central to ferroptosis suggests that this form of cell death could
be integral to the pathophysiology of schizophrenia. Understanding
the intricate balance of iron metabolism and the oxidative stress
response within neural circuits could reveal novel therapeutic
targets, potentially revolutionizing the management of
schizophrenia. Future research should focus on the validation of
ferroptosis markers in clinical settings and the development of
targeted therapies that modulate iron homeostasis and antioxidant
systems. By integrating the mechanisms of ferroptosis with existing
knowledge of schizophrenia, researchers can pave the way for
innovative treatments that not only alleviate symptoms but also
address fundamental pathological processes, ultimately enhancing
patient outcomes and quality of life.
Not applicable.
Funding: No funding was received.
Not applicable.
SL contributed to the writing and editing of the
present review. CL collected the information from the literature.
Data authentication is not applicable. Both authors read and
approved the final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Solmi M, Seitidis G, Mavridis D, Correll
CU, Dragioti E, Guimond S, Tuominen L, Dargél A, Carvalho AF,
Fornaro M, et al: Incidence, prevalence, and global burden of
schizophrenia-data, with critical appraisal, from the global burden
of disease (GBD) 2019. Mol Psychiatry. 28:5319–5327. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Schultz SH, North SW and Shields CG:
Schizophrenia: A review. Am Fam Physician. 75:1821–1829.
2007.PubMed/NCBI
|
|
3
|
Bassett AS, Collins EJ, Nuttall SE and
Honer WG: Positive and negative symptoms in families with
schizophrenia. Schizophr Res. 11:9–19. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Azorin JM, Belzeaux R and Adida M:
Negative symptoms in schizophrenia: where we have been and where we
are heading. CNS Neurosci Ther. 20:801–808. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Marder SR and Umbricht D: Negative
symptoms in schizophrenia: Newly emerging measurements, pathways,
and treatments. Schizophr Res. 258:71–77. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
McCutcheon RA, Keefe RSE and McGuire PK:
Cognitive impairment in schizophrenia: Aetiology, pathophysiology,
and treatment. Mol Psychiatry. 28:1902–1918. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bobes J, Arango C, Garcia-Garcia M and
Rejas J; CLAMORS Study Collaborative Group, : Prevalence of
negative symptoms in outpatients with schizophrenia spectrum
disorders treated with antipsychotics in routine clinical practice:
Findings from the CLAMORS study. J Clin Psychiatry. 71:280–286.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Austin SF, Mors O, Budtz-Jørgensen E,
Secher RG, Hjorthøj CR, Bertelsen M, Jeppesen P, Petersen L, Thorup
A and Nordentoft M: Long-term trajectories of positive and negative
symptoms in first episode psychosis: A 10 year follow-up study in
the OPUS cohort. Schizophr Res. 168:84–91. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pandey A and Kalita KN:
Treatment-resistant schizophrenia: How far have we traveled? Front
Psychiatry. 13:9944252022. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ochoa S, Usall J, Cobo J, Labad X and
Kulkarni J: Gender differences in schizophrenia and first-episode
psychosis: A comprehensive literature review. Schizophr Res
Treatment. 2012:9161982012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Andriopoulos I, Ellul J, Skokou M and
Beratis S: Suicidality in the ‘prodromal’ phase of schizophrenia.
Compr Psychiatry. 52:479–485. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Donnelly L, Rathbone J and Adams CE:
Haloperidol dose for the acute phase of schizophrenia. Cochrane
Database Syst Rev. CD0019512013.PubMed/NCBI
|
|
13
|
Charlson FJ, Ferrari AJ, Santomauro DF,
Diminic S, Stockings E, Scott JG, McGrath JJ and Whiteford HA:
Global epidemiology and burden of schizophrenia: Findings from the
global burden of disease study 2016. Schizophr Bull. 44:1195–1203.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Saito Y, Sakurai H, Kane JM, Schooler NR,
Suzuki T, Mimura M and Uchida H: Predicting relapse with residual
symptoms in schizophrenia: A secondary analysis of the PROACTIVE
trial. Schizophr Res. 215:173–180. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Patel KR, Cherian J, Gohil K and Atkinson
D: Schizophrenia: Overview and treatment options. P T. 39:638–645.
2014.PubMed/NCBI
|
|
16
|
Howes OD, Bukala BR and Beck K:
Schizophrenia: From neurochemistry to circuits, symptoms and
treatments. Nat Rev Neurol. 20:22–35. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Butler AC, Chapman JE, Forman EM and Beck
AT: The empirical status of cognitive-behavioral therapy: A review
of meta-analyses. Clin Psychol Rev. 26:17–31. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kim SH and Park S: Effectiveness of family
interventions for patients with schizophrenia: A systematic review
and meta-analysis. Int J Ment Health Nurs. 32:1598–1615. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sinclair DJM, Zhao S, Qi F, Nyakyoma K,
Kwong JSW and Adams CE: Electroconvulsive therapy for
treatment-resistant schizophrenia. Schizophr Bull. 45:730–732.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yang WS and Stockwell BR: Ferroptosis:
Death by lipid peroxidation. Trends Cell Biol. 26:165–176. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu Y, Wan Y, Jiang Y, Zhang L and Cheng
W: GPX4: The hub of lipid oxidation, ferroptosis, disease and
treatment. Biochim Biophys Acta Rev Cancer. 1878:1888902023.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang XD, Liu ZY, Wang MS, Guo YX, Wang
XK, Luo K, Huang S and Li RF: Mechanisms and regulations of
ferroptosis. Front Immunol. 14:12694512023. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bersuker K, Hendricks JM, Li Z, Magtanong
L, Ford B, Tang PH, Roberts MA, Tong B, Maimone TJ, Zoncu R, et al:
The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit
ferroptosis. Nature. 575:688–692. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Doll S, Freitas FP, Shah R, Aldrovandi M,
da Silva MC, Ingold I, Goya Grocin A, Xavier da Silva TN, Panzilius
E, Scheel CH, et al: FSP1 is a glutathione-independent ferroptosis
suppressor. Nature. 575:693–698. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen X, Comish PB, Tang D and Kang R:
Characteristics and biomarkers of ferroptosis. Front Cell Dev Biol.
9:6371622021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Friedmann Angeli JP, Schneider M, Proneth
B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch
A, Eggenhofer E, et al: Inactivation of the ferroptosis regulator
Gpx4 triggers acute renal failure in mice. Nat Cell Biol.
16:1180–1191. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yagoda N, von Rechenberg M, Zaganjor E,
Bauer AJ, Yang WS, Fridman DJ, Wolpaw AJ, Smukste I, Peltier JM,
Boniface JJ, et al: RAS-RAF-MEK-dependent oxidative cell death
involving voltage-dependent anion channels. Nature. 447:864–868.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Riegman M, Sagie L, Galed C, Levin T,
Steinberg N, Dixon SJ, Wiesner U, Bradbury MS, Niethammer P,
Zaritsky A and Overholtzer M: Ferroptosis occurs through an osmotic
mechanism and propagates independently of cell rupture. Nat Cell
Biol. 22:1042–1048. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Demuynck R, Efimova I, Naessens F and
Krysko DV: Immunogenic ferroptosis and where to find it? J
Immunother Cancer. 9:e0034302021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jiang X, Stockwell BR and Conrad M:
Ferroptosis: Mechanisms, biology and role in disease. Nat Rev Mol
Cell Biol. 22:266–282. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liang D, Minikes AM and Jiang X:
Ferroptosis at the intersection of lipid metabolism and cellular
signaling. Mol Cell. 82:2215–2227. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tang D, Chen X, Kang R and Kroemer G:
Ferroptosis: Molecular mechanisms and health implications. Cell
Res. 31:107–125. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen X, Kang R, Kroemer G and Tang D:
Broadening horizons: The role of ferroptosis in cancer. Nat Rev
Clin Oncol. 18:280–296. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang Y, Wu S, Li Q, Sun H and Wang H:
Pharmacological inhibition of ferroptosis as a therapeutic target
for neurodegenerative diseases and strokes. Adv Sci (Weinh).
