Introduction
Non-alcoholic fatty liver disease (NAFLD) has become
the leading cause of chronic liver disease worldwide, with ~25% of
the global population suffering from NAFLD (1). NAFLD includes non-alcoholic fatty
liver (NAFL), non-alcoholic steatohepatitis (NASH), liver fibrosis
and cirrhosis, and the main early manifestation of NAFLD is hepatic
steatosis. As the disease progresses, hepatic steatosis can
progress to NASH with liver fibrosis (2,3).
After NASH progresses to the middle or advanced stages, cirrhosis
or even liver cancer may occur (4). Previous studies have shown that NASH
is a pathogenic factor of cirrhosis, hepatocellular carcinoma and
other end-stage liver diseases (5,6).
Although the aetiology and pathogenesis of NAFLD are
still unknown, a number of studies have indicated that, in addition
to insulin resistance and hepatocellular inflammation, changes in
the intestinal microflora and liver metabolic genes may be key
factors in the occurrence and progression of NAFLD (7,8). The
intestine and liver are closely connected, and enterohepatic axis
dysbiosis is closely related to the occurrence of numerous liver
diseases, including NAFL, NASH, liver fibrosis and cirrhosis
(9–11). The metabolites of the intestinal
microflora include bile acids, choline, short-chain fatty acids and
endogenous intestinal ethanol. Previous studies have shown that
abnormal intestinal microflora metabolites can induce the
occurrence and development of NAFLD (12–15).
Therefore, the treatment of NAFLD by regulating the intestinal
microflora and its metabolites has garnered interest (14).
Rifaximin is an intestinal-specific broad-spectrum
antibiotic, which is not absorbed by the intestine and is not
metabolized by the liver. Rifaximin can improve the intestinal
microflora and has been approved by the U.S. Food and Drug
Administration (FDA) for treating traveller's diarrhoea and hepatic
encephalopathy (16,17). It can also effectively improve the
abdominal distension and stool characteristics of patients with
irritable bowel syndrome (18,19).
Clinical studies have confirmed that rifaximin can ameliorate serum
endotoxaemia, inhibit proinflammatory factor expression and improve
liver function in patients with NASH (20,21).
However, the underlying molecular biological mechanisms of
rifaximin in treating NASH are still unclear.
The present study aimed to investigate the anti-NASH
effect of the nonabsorbable antibiotic rifaximin and its specific
molecular mechanisms.
Materials and methods
Ethics approval
Animal health and behaviour were monitored once
every 2 days. The mice with methionine-choline deficient (MCD)
diet-induced NASH, mice with NASH and intestinal decontamination,
and Hnf1α knockout (Hnf1αH-KO) mice with NASH were
sacrificed at the end of 6, 4 and 24 weeks, respectively, and blood
(from cardiac puncture), liver, terminal ileum and caecal content
samples were collected. The mice were sacrificed by cervical
dislocation following the intraperitoneal injection of
pentobarbital sodium (>120 mg/kg). Animal welfare was
considered, including minimizing suffering and distress, and using
the most appropriate dose of anaesthetic. In addition, death was
verified by respiratory and cardiac arrest. All animal experiments
were approved by the Naval Medical University (approval no.
SYXK2021-0075) and Nanchang University (approval no.
LL-202303280001).
NASH mouse model and rifaximin
treatment
A total of 24 male C57BL/6 mice (age, 6 weeks;
weight, ~20 g) were purchased from the Shanghai Experimental Center
of the Chinese Academy of Sciences and were housed in the
Experimental Animal Center of the Second Military Medical
University (also known as Naval Medical University, Shanghai,
China) or Nanchang University (Nanchang, China) in a specific
pathogen-free environment at 24°C and 50% humidity under a 12-h
light/dark cycle. The mice were randomly divided into two groups:
One group was fed a methionine-choline sufficient (MCS) diet ad
libitum (n=8), and the other group was fed an MCD diet (Trophic
Animal Feed High-tech Co.) ad libitum (n=16). After 2 weeks,
the mice fed the MCD diet were randomly separated into two groups:
The MCD group (n=8) and the MCD + rifaximin group (n=8). The mice
in the MCD + rifaximin group were treated with rifaximin
(MedChemExpress) by oral gavage (100 mg/kg/day) for 4 weeks
(Fig. S1A). The rifaximin dose
was chosen based on a previous report (22). The mice in the MCS and MCD groups
were fed water according to their body weight for 4 weeks.
Intestinal decontamination of mice
with NASH and rifaximin treatment
A total of 32 male C57BL/6 mice (age, 6 weeks;
weight, ~20 g) were purchased from the Shanghai Experimental Center
of the Chinese Academy of Sciences and were housed in the
Experimental Animal Center of the Second Military Medical
University or Nanchang University in a specific pathogen-free
environment at 24°C and 50% humidity under a 12-h light/dark cycle.
The mice were randomly divided into the following four groups: MCD
group (n=8), MCD + rifaximin group (n=8), MCD + Abx group (n=8) and
MCD + Abx + rifaximin group (n=8). To determine whether rifaximin
relies on the intestinal microflora to serve a biological role in
mice with NASH, after 1 week of treatment with broad-spectrum
antibiotics (ampicillin 1 g/l, neomycin sulfate 1 g/l,
metronidazole 1 g/l and vancomycin 0.5 g/l; MedChemExpress), which
were added to the drinking water as described previously (23), 6-week-old male C57BL/6 mice in the
MCD + Abx + rifaximin group were administered rifaximin (100
mg/kg/day) by oral gavage for 3 weeks. The mice in the MCD + Abx
group were administered broad-spectrum antibiotics in their
drinking water for 1 week and were then treated with water by oral
gavage according to their body weight for 3 weeks. The mice in the
MCD group were administered water for 1 week and were then treated
with water by oral gavage according to their body weight for 3
weeks. The mice in the MCD + rifaximin group were administered
water for 1 week and were then treated with rifaximin (100
mg/kg/day) by oral gavage for 3 weeks. All mice were fed an MCD
diet for 4 weeks. Animal health and behaviour were monitored once
every 2 days. All mice were sacrificed after being fed an MCD diet
for 4 weeks, and blood (from cardiac puncture), liver and terminal
ileum samples were collected.
Generation of hepatocyte-specific
Hnf1αH-KO mice and rifaximin treatment
Hnf1αH-KO mice were generated by Shanghai
Model Organisms Centre, Inc. by crossing mice homozygous for floxed
Hnf1α (Hnf1αf/f) with Alb-Cre transgenic mice as
described in our previous research (24). Male Hnf1αH-KO mice (age,
6 weeks; weight, ~20 g, n=10) were fed a normal chow diet ad
libitum in a specific pathogen-free environment at 24°C under a
12-h light/dark cycle for 20 weeks to induce NASH, as described in
our previous research (24). In
addition, the male mice in the Hnf1αf/f group (age, 6
weeks; weight, ~20 g, n=5) were fed a normal chow diet ad
libitum for 20 weeks. Subsequently, Hnf1αH-KO mice
were randomly separated into two groups: The Hnf1αH-KO
group (n=5) and the Hnf1αH-KO + rifaximin group (n=5).
The mice in Hnf1αH-KO + rifaximin group were
administered rifaximin (100 mg/kg/day) by oral gavage for 4 weeks.
The mice in the Hnf1αf/f group and Hnf1αH-KO
group were treated with water according to their body weight for 4
weeks. Animal health and behaviour were monitored once every 2
days. All mice were sacrificed at the end of 24 weeks, and blood
(from cardiac puncture), liver and terminal ileum samples were
collected.
Histology and
immunohistochemistry
Mouse livers that were fixed in 4% paraformaldehyde
at 4°C for 24 h were used for frozen sectioning for Oil Red O
staining, and those fixed in 10% formalin at room temperature for
24 h were used for paraffin sectioning according to standard
procedures. Paraffin-embedded liver sections (4 µm) were stained
with haematoxylin and eosin (H&E; haematoxylin staining for 5
min at room temperature and eosin staining for 15 sec at room
temperature) for histopathological examination. Oil Red O
(Sigma-Aldrich; Merck KGaA) staining was performed for 20 min at
room temperature to evaluate the degree of hepatocyte steatosis.
Sirius red (Leagene; Beijing Regen Biotechnology Co., Ltd.)
staining was performed for 1 h at room temperature for collagen
detection. Images were captured using a light microscope
(magnifications, ×10, ×20 and ×40). Liver histology was estimated
according to the steatosis score, ballooning score, inflammation
score and NAFLD activity score (NAS) designed and validated by the
NASH-Clinical Research Network (25). Image analysis software Image-Pro
Plus 6.0 (Media Cybernetics, Inc.) was used to semi-quantify the
intensity of steatosis or collagen deposition according to the
percentage of the positive area of Oil Red O staining or Sirius red
staining in the corresponding field of liver tissue.
The paraffin-embedded liver sections underwent
immunohistochemical staining according to standard procedures.