10:e23003252023. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bitanihirwe BKY and Woo TUW: Oxidative
stress in schizophrenia: An integrated approach. Neurosci Biobehav
Rev. 35:878–893. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lotan A, Luza S, Opazo CM, Ayton S, Lane
DJR, Mancuso S, Pereira A, Sundram S, Weickert CS, Bousman C, et
al: Perturbed iron biology in the prefrontal cortex of people with
schizophrenia. Mol Psychiatry. 28:2058–2070. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Götz ME, Künig G, Riederer P and Youdim
MB: Oxidative stress: Free radical production in neural
degeneration. Pharmacol Ther. 63:37–122. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
David S, Jhelum P, Ryan F, Jeong SY and
Kroner A: Dysregulation of iron homeostasis in the central nervous
system and the role of ferroptosis in neurodegenerative disorders.
Antioxid Redox Signal. 37:150–170. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Qin Y, He Y, She Q, Larese-Casanova P, Li
P and Chai Y: Heterogeneity in respiratory electron transfer and
adaptive iron utilization in a bacterial biofilm. Nat Commun.
10:37022019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gomme PT, McCann KB and Bertolini J:
Transferrin: Structure, function and potential therapeutic actions.
Drug Discov Today. 10:267–273. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kakhlon O and Cabantchik ZI: The labile
iron pool: Characterization, measurement, and participation in
cellular processes(1). Free Radic Biol Med. 33:1037–1046. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Worwood M: Ferritin. Blood Rev. 4:259–269.
1990. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
MacKenzie EL, Iwasaki K and Tsuji Y:
Intracellular iron transport and storage: From molecular mechanisms
to health implications. Antioxid Redox Signal. 10:997–1030. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Reinert A, Morawski M, Seeger J, Arendt T
and Reinert T: Iron concentrations in neurons and glial cells with
estimates on ferritin concentrations. BMC Neurosci. 20:252019.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sunkara S, Radulović S, Lipovšek S, Birkl
C, Eggenreich S, Birkl-Toeglhofer AM, Schinagl M, Funk D,
Stöger-Pollach M, Haybaeck J, et al: Autolysis affects the iron
cargo of ferritins in neurons and glial cells at different rates in
the human brain. Cell Mol Neurobiol. 43:2909–2923. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Hansen TM, Nielsen H, Bernth N and Moos T:
Expression of ferritin protein and subunit mRNAs in normal and iron
deficient rat brain. Brain Res Mol Brain Res. 65:186–197. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wang P, Ren Q, Shi M, Liu Y, Bai H and
Chang YZ: Overexpression of mitochondrial ferritin enhances
blood-brain barrier integrity following ischemic stroke in mice by
maintaining iron homeostasis in endothelial cells. Antioxidants
(Basel). 11:12572022. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Wu H, Liu Q, Shan X, Gao W and Chen Q: ATM
orchestrates ferritinophagy and ferroptosis by phosphorylating
NCOA4. Autophagy. 19:2062–2077. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhang DL and Rouault TA: How does hepcidin
hinder ferroportin activity? Blood. 131:840–842. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Fang X, Ardehali H, Min J and Wang F: The
molecular and metabolic landscape of iron and ferroptosis in
cardiovascular disease. Nat Rev Cardiol. 20:7–23. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Delaby C, Pilard N, Gonçalves AS, Beaumont
C and Canonne-Hergaux F: Presence of the iron exporter ferroportin
at the plasma membrane of macrophages is enhanced by iron loading
and down-regulated by hepcidin. Blood. 106:3979–3984. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Ganz T: Hepcidin and its role in
regulating systemic iron metabolism. Hematology Am Soc Hematol Educ
Program. 2006:29–35. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ramey G, Deschemin JC, Durel B,
Canonne-Hergaux F, Nicolas G and Vaulont S: Hepcidin targets
ferroportin for degradation in hepatocytes. Haematologica.
95:501–504. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Qiu B, Zandkarimi F, Bezjian CT, Reznik E,
Soni RK, Gu W, Jiang X and Stockwell BR: Phospholipids with two
polyunsaturated fatty acyl tails promote ferroptosis. Cell.
187:1177–1190.e18. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Yang WS, Kim KJ, Gaschler MM, Patel M,
Shchepinov MS and Stockwell BR: Peroxidation of polyunsaturated
fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci
USA. 113:E4966–E4975. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Talvenmäki H, Lallukka N, Survo S and
Romantschuk M: Fenton's reaction-based chemical oxidation in
suboptimal conditions can lead to mobilization of oil hydrocarbons
but also contribute to the total removal of volatile compounds.
Environ Sci Pollut Res Int. 26:34670–34684. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Henning Y, Blind US, Larafa S, Matschke J
and Fandrey J: Hypoxia aggravates ferroptosis in RPE cells by
promoting the Fenton reaction. Cell Death Dis. 13:6622022.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Abe C and Miyazawa T and Miyazawa T:
Current use of Fenton reaction in drugs and food. Molecules.
27:54512022. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Valgimigli L: Lipid peroxidation and
antioxidant protection. Biomolecules. 13:12912023. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Minotti G and Aust SD: The role of iron in
oxygen radical mediated lipid peroxidation. Chem Biol Interact.
71:1–19. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Gaschler MM and Stockwell BR: Lipid
peroxidation in cell death. Biochem Biophys Res Commun.
482:419–425. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Janero DR: Malondialdehyde and
thiobarbituric acid-reactivity as diagnostic indices of lipid
peroxidation and peroxidative tissue injury. Free Radic Biol Med.
9:515–540. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Mas-Bargues C, Escrivá C, Dromant M,
Borrás C and Viña J: Lipid peroxidation as measured by
chromatographic determination of malondialdehyde. Human plasma
reference values in health and disease. Arch Biochem Biophys.
709:1089412021. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Li Y, Zhao T, Li J, Xia M, Li Y, Wang X,
Liu C, Zheng T, Chen R, Kan D, et al: Oxidative stress and
4-hydroxy-2-nonenal (4-HNE): Implications in the pathogenesis and
treatment of aging-related diseases. J Immunol Res.
2022:22339062022.PubMed/NCBI
|
|
66
|
Dalleau S, Baradat M, Guéraud F and Huc L:
Cell death and diseases related to oxidative stress:
4-Hydroxynonenal (HNE) in the balance. Cell Death Differ.
20:1615–1630. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Zhang X, Hou L, Guo Z, Wang G, Xu J, Zheng
Z, Sun K and Guo F: Lipid peroxidation in osteoarthritis: Focusing
on 4-hydroxynonenal, malondialdehyde, and ferroptosis. Cell Death
Discov. 9:3202023. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Butterfield DA: Brain lipid peroxidation
and alzheimer disease: Synergy between the Butterfield and Mattson
laboratories. Ageing Res Rev. 64:1010492020. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Villalón-García I, Povea-Cabello S,
Álvarez-Córdoba M, Talaverón-Rey M, Suárez-Rivero JM,
Suárez-Carrillo A, Munuera-Cabeza M, Reche-López D,
Cilleros-Holgado P, Piñero-Pérez R and Sánchez-Alcázar JA: Vicious
cycle of lipid peroxidation and iron accumulation in
neurodegeneration. Neural Regen Res. 18:1196–1202. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Shichiri M: The role of lipid peroxidation
in neurological disorders. J Clin Biochem Nutr. 54:151–160. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Dietrich-Muszalska A and Kontek B: Lipid
peroxidation in patients with schizophrenia. Psychiatry Clin
Neurosci. 64:469–475. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Bošković M, Vovk T, Kores Plesničar B and
Grabnar I: Oxidative stress in schizophrenia. Curr Neuropharmacol.