Briefly, liver sections were deparaffinized in xylene and
rehydrated in a series of alcohol concentrations. Subsequently,
liver sections underwent antigen retrieval (alkali repair; Tris
base 0.3 g, EDTA 0.1 g, H2O 250 ml) at 100°C for 20 min.
They were then soaked in 3% H2O2 for 10 min
at room temperature to remove endogenous peroxidase. Liver sections
were blocked with 10% goat serum (cat. no. c0265; Beyotime
Institute of Biotechnology) at room temperature for 1 h. The slides
were incubated overnight at 4°C with primary antibodies against
α-smooth muscle actin (α-SMA; 1:1,000; cat. no. ab5694; Abcam) and
collagen type 1 α1 (Col1A1; 1:1,000; cat. no. BA0325; Wuhan Boster
Biological Technology, Ltd.), followed by incubation with a
horseradish peroxidase-linked immunoglobulin G secondary antibody
[cat. no. GK500710; reagent A; 1:500; Gene Technology (Shanghai)
Co., Ltd.] at room temperature for 1 h. Finally, an
GTVision™ III Detection Rabbit/Mouse Kit [cat. no.
GK500710; reagent B1; Gene Technology (Shanghai) Co., Ltd.] was
used for staining for 3 min at room temperature. Images were
captured using a light microscope (magnifications, ×10, ×20 and
×40).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from the liver and distal
ileum tissues following the standard TRIzol®
(Invitrogen; Thermo Fisher Scientific, Inc.) method. Subsequently,
1 µg total RNA was used as a template for cDNA synthesis using
PrimeScript RT Master Mix (Takara Bio, Inc.) at 37°C for 15 min and
85°C for 30 sec. The transcript levels were detected by qPCR with a
SYBR Green PCR Kit (Takara Bio, Inc.). qPCR conditions were as
follows: 95°C for 5 min, followed by 40 cycles at 95°C for 30 sec
and 60°C for 30 sec. mRNA expression levels were calculated using
the 2−ΔΔCq method and Gapdh was used as the internal
reference (26). The primers used
are listed in Table I. All
reactions were repeated three times.
 | Table I.Primer sequences used for reverse
transcription-quantitative PCR. |
Table I.
Primer sequences used for reverse
transcription-quantitative PCR.
| Gene | Primer
sequence |
|---|
| Pparγ | Forward:
5′-TCGCTGATGCACTGCCTATG-3′ |
|
| Reverse:
5′-GAGAGGTCCACAGAGCTGATT-3′ |
| CD36 | Forward:
5′-GTGGCCTTGCACTCTCTCAT-3′ |
|
| Reverse:
5′-CATCCACCAGTTGCTCCACA-3′ |
| Srebp1c | Forward:
5′-GAAGCTGTCGGGGTAGCGTCT-3′ |
|
| Reverse:
5′-CTCTCAGGAGAGTTGGCACCTG-3′ |
| α-Sma | Forward:
5′-CTGTCCCTCTATGCCTCTGG-3′ |
|
| Reverse:
5′-AGGGCTGTGATCTCCTTCTG-3′ |
| Col1a1 | Forward:
5′-TAAAGGGTCATCGTGGCTTC-3′ |
|
| Reverse:
5′-GACGGCTGAGTAGGGAACAC-3′ |
| Fxr | Forward:
5′-TGGGCTCCGAATCCTCTTAGA-3′ |
|
| Reverse:
5′-TGGTCCTCAAATAAGATCCTTGG-3′ |
| Fgf15 | Forward:
5′-GCCATCAAGGACGTCAGCA-3′ |
|
| Reverse:
5′-CTTCCTCCGAGTAGCGAATCAG-3′ |
| Shp | Forward:
5′-TCTGCAGGTCGTCCGACTATTC-3′ |
|
| Reverse:
5′-AGGCAGTGGCTGTGAGATGC-3′ |
| Cyp7a1 | Forward:
5′-TCATTGCTTCAGGGCTCCTG-3′ |
|
| Reverse:
5′-TGGGCATCTCAAGCAAACAC-3′ |
| Cyp7b1 | Forward:
5′-TAGGCATGACGATCCTGAAA-3′ |
|
| Reverse:
5′-TCTCTGGTGAAGTGGACTGAAA-3′ |
| Cyp8b1 | Forward:
5′-GATCCGTCGCGGAGATAAGG-3′ |
|
| Reverse:
5′-CGGGTTGAGGAACCGATCAT-3′ |
| Cyp27a1 | Forward:
5′-TCTGGCTACCTGCACTTCCT-3′ |
|
| Reverse:
5′-GTGTGTTGGATGTCGTGTCC-3′ |
| Hnf1α | Forward:
5′-ATGACACGGATGACGATGGG-3′ |
|
| Reverse:
5′-GCCATGGGTCCTCCTGAAG-3′ |
| Gapdh | Forward:
5′-ACCCTTAAGAGGGATGCTGC-3′ |
|
| Reverse:
5′-CCCAATACGGCCAAATCCGT-3′ |
Western blot analysis
Total protein was extracted from the liver tissues
using lysis buffer [Tris-HCl (pH 6.8), 5 ml; 20% SDS, 10 ml;
glycerol, 9.9 ml] supplemented with phenylmethanesulfonyl fluoride
(Beyotime Institute of Biotechnology). The protein concentration
was determined using a BCA protein assay kit (Beyotime Institute of
Biotechnology). Protein samples were electrophoresed on 10%
SDS-PAGE gels and were then electrotransferred onto nitrocellulose
membranes (cat. no. HATF29325; MilliporeSigma). After blocking with
5% skim milk in PBS-0.1% Tween, the membranes were incubated with
specific primary antibodies overnight at 4°C, followed by
incubation with donkey-anti-mouse or donkey-anti-rabbit secondary
antibodies (1:3,000; cat. nos. 926-32212 and 926-32213; IRDye 700-
or 800-conjugated; LI-COR Biosciences) for 1 h at room temperature.