9:301–312. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Chen P, Wang D, Xiu M, Chen D, Lackey B,
Wu HE, Wang L and Zhang X: Association of transferrin gene
polymorphism with cognitive deficits and psychiatric symptoms in
patients with chronic schizophrenia. J Clin Med. 11:64142022.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Yeh S and Chang C: Cloning and
characterization of a specific coactivator, ARA70, for the androgen
receptor in human prostate cells. Proc Natl Acad Sci USA.
93:5517–5521. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Moore JMR, Galicia SJ, McReynolds AC,
Nguyen NH, Scanlan TS and Guy RK: Quantitative proteomics of the
thyroid hormone receptor-coregulator interactions. J Biol Chem.
279:27584–27590. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Kollara A and Brown TJ: Expression and
function of nuclear receptor co-activator 4: Evidence of a
potential role independent of co-activator activity. Cell Mol Life
Sci. 69:3895–3909. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Heinlein CA, Ting HJ, Yeh S and Chang C:
Identification of ARA70 as a ligand-enhanced coactivator for the
peroxisome proliferator-activated receptor gamma. J Biol Chem.
274:16147–16152. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Zhou ZX, He B, Hall SH, Wilson EM and
French FS: Domain interactions between coregulator ARA(70) and the
androgen receptor (AR). Mol Endocrinol. 16:287–300. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Mancias JD, Pontano Vaites L, Nissim S,
Biancur DE, Kim AJ, Wang X, Liu Y, Goessling W, Kimmelman AC and
Harper JW: Ferritinophagy via NCOA4 is required for erythropoiesis
and is regulated by iron dependent HERC2-mediated proteolysis.
Elife. 4:e103082015. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Federico G, Carrillo F, Dapporto F,
Chiariello M, Santoro M, Bellelli R and Carlomagno F: NCOA4 links
iron bioavailability to DNA metabolism. Cell Rep. 40:1112072022.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Kuno S, Fujita H, Tanaka YK, Ogra Y and
Iwai K: Iron-induced NCOA4 condensation regulates ferritin fate and
iron homeostasis. EMBO Rep. 23:e542782022. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Bellelli R, Federico G, Matte' A,
Colecchia D, Iolascon A, Chiariello M, Santoro M, De Franceschi L
and Carlomagno F: NCOA4 deficiency impairs systemic iron
homeostasis. Cell Rep. 14:411–421. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Arcos M, Liu Z, Villareal LB, Velez PK,
Desai SP, Noureddine A, Zheng H, Martin DR, Brinker J, Zhang D and
Xue X: Myeloid NCOA4 sequesters KEAP1 to reduce ferroptosis for
protection against salmonellosis in mice. Res Sq [Preprint].
rs.3.rs-4278310. 2024.PubMed/NCBI
|
|
84
|
Mancias JD, Wang X, Gygi SP, Harper JW and
Kimmelman AC: Quantitative proteomics identifies NCOA4 as the cargo
receptor mediating ferritinophagy. Nature. 509:105–109. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Kuno S and Iwai K: Oxygen modulates iron
homeostasis by switching iron sensing of NCOA4. J Biol Chem.
299:1047012023. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Gryzik M, Srivastava A, Longhi G, Bertuzzi
M, Gianoncelli A, Carmona F, Poli M and Arosio P: Expression and
characterization of the ferritin binding domain of nuclear receptor
coactivator-4 (NCOA4). Biochim Biophys Acta Gen Subj.
1861:2710–2716. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Zhao H, Lu Y, Zhang J, Sun Z, Cheng C, Liu
Y, Wu L, Zhang M, He W, Hao S and Li K: NCOA4 requires a [3Fe-4S]
to sense and maintain the iron homeostasis. J Biol Chem.
300:1056122024. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Li HY, Wei TT, Zhuang M, Tan CY, Xie TH,
Cai J, Yao Y and Zhu L: Iron derived from NCOA4-mediated
ferritinophagy causes cellular senescence via the cGAS-STING
pathway. Cell Death Discov. 9:4192023. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Hoelzgen F, Nguyen TTP, Klukin E, Boumaiza
M, Srivastava AK, Kim EY, Zalk R, Shahar A, Cohen-Schwartz S,
Meyron-Holtz EG, et al: Structural basis for the intracellular
regulation of ferritin degradation. Nat Commun. 15:38022024.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Yuan L, Sun Y, Zhou N, Wu W, Zheng W and
Wang Y: Dihydroquercetin attenuates silica-induced pulmonary
fibrosis by inhibiting ferroptosis signaling pathway. Front
Pharmacol. 13:8456002022. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Lahiri V, Hawkins WD and Klionsky DJ:
Watch what you (self-) eat: Autophagic mechanisms that modulate
metabolism. Cell Metab. 29:803–826. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Vermot A, Petit-Härtlein I, Smith SME and
Fieschi F: NADPH oxidases (NOX): An overview from discovery,
molecular mechanisms to physiology and pathology. Antioxidants
(Basel). 10:8902021. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Nazari B, Jaquet V and Krause KH: NOX
family NADPH oxidases in mammals: Evolutionary conservation and
isoform-defining sequences. Redox Biol. 66:1028512023. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Lambeth JD: Nox enzymes, ROS, and chronic
disease: An example of antagonistic pleiotropy. Free Radic Biol
Med. 43:332–347. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Chen F, Haigh S, Barman S and Fulton DJR:
From form to function: The role of Nox4 in the cardiovascular
system. Front Physiol. 3:4122012. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Altenhöfer S, Radermacher KA, Kleikers
PWM, Wingler K and Schmidt HHHW: Evolution of NADPH oxidase
inhibitors: Selectivity and mechanisms for target engagement.
Antioxid Redox Signal. 23:406–427. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Martyn KD, Frederick LM, von Loehneysen K,
Dinauer MC and Knaus UG: Functional analysis of Nox4 reveals unique
characteristics compared to other NADPH oxidases. Cell Signal.
18:69–82. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Nisimoto Y, Diebold BA, Cosentino-Gomes D
and Lambeth JD: Nox4: A hydrogen peroxide-generating oxygen sensor.
Biochemistry. 53:5111–5120. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Serrander L, Cartier L, Bedard K, Banfi B,
Lardy B, Plastre O, Sienkiewicz A, Fórró L, Schlegel W and Krause
KH: NOX4 activity is determined by mRNA levels and reveals a unique
pattern of ROS generation. Biochem J. 406:105–114. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Di Marzo N, Chisci E and Giovannoni R: The
role of hydrogen peroxide in redox-dependent signaling: Homeostatic
and pathological responses in mammalian cells. Cells. 7:1562018.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Feng S, Tang D, Wang Y, Li X, Bao H, Tang
C, Dong X, Li X, Yang Q, Yan Y, et al: The mechanism of ferroptosis
and its related diseases. Mol Biomed. 4:332023. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Rada B and Leto TL: Oxidative innate
immune defenses by Nox/Duox family NADPH oxidases. Contrib
Microbiol. 15:164–187. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Konno T, Melo EP, Chambers JE and Avezov
E: Intracellular sources of ROS/H2O2 in
health and neurodegeneration: Spotlight on endoplasmic reticulum.
Cells. 10:2332021. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Yin H, Xu L and Porter NA: Free radical
lipid peroxidation: Mechanisms and analysis. Chem Rev.
111:5944–5972. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Rochette L, Dogon G, Rigal E, Zeller M,
Cottin Y and Vergely C: Lipid peroxidation and iron metabolism: Two
corner stones in the homeostasis control of ferroptosis. Int J Mol
Sci. 24:4492022. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Zhang H, Wang A, Li G, Zhai Q, Huang Z,
Wang X, Cao Z, Liu L, Liu G, Chen B, et al: Osteoporotic bone loss
from excess iron accumulation is driven by NOX4-triggered
ferroptosis in osteoblasts. Free Radic Biol Med. 198:123–136. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Mancardi D, Mezzanotte M, Arrigo E,
Barinotti A and Roetto A: Iron overload, oxidative stress, and
ferroptosis in the failing heart and liver. Antioxidants (Basel).
10:18642021. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Orino K, Lehman L, Tsuji Y, Ayaki H, Torti
SV and Torti FM: Ferritin and the response to oxidative stress.
Biochem J. 357:241–247. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Zhu J, Xiong Y, Zhang Y, Wen J, Cai N,
Cheng K, Liang H and Zhang W: The Molecular mechanisms of
regulating oxidative stress-induced ferroptosis and therapeutic
strategy in tumors. Oxid Med Cell Longev. 2020:88107852020.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Cipriano A, Viviano M, Feoli A, Milite C,
Sarno G, Castellano S and Sbardella G: NADPH oxidases: From
molecular mechanisms to current inhibitors. J Med Chem.
66:11632–11655. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Yang WS, SriRamaratnam R, Welsch ME,
Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji
AF, Clish CB, et al: Regulation of ferroptotic cancer cell death by
GPX4. Cell. 156:317–331. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Wu J, Feng Z, Chen L, Li Y, Bian H, Geng
J, Zheng ZH, Fu X, Pei Z, Qin Y, et al: TNF antagonist sensitizes
synovial fibroblasts to ferroptotic cell death in collagen-induced
arthritis mouse models. Nat Commun. 13:6762022. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Wang L, Liu Y, Du T, Yang H, Lei L, Guo M,
Ding HF, Zhang J, Wang H, Chen X and Yan C: ATF3 promotes
erastin-induced ferroptosis by suppressing system Xc. Cell Death
Differ. 27:662–675. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Inaba Y, Hashiuchi E, Watanabe H, Kimura
K, Oshima Y, Tsuchiya K, Murai S, Takahashi C, Matsumoto M,
Kitajima S, et al: The transcription factor ATF3 switches cell
death from apoptosis to necroptosis in hepatic steatosis in male
mice. Nat Commun. 14:1672023. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Liu S, Li Z, Lan S, Hao H, Baz AA, Yan X,
Gao P, Chen S and Chu Y: The dual roles of activating transcription
factor 3 (ATF3) in inflammation, apoptosis, ferroptosis, and
pathogen infection responses. Int J Mol Sci. 25:8242024. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Wu J, Huang Y, Zhou X, Xiang Z, Yang Z,
Meng D, Wu D, Zhang J and Yang J: ATF3 and its emerging role in
atherosclerosis: A narrative review. Cardiovasc Diagn Ther.
12:926–942. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Shao CJ, Zhou HL, Gao XZ and Xu CF:
Downregulation of miR-221-3p promotes the ferroptosis in gastric
cancer cells via upregulation of ATF3 to mediate the transcription
inhibition of GPX4 and HRD1. Transl Oncol. 32:1016492023.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Feng Z, Cao K, Sun H and Liu X: SEH1L
siliencing induces ferroptosis and suppresses hepatocellular
carcinoma progression via ATF3/HMOX1/GPX4 axis. Apoptosis.
29:1723–1737. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Lu H, Zheng Y and Wang D: ATF3 affects
osteogenic differentiation in inflammatory hPDLSCs by mediating
ferroptosis via regulating the Nrf2/HO-1 signaling pathway. Tissue
Cell. 89:1024472024. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Lei P, Bai T and Sun Y: Mechanisms of
ferroptosis and relations with regulated cell death: A review.
Front Physiol. 10:1392019. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Wu Y, Zhang S, Gong X, Tam S, Xiao D, Liu
S and Tao Y: The epigenetic regulators and metabolic changes in
ferroptosis-associated cancer progression. Mol Cancer. 19:392020.
View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Wei S, Wang H, Lu C, Malmut S, Zhang J,
Ren S, Yu G, Wang W, Tang DD and Yan C: The activating
transcription factor 3 protein suppresses the oncogenic function of
mutant p53 proteins. J Biol Chem. 289:8947–8959. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Li X, Guo M, Cai L, Du T, Liu Y, Ding HF,
Wang H, Zhang J, Chen X and Yan C: Competitive ubiquitination
activates the tumor suppressor p53. Cell Death Differ.
27:1807–1818. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Zhao J, Li X, Guo M, Yu J and Yan C: The
common stress responsive transcription factor ATF3 binds genomic
sites enriched with p300 and H3K27ac for transcriptional
regulation. BMC Genomics. 17:3352016. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Akbarpour Arsanjani A, Abuei H,
Behzad-Behbahani A, Bagheri Z, Arabsolghar R and Farhadi A:
Activating transcription factor 3 inhibits NF-κB p65 signaling
pathway and mediates apoptosis and cell cycle arrest in cervical
cancer cells. Infect Agent Cancer. 17:622022. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Kwon JW, Kwon HK, Shin HJ, Choi YM, Anwar
MA and Choi S: Activating transcription factor 3 represses
inflammatory responses by binding to the p65 subunit of NF-κB. Sci
Rep. 5:144702015. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Liu J, Zhang C, Wang J, Hu W and Feng Z:
The regulation of ferroptosis by tumor suppressor p53 and its
pathway. Int J Mol Sci. 21:83872020. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Kang R, Kroemer G and Tang D: The tumor
suppressor protein p53 and the ferroptosis network. Free Radic Biol
Med. 133:162–168. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Jiang L, Kon N, Li T, Wang SJ, Su T,
Hibshoosh H, Baer R and Gu W: Ferroptosis as a p53-mediated
activity during tumour suppression. Nature. 520:57–62. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Comporti M: Glutathione depleting agents
and lipid peroxidation. Chem Phys Lipids. 45:143–169. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Ou Y, Wang SJ, Li D, Chu B and Gu W:
Activation of SAT1 engages polyamine metabolism with p53-mediated
ferroptotic responses. Proc Natl Acad Sci USA. 113:E6806–E6812.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Suzuki S, Tanaka T, Poyurovsky MV, Nagano
H, Mayama T, Ohkubo S, Lokshin M, Hosokawa H, Nakayama T, Suzuki Y,
et al: Phosphate-activated glutaminase (GLS2), a p53-inducible
regulator of glutamine metabolism and reactive oxygen species. Proc
Natl Acad Sci USA. 107:7461–7466. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Hu W, Zhang C, Wu R, Sun Y, Levine A and
Feng Z: Glutaminase 2, a novel p53 target gene regulating energy
metabolism and antioxidant function. Proc Natl Acad Sci USA.
107:7455–7460. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Engeland K: Cell cycle regulation:
p53-p21-RB signaling. Cell Death Differ. 29:946–960. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Engeland K: Cell cycle arrest through
indirect transcriptional repression by p53: I have a DREAM. Cell
Death Differ. 25:114–132. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Chen W, Sun Z, Wang XJ, Jiang T, Huang Z,
Fang D and Zhang DD: Direct interaction between Nrf2 and
p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response.
Mol Cell. 34:663–673. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Pouremamali F, Pouremamali A, Dadashpour
M, Soozangar N and Jeddi F: An update of Nrf2 activators and
inhibitors in cancer prevention/promotion. Cell Commun Signal.
20:1002022. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Villeneuve NF, Sun Z, Chen W and Zhang DD:
Nrf2 and p21 regulate the fine balance between life and death by
controlling ROS levels. Cell Cycle. 8:3255–3256. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Franklin CC, Backos DS, Mohar I, White CC,
Forman HJ and Kavanagh TJ: Structure, function, and
post-translational regulation of the catalytic and modifier
subunits of glutamate cysteine ligase. Mol Aspects Med. 30:86–98.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Lu SC: Regulation of glutathione
synthesis. Mol Aspects Med. 30:42–59. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Dahl EL and Mulcahy RT: Cell-type specific
differences in glutamate cysteine ligase transcriptional regulation
demonstrate independent subunit control. Toxicol Sci. 61:265–272.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Wang X, Shen T, Lian J, Deng K, Qu C, Li
E, Li G, Ren Y, Wang Z, Jiang Z, et al: Resveratrol reduces
ROS-induced ferroptosis by activating SIRT3 and compensating the
GSH/GPX4 pathway. Mol Med. 29:1372023. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Weaver K and Skouta R: The selenoprotein
glutathione peroxidase 4: From molecular mechanisms to novel
therapeutic opportunities. Biomedicines. 10:8912022. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Xie Y, Kang R, Klionsky DJ and Tang D:
GPX4 in cell death, autophagy, and disease. Autophagy.