Finally, the signals were visualized using an Odyssey infrared
imaging system (LI-COR Biosciences) at a wavelength of 700 or 800
nm. The densities of the protein bands were semi-quantified using
ImageJ 1.8.0 software (National Institutes of Health). Gapdh was
used as the internal control. The primary antibodies used were as
follows: Pparγ (cat. no. sc-7273; Santa Cruz Biotechnology, Inc.),
sterol regulatory element-binding protein 1 (Srebp1; cat. no.
ab28481; Abcam), α-Sma (cat. no. ab5694; Abcam), Hnf1α (cat. no.
ab272693; Abcam) and Gapdh (cat. no. BSAP0063; Bioworld Technology,
Inc.).
Measurement of hydroxyproline
content
The liver hydroxyproline content was measured
according to the protocol of the Hydroxyproline Detection Kit (cat.
no. A030-2; Nanjing Jiancheng Bioengineering Institute) as
described in our previous study (27).
Bacterial 16S rRNA amplicon sequencing
and analysis
Caecal contents were collected from the mice and
immediately frozen at −80°C. Microbial DNA was extracted from the
caecal contents using the PF Mag-Bind Stool DNA Kit (cat. no.
M9016-02, Omega Bio-Tek, Inc.) according to the manufacturer's
instructions. Briefly, 150 mg caecal contents were added to
microcentrifuge tubes containing lysis buffer, and 200 µl Buffer AL
was added to the sample and mixed. The 1.5-ml microcentrifuge tube
was used for collecting extracted DNA (centrifugation, 4°C, 13,000
× g, 5 min). The DNA quality was checked by 1% agarose gel
electrophoresis. The V3-V4 region of the 16S rRNA gene sequence was
PCR-amplified with primers (338 forward,
5′-ACTCCTACGGGAGGCAGCAG-3′; 806 reverse,
5′-GGACTACHVGGGTWTCTAAT-3′). The PCR conditions were as follows:
95°C for 3 min, followed by 27 cycles at 95°C for 30 sec and 55°C
for 30 sec. DNA (100 ng) underwent paired end sequencing
(sequencing kit: cat. no. NOVA-5144; NEXTflex Rapid DNA-Seq kit;
Bioo Scientific Corporation) on the Illumina MiSeq PE300 platform
(Illumina, Inc.) using standard protocols by Shanghai Majorbio
Bio-pharm Technology Co., Ltd.
The initial raw sequences were clustered into
operational taxonomic units (OTUs) of a 97% identity threshold
using Usearch (version 7.1) (28).
In addition, α diversity analysis was calculated with mothur index
analysis (version v.1.30.2; http://mothur.org/wiki/calculators). β diversity
analysis was performed using R software (version 3.3.1) (29) for mapping, using FastTree (version
2.1.3 http://www.microbesonline.org/fasttree/) for
constructing the evolutionary tree, and using FastUniFrac
(http://github.com/biocore/unifrac)
for analysing the distance matrix between samples. α diversity
analysis and β diversity analysis were used to assess the relative
abundance of intestinal microflora among the MCS group, MCD group
and MCD + rifaximin group. Non-metric multidimensional scaling
(NMDS) analysis, Adonis analysis and partial least squares
discriminant analysis (PLS-DA) based on amplicon sequence variant
were used to show that the overall composition of the intestinal
microbiome was significantly