19:2621–2638. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Labrecque CL and Fuglestad B:
Electrostatic drivers of GPx4 interactions with membrane, lipids,
and DNA. Biochemistry. 60:2761–2772. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Yan HF, Zou T, Tuo QZ, Xu S, Li H, Belaidi
AA and Lei P: Ferroptosis: Mechanisms and links with diseases.
Signal Transduct Target Ther. 6:492021. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Sugezawa K, Morimoto M, Yamamoto M,
Matsumi Y, Nakayama Y, Hara K, Uejima C, Kihara K, Matsunaga T,
Tokuyasu N, et al: GPX4 regulates tumor cell proliferation via
suppressing ferroptosis and exhibits prognostic significance in
gastric cancer. Anticancer Res. 42:5719–5729. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Li J, Liu J, Zhou Z, Wu R, Chen X, Yu C,
Stockwell B, Kroemer G, Kang R and Tang D: Tumor-specific GPX4
degradation enhances ferroptosis-initiated antitumor immune
response in mouse models of pancreatic cancer. Sci Transl Med.
15:eadg30492023. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Li H, Sun Y, Yao Y, Ke S, Zhang N, Xiong
W, Shi J, He C, Xiao X, Yu H, et al: USP8-governed GPX4 homeostasis
orchestrates ferroptosis and cancer immunotherapy. Proc Natl Acad
Sci USA. 121:e23155411212024. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Shimizu J, Murao A, Nofi C, Wang P and
Aziz M: Extracellular CIRP promotes GPX4-mediated ferroptosis in
sepsis. Front Immunol. 13:9038592022. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Niu B, Liao K, Zhou Y, Wen T, Quan G, Pan
X and Wu C: Application of glutathione depletion in cancer therapy:
Enhanced ROS-based therapy, ferroptosis, and chemotherapy.
Biomaterials. 277:1211102021. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Wu M, Xu LG, Li X, Zhai Z and Shu HB:
AMID, an apoptosis-inducing factor-homologous
mitochondrion-associated protein, induces caspase-independent
apoptosis. J Biol Chem. 277:25617–25623. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Ohiro Y, Garkavtsev I, Kobayashi S,
Sreekumar KR, Nantz R, Higashikubo BT, Duffy SL, Higashikubo R,
Usheva A, Gius D, et al: A novel p53-inducible apoptogenic gene,
PRG3, encodes a homologue of the apoptosis-inducing factor (AIF).
FEBS Lett. 524:163–171. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Nakamura T, Mishima E, Yamada N, Mourão
ASD, Trümbach D, Doll S, Wanninger J, Lytton E, Sennhenn P, Nishida
Xavier da Silva T, et al: Integrated chemical and genetic screens
unveil FSP1 mechanisms of ferroptosis regulation. Nat Struct Mol
Biol. 30:1806–1815. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Lv Y, Liang C, Sun Q, Zhu J, Xu H, Li X,
Li YY, Wang Q, Yuan H, Chu B and Zhu D: Structural insights into
FSP1 catalysis and ferroptosis inhibition. Nat Commun. 14:59332023.
View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Deshwal S, Onishi M, Tatsuta T, Bartsch T,
Cors E, Ried K, Lemke K, Nolte H, Giavalisco P and Langer T:
Mitochondria regulate intracellular coenzyme Q transport and
ferroptotic resistance via STARD7. Nat Cell Biol. 25:246–257.
2023.PubMed/NCBI
|
|
157
|
Frei B, Kim MC and Ames BN: Ubiquinol-10
is an effective lipid-soluble antioxidant at physiological
concentrations. Proc Natl Acad Sci USA. 87:4879–4883. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Ernster L, Forsmark P and Nordenbrand K:
The mode of action of lipid-soluble antioxidants in biological
membranes. Relationship between the effects of ubiquinol and
vitamin E as inhibitors of lipid peroxidation in submitochondrial
particles. J Nutr Sci Vitaminol (Tokyo) Spec No. 548–551. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Sharifi-Rad M, Anil Kumar NV, Zucca P,
Varoni EM, Dini L, Panzarini E, Rajkovic J, Tsouh Fokou PV, Azzini
E, Peluso I, et al: Lifestyle, oxidative stress, and antioxidants:
Back and forth in the pathophysiology of chronic diseases. Front
Physiol. 11:6942020. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Wang Y and Hekimi S: Understanding
ubiquinone. Trends Cell Biol. 26:367–378. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Naguib YW, Saha S, Skeie JM, Acri T, Ebeid
K, Abdel-Rahman S, Kesh S, Schmidt GA, Nishimura DY, Banas JA, et
al: Solubilized ubiquinol for preserving corneal function.
Biomaterials. 275:1208422021. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Zameitat E, Freymark G, Dietz CD, Löffler
M and Bölker M: Functional expression of human dihydroorotate
dehydrogenase (DHODH) in pyr4 mutants of ustilago maydis allows
target validation of DHODH inhibitors in vivo. Appl Environ
Microbiol. 73:3371–3379. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Reis RAG, Calil FA, Feliciano PR, Pinheiro
MP and Nonato MC: The dihydroorotate dehydrogenases: Past and
present. Arch Biochem Biophys. 632:175–191. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Evans DR and Guy HI: Mammalian pyrimidine
biosynthesis: Fresh insights into an ancient pathway. J Biol Chem.
279:33035–33038. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Leban J, Kralik M, Mies J, Gassen M,
Tentschert K and Baumgartner R: SAR, species specificity, and
cellular activity of cyclopentene dicarboxylic acid amides as DHODH
inhibitors. Bioorg Med Chem Lett. 15:4854–4857. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Vyas VK and Ghate M: Recent developments
in the medicinal chemistry and therapeutic potential of
dihydroorotate dehydrogenase (DHODH) inhibitors. Mini Rev Med Chem.
11:1039–1055. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Rodriguez JMO, Krupinska E, Wacklin-Knecht
H and Knecht W: Preparation of human dihydroorotate dehydrogenase
for interaction studies with lipid bilayers. Nucleosides
Nucleotides Nucleic Acids. 39:1306–1319. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Peeters MJW, Aehnlich P, Pizzella A,
Mølgaard K, Seremet T, Met Ö, Rasmussen LJ, Thor Straten P and
Desler C: Mitochondrial-linked de novo pyrimidine biosynthesis
dictates human T-cell proliferation but not expression of effector
molecules. Front Immunol. 12:7188632021. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Martínez-Reyes I, Cardona LR, Kong H,
Vasan K, McElroy GS, Werner M, Kihshen H, Reczek CR, Weinberg SE,
Gao P, et al: Mitochondrial ubiquinol oxidation is necessary for
tumour growth. Nature. 585:288–292. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Yang C, Zhao Y, Wang L, Guo Z, Ma L, Yang
R, Wu Y, Li X, Niu J, Chu Q, et al: De novo pyrimidine biosynthetic
complexes support cancer cell proliferation and ferroptosis
defence. Nat Cell Biol. 25:836–847. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee
H, Koppula P, Wu S, Zhuang L, Fang B, et al: DHODH-mediated
ferroptosis defence is a targetable vulnerability in cancer.
Nature. 593:586–590. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Ding Q, Tang W, Li X, Ding Y, Chen X, Cao
W, Wang X, Mo W, Su Z, Zhang Q and Guo H: Mitochondrial-targeted
brequinar liposome boosted mitochondrial-related ferroptosis for
promoting checkpoint blockade immunotherapy in bladder cancer. J
Control Release. 363:221–234. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Wang F and Min J: DHODH tangoing with GPX4
on the ferroptotic stage. Signal Transduct Target Ther. 6:2442021.
View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Diao J, Jia Y, Dai E, Liu J, Kang R, Tang
D, Han L, Zhong Y and Meng L: Ferroptotic therapy in cancer:
Benefits, side effects, and risks. Mol Cancer. 23:892024.
View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Westphal S and Kalthoff H: Apoptosis:
Targets in pancreatic cancer. Mol Cancer. 2:62003. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Todaro M, Lombardo Y, Francipane MG, Alea
MP, Cammareri P, Iovino F, Di Stefano AB, Di Bernardo C, Agrusa A,
Condorelli G, et al: Apoptosis resistance in epithelial tumors is
mediated by tumor-cell-derived interleukin-4. Cell Death Differ.
15:762–772. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Miao Z, Xu L, Gu W, Ren Y, Li R, Zhang S,
Chen C, Wang H, Ji J and Chen J: A targetable PRR11-DHODH axis
drives ferroptosis- and temozolomide-resistance in glioblastoma.
Redox Biol. 73:1032202024. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Fanet H, Capuron L, Castanon N, Calon F
and Vancassel S: Tetrahydrobioterin (BH4) pathway: From metabolism
to neuropsychiatry. Curr Neuropharmacol. 19:591–609. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Feng Y, Feng Y, Gu L, Liu P, Cao J and
Zhang S: The critical role of tetrahydrobiopterin (BH4) metabolism
in modulating radiosensitivity: BH4/NOS axis as an angel or a
devil. Front Oncol. 11:7206322021. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Burg AW and Brown GM: The biosynthesis of
folic acid. 8. Purification and properties of the enzyme that
catalyzes the production of formate from carbon atom 8 of guanosine
triphosphate. J Biol Chem. 243:2349–2358. 1968. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Niederwieser A, Staudenmann W and Wetzel
E: High-performance liquid chromatography with column switching for
the analysis of biogenic amine metabolites and pterins. J
Chromatogr. 290:237–246. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Jiang Y, Zhao J, Li R, Liu Y, Zhou L, Wang
C, Lv C, Gao L and Cui D: CircLRFN5 inhibits the progression of
glioblastoma via PRRX2/GCH1 mediated ferroptosis. J Exp Clin Cancer
Res. 41:3072022. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Liang D, Shu R, Jiang S, Gan Q, Wu S, Zhao
Y, Yang L, Xu M, Gao J and Meng Y: Expression of BmDHFR is
up-regulated to trigger an increase in the BH4/BH2 ratio when the
de novo synthesis of BH4 is blocked in silkworm, Bombyx mori. Int J
Biol Macromol. 225:625–633. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Ebenhoch R, Bauer M, Reinert D, Kersting
A, Huber S, Schmid A, Hinz I, Feiler M, Müller K and Nar H:
Biophysical and structural investigation of the regulation of human
GTP cyclohydrolase I by its regulatory protein GFRP. J Struct Biol.
213:1076912021. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Kraft VAN, Bezjian CT, Pfeiffer S,
Ringelstetter L, Müller C, Zandkarimi F, Merl-Pham J, Bao X,
Anastasov N, Kössl J, et al: GTP cyclohydrolase
1/tetrahydrobiopterin counteract ferroptosis through lipid
remodeling. ACS Cent Sci. 6:41–53. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Wang D, Liang W, Huo D, Wang H, Wang Y,
Cong C, Zhang C, Yan S, Gao M, Su X, et al: SPY1 inhibits neuronal
ferroptosis in amyotrophic lateral sclerosis by reducing lipid
peroxidation through regulation of GCH1 and TFR1. Cell Death
Differ. 30:369–382. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Vasquez-Vivar J, Shi Z and Tan S:
Tetrahydrobiopterin in cell function and death mechanisms. Antioxid
Redox Signal. 37:171–183. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Eichwald T, da Silva LDB, Staats Pires AC,
Niero L, Schnorrenberger E, Filho CC, Espíndola G, Huang WL,
Guillemin GJ, Abdenur JE and Latini A: Tetrahydrobiopterin: Beyond
its traditional role as a cofactor. Antioxidants (Basel).
12:10372023. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Villaume WA: Marginal BH4 deficiencies,
iNOS, and self-perpetuating oxidative stress in post-acute sequelae
of Covid-19. Med Hypotheses. 163:1108422022. View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Soula M, Weber RA, Zilka O, Alwaseem H, La
K, Yen F, Molina H, Garcia-Bermudez J, Pratt DA and Birsoy K:
Metabolic determinants of cancer cell sensitivity to canonical
ferroptosis inducers. Nat Chem Biol. 16:1351–1360. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Xiao Y, Yuan Y, Yang Y, Liu B, Ding Z, Luo
J, Chen S and Yu L: GCH1 reduces LPS-induced alveolar macrophage
polarization and inflammation by inhibition of ferroptosis. Inflamm
Res. 72:1941–1955. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Famitafreshi H and Karimian M: Paradoxical
regulation of iron in hippocampus and prefrontal cortex induces
schizophrenic-like symptoms in male rats. Int J Neurosci.
130:384–390. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
193
|
Kim SW, Stewart R, Park WY, Jhon M, Lee
JY, Kim SY, Kim JM, Amminger P, Chung YC and Yoon JS: Latent iron
deficiency as a marker of negative symptoms in patients with
first-episode schizophrenia spectrum disorder. Nutrients.
10:17072018. View Article : Google Scholar : PubMed/NCBI
|
|
194
|
Wei N, Ju M, Su X, Zhang Y, Huang Y, Rao
X, Cui L, Lin Z and Dong Y: Transplantation of gut microbiota
derived from patients with schizophrenia induces schizophrenia-like
behaviors and dysregulated brain transcript response in mice.
Schizophrenia (Heidelb). 10:442024. View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Feng S, Chen J, Qu C, Yang L, Wu X, Wang
S, Yang T, Liu H, Fang Y and Sun P: Identification of
ferroptosis-related genes in schizophrenia based on bioinformatic
analysis. Genes (Basel). 13:21682022. View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Liu X, Fan L, Lu C, Yin S and Hu H:
Functional role of p53 in the regulation of chemical-induced
oxidative stress. Oxid Med Cell Longev. 2020:60397692020.PubMed/NCBI
|
|
197
|
Byeon SH, Lee SC, Choi SH, Lee HK, Lee JH,
Chu YK and Kwon OW: Vascular endothelial growth factor as an
autocrine survival factor for retinal pigment epithelial cells
under oxidative stress via the VEGF-R2/PI3K/Akt. Invest Ophthalmol
Vis Sci. 51:1190–1197. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Martín-Vázquez E, Cobo-Vuilleumier N,
López-Noriega L, Lorenzo PI and Gauthier BR: The
PTGS2/COX2-PGE2 signaling cascade in inflammation: Pro
or anti? A case study with type 1 diabetes mellitus. Int J Biol
Sci. 19:4157–4165. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
199
|
Lian K, Li Y, Yang W, Ye J, Liu H, Wang T,
Yang G, Cheng Y and Xu X: Hub genes, a diagnostic model, and immune
infiltration based on ferroptosis-linked genes in schizophrenia.
IBRO Neurosci Rep. 16:317–328. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Feng Y and Shen J: Machine learning-based
predictive models and drug prediction for schizophrenia in multiple
programmed cell death patterns. Front Mol Neurosci. 16:11237082023.
View Article : Google Scholar : PubMed/NCBI
|
|
201
|
Zhang D, Wu X, Xue X, Li W, Zhou P, Lv Z,
Zhao K and Zhu F: Ancient dormant virus remnant ERVW-1 drives
ferroptosis via degradation of GPX4 and SLC3A2 in schizophrenia.
Virol Sin. 39:31–43. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
202
|
Ursini F and Maiorino M: Lipid
peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic
Biol Med. 152:175–185. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
203
|
Forcina GC and Dixon SJ: GPX4 at the
crossroads of lipid homeostasis and ferroptosis. Proteomics.
19:e18003112019. View Article : Google Scholar : PubMed/NCBI
|
|
204
|
Casanova MF, Crapanzano KA, Mannheim G and
Kruesi M: Sydenham's chorea and schizophrenia: A case report.
Schizophr Res. 16:73–76. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
205
|
Casanova MF, Comparini SO, Kim RW and
Kleinman JE: Staining intensity of brain iron in patients with
schizophrenia: A postmortem study. J Neuropsychiatry Clin Neurosci.
4:36–41. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
206
|
Madden DJ and Merenstein JL: Quantitative
susceptibility mapping of brain iron in healthy aging and
cognition. Neuroimage. 282:1204012023. View Article : Google Scholar : PubMed/NCBI
|
|
207
|
Ravanfar P, Syeda WT, Jayaram M, Rushmore
RJ, Moffat B, Lin AP, Lyall AE, Merritt AH, Yaghmaie N, Laskaris L,
et al: In vivo 7-tesla MRI investigation of brain iron and its
metabolic correlates in chronic schizophrenia. Schizophrenia
(Heidelb). 8:862022. View Article : Google Scholar : PubMed/NCBI
|
|
208
|
Sonnenschein SF, Parr AC, Larsen B,
Calabro FJ, Foran W, Eack SM, Luna B and Sarpal DK: Subcortical
brain iron deposition in individuals with schizophrenia. J
Psychiatr Res. 151:272–278. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
209
|
Cao B, Yan L, Ma J, Jin M, Park C, Nozari
Y, Kazmierczak OP, Zuckerman H, Lee Y, Pan Z, et al: Comparison of
serum essential trace metals between patients with schizophrenia
and healthy controls. J Trace Elem Med Biol. 51:79–85. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
210
|
Santa Cruz EC, Madrid KC, Arruda MAZ and
Sussulini A: Association between trace elements in serum from
bipolar disorder and schizophrenia patients considering treatment
effects. J Trace Elem Med Biol. 59:1264672020. View Article : Google Scholar : PubMed/NCBI
|
|
211
|
Owiredu WKBA, Brenya PK, Osei Y, Laing EF,
Okrah CO, Obirikorang C, Anto EO, Acheampong E and Donkor S:
Evaluation of serum iron overload, AST:ALT ratio and
log10ferritin:AST ratio among schizophrenia patients in
the Kumasi Metropolis, Ghana: A case-control study. BMC Res Notes.
12:8022019. View Article : Google Scholar : PubMed/NCBI
|
|
212
|
Kumar J, Liddle EB, Fernandes CC,
Palaniyappan L, Hall EL, Robson SE, Simmonite M, Fiesal J, Katshu
MZ, Qureshi A, et al: Glutathione and glutamate in schizophrenia: A
7T MRS study. Mol Psychiatry. 25:873–882. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
213
|
Nucifora LG, Tanaka T, Hayes LN, Kim M,
Lee BJ, Matsuda T, Nucifora FC Jr, Sedlak T, Mojtabai R, Eaton W
and Sawa A: Reduction of plasma glutathione in psychosis associated
with schizophrenia and bipolar disorder in translational
psychiatry. Transl Psychiatry. 7:e12152017. View Article : Google Scholar : PubMed/NCBI
|
|
214
|
Tenório MCDS, Graciliano NG, Moura FA, de
Oliveira ACM and Goulart MOF: N-acetylcysteine (NAC): Impacts on
human health. Antioxidants (Basel). 10:9672021. View Article : Google Scholar : PubMed/NCBI
|
|
215
|
Raghu G, Berk M, Campochiaro PA, Jaeschke
H, Marenzi G, Richeldi L, Wen FQ, Nicoletti F and Calverley PMA:
The multifaceted therapeutic role of N-acetylcysteine (NAC) in
disorders characterized by oxidative stress. Curr Neuropharmacol.
19:1202–1224. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
216
|
das Neves Duarte JM, Kulak A,
Gholam-Razaee MM, Cuenod M, Gruetter R and Do KQ: N-acetylcysteine
normalizes neurochemical changes in the glutathione-deficient
schizophrenia mouse model during development. Biol Psychiatry.
71:1006–1014. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
217
|
Berk M, Copolov DL, Dean O, Lu K, Jeavons
S, Schapkaitz I, Anderson-Hunt M and Bush AI: N-acetyl cysteine for
depressive symptoms in bipolar disorder-a double-blind randomized
placebo-controlled trial. Biol Psychiatry. 64:468–475. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
218
|
Conus P, Seidman LJ, Fournier M, Xin L,
Cleusix M, Baumann PS, Ferrari C, Cousins A, Alameda L,
Gholam-Rezaee M, et al: N-acetylcysteine in a double-blind
randomized placebo-controlled trial: Toward biomarker-guided
treatment in early psychosis. Schizophr Bull. 44:317–327. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
219
|
Rapado-Castro M, Dodd S, Bush AI, Malhi
GS, Skvarc DR, On ZX, Berk M and Dean OM: Cognitive effects of
adjunctive N-acetyl cysteine in psychosis. Psychol Med. 47:866–876.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
220
|
Sepehrmanesh Z and Heidary M, Akasheh N,
Akbari H and Heidary M: Therapeutic effect of adjunctive N-acetyl
cysteine (NAC) on symptoms of chronic schizophrenia: A
double-blind, randomized clinical trial. Prog Neuropsychopharmacol
Biol Psychiatry. 82:289–296. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
221
|
Farokhnia M, Azarkolah A, Adinehfar F,
Khodaie-Ardakani MR, Hosseini SM, Yekehtaz H, Tabrizi M, Rezaei F,
Salehi B, Sadeghi SM, et al: N-acetylcysteine as an adjunct to
risperidone for treatment of negative symptoms in patients with
chronic schizophrenia: A randomized, double-blind,
placebo-controlled study. Clin Neuropharmacol. 36:185–192. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
222
|
Breier A, Liffick E, Hummer TA, Vohs JL,
Yang Z, Mehdiyoun NF, Visco AC, Metzler E, Zhang Y and Francis MM:
Effects of 12-month, double-blind N-acetyl cysteine on symptoms,
cognition and brain morphology in early phase schizophrenia
spectrum disorders. Schizophr Res. 199:395–402. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
223
|
Medvedev ON, Berk M, Dean OM, Brown E,
Sandham MH, Dipnall JF, McNamara RK, Sumich A, Krägeloh CU,
Narayanan A and Siegert RJ: A novel way to quantify schizophrenia
symptoms in clinical trials. Eur J Clin Invest. 51:e133982021.
View Article : Google Scholar : PubMed/NCBI
|
|
224
|
Rossell SL, Francis PS, Galletly C, Harris
A, Siskind D, Berk M, Bozaoglu K, Dark F, Dean O, Liu D, et al:
N-acetylcysteine (NAC) in schizophrenia resistant to clozapine: A
double blind randomised placebo controlled trial targeting negative
symptoms. BMC Psychiatry. 16:3202016. View Article : Google Scholar : PubMed/NCBI
|
|
225
|
Lavoie S, Murray MM, Deppen P, Knyazeva
MG, Berk M, Boulat O, Bovet P, Bush AI, Conus P, Copolov D, et al:
Glutathione precursor, N-acetyl-cysteine, improves mismatch
negativity in schizophrenia patients. Neuropsychopharmacology.
33:2187–2199. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
226
|
Carmeli C, Knyazeva MG, Cuénod M and Do
KQ: Glutathione precursor N-acetyl-cysteine modulates EEG
synchronization in schizophrenia patients: A double-blind,
randomized, placebo-controlled trial. PLoS One. 7:e293412012.
View Article : Google Scholar : PubMed/NCBI
|
|
227
|
Naguy A and Naguy C: N-acetyl-cysteine in
schizophrenia-there is more than meets the eyes! CNS Spectr.
26:446–447. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
228
|
Deepmala Slattery J, Kumar N, Delhey L,
Berk M, Dean O, Spielholz C and Frye R: Clinical trials of
N-acetylcysteine in psychiatry and neurology: A systematic review.
Neurosci Biobehav Rev. 55:294–321. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
229
|
Dean O, Giorlando F and Berk M:
N-acetylcysteine in psychiatry: Current therapeutic evidence and
potential mechanisms of action. J Psychiatry Neurosci. 36:78–86.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
230
|
Klauser P, Xin L, Fournier M, Griffa A,
Cleusix M, Jenni R, Cuenod M, Gruetter R, Hagmann P, Conus P, et
al: N-acetylcysteine add-on treatment leads to an improvement of
fornix white matter integrity in early psychosis: A double-blind
randomized placebo-controlled trial. Transl Psychiatry. 8:2202018.
View Article : Google Scholar : PubMed/NCBI
|
|
231
|
Tharoor H, Mara S and Gopal S: Role of
novel dietary supplement N-acetyl cysteine in treating negative
symptoms in schizophrenia: A 6-month follow-up study. Indian J
Psychol Med. 40:139–142. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
232
|
Tiihonen J, Tanskanen A and Taipale H:
20-Year nationwide follow-up study on discontinuation of
antipsychotic treatment in first-episode schizophrenia. Am J
Psychiatry. 175:765–773. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
233
|
Bjørklund G, Shanaida M, Lysiuk R,
Antonyak H, Klishch I, Shanaida V and Peana M: Selenium: An
antioxidant with a critical role in anti-aging. Molecules.
27:66132022. View Article : Google Scholar : PubMed/NCBI
|
|
234
|
Brown JS Jr: Role of selenium and other
trace elements in the geography of schizophrenia. Schizophr Bull.
20:387–398. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
235
|
Deng MG, Cui HT, Nie JQ, Liang Y and Chai
C: Genetic association between circulating selenium level and the
risk of schizophrenia in the European population: A two-sample
Mendelian randomization study. Front Nutr. 9:9698872022. View Article : Google Scholar : PubMed/NCBI
|
|
236
|
Cai L, Chen T, Yang J, Zhou K, Yan X, Chen
W, Sun L, Li L, Qin S, Wang P, et al: Serum trace element
differences between schizophrenia patients and controls in the Han
Chinese population. Sci Rep. 5:150132015. View Article : Google Scholar : PubMed/NCBI
|
|
237
|
Mattmiller SA, Carlson BA and Sordillo LM:
Regulation of inflammation by selenium and selenoproteins: Impact
on eicosanoid biosynthesis. J Nutr Sci. 2:e282013. View Article : Google Scholar : PubMed/NCBI
|
|
238
|
Soares de Oliveira AR, Jayanne Clímaco
Cruz K, Beatriz Silva Morais J, Rocha Dos Santos L, Rodrigues de
Sousa Melo S, Fontenelle LC, Santos de Sousa G, Costa Maia CS,
Oliveira Duarte de Araújo C, Leal Mendes I, et al: Selenium status
and oxidative stress in obese: Influence of adiposity. Eur J Clin
Invest. 51:e135382021. View Article : Google Scholar : PubMed/NCBI
|
|
239
|
Rayman MP: The importance of selenium to
human health. Lancet. 356:233–241. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
240
|
Li Z, Liu Y, Li X, Ju W, Wu G, Yang X, Fu
X and Gao X: Association of elements with schizophrenia and
intervention of selenium supplements. Biol Trace Elem Res.
183:16–21. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
241
|
Jamilian H and Ghaderi A: The effects of
probiotic and selenium co-supplementation on clinical and metabolic
scales in chronic schizophrenia: A randomized, double-blind,
placebo-controlled trial. Biol Trace Elem Res. 199:4430–4438. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
242
|
Calder PC: Omega-3 fatty acids and
inflammatory processes. Nutrients. 2:355–374. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
243
|
Djuricic I and Calder PC: Beneficial
outcomes of omega-6 and omega-3 polyunsaturated fatty acids on
human health: An update for 2021. Nutrients. 13:24212021.
View Article : Google Scholar : PubMed/NCBI
|
|
244
|
Tang W, Wang Y, Xu F, Fan W, Zhang Y, Fan
K, Wang W, Zhang Y and Zhang C: Omega-3 fatty acids ameliorate
cognitive dysfunction in schizophrenia patients with metabolic
syndrome. Brain Behav Immun. 88:529–534. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
245
|
Hsu MC, Huang YS and Ouyang WC: Beneficial
effects of omega-3 fatty acid supplementation in schizophrenia:
Possible mechanisms. Lipids Health Dis. 19:1592020. View Article : Google Scholar : PubMed/NCBI
|
|
246
|
Hsu MC and Ouyang WC: A systematic review
of effectiveness of omega-3 fatty acid supplementation on symptoms,
social functions, and neurobiological variables in schizophrenia.
Biol Res Nurs. 23:723–737. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
247
|
Goh KK, Chen CYA, Chen CH and Lu ML:
Effects of omega-3 polyunsaturated fatty acids supplements on
psychopathology and metabolic parameters in schizophrenia: A
meta-analysis of randomized controlled trials. J Psychopharmacol.
35:221–235. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
248
|
Surette ME: The science behind dietary
omega-3 fatty acids. CMAJ. 178:177–180. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
249
|
Fuentes NR, Kim E, Fan YY and Chapkin RS:
Omega-3 fatty acids, membrane remodeling and cancer prevention. Mol
Aspects Med. 64:79–91. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
250
|
Joffre C, Rey C and Layé S: N-3
polyunsaturated fatty acids and the resolution of
neuroinflammation. Front Pharmacol. 10:10222019. View Article : Google Scholar : PubMed/NCBI
|
|
251
|
Chen Y, Fang ZM, Yi X, Wei X and Jiang DS:
The interaction between ferroptosis and inflammatory signaling
pathways. Cell Death Dis. 14:2052023. View Article : Google Scholar : PubMed/NCBI
|
|
252
|
Simopoulos AP: Omega-3 fatty acids in
inflammation and autoimmune diseases. J Am Coll Nutr. 21:495–505.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
253
|
Gutiérrez S, Svahn SL and Johansson ME:
Effects of omega-3 fatty acids on immune cells. Int J Mol Sci.
20:50282019. View Article : Google Scholar : PubMed/NCBI
|
|
254
|
Nuñez MT and Chana-Cuevas P: New
perspectives in iron chelation therapy for the treatment of
neurodegenerative diseases. Pharmaceuticals (Basel). 11:1092018.
View Article : Google Scholar : PubMed/NCBI
|
|
255
|
Devos D, Labreuche J, Rascol O, Corvol JC,
Duhamel A, Guyon Delannoy P, Poewe W, Compta Y, Pavese N, Růžička
E, et al: Trial of deferiprone in Parkinson's disease. N Engl J
Med. 387:2045–2055. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
256
|
Urrutia PJ, Bórquez DA and Núñez MT:
Inflaming the brain with Iron. Antioxidants (Basel). 10:612021.
View Article : Google Scholar : PubMed/NCBI
|
|
257
|
Costa I, Barbosa DJ, Benfeito S, Silva V,
Chavarria D, Borges F, Remião F and Silva R: Molecular mechanisms
of ferroptosis and their involvement in brain diseases. Pharmacol
Ther. 244:1083732023. View Article : Google Scholar : PubMed/NCBI
|
|
258
|
Ramadhan MIA, Sitanaya SN, Hakim AHW and
Ramli Y: The role of iron-chelating therapy in improving
neurological outcome in patients with intracerebral hemorrhage:
Evidence-based case report. Medicina (Kaunas). 59:4532023.
View Article : Google Scholar : PubMed/NCBI
|
|
259
|
Dusek P, Schneider SA and Aaseth J: Iron
chelation in the treatment of neurodegenerative diseases. J Trace
Elem Med Biol. 38:81–92. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
260
|
Cutler P: Iron overload and psychiatric
illness. Can J Psychiatry. 39:8–11. 1994. View Article : Google Scholar : PubMed/NCBI
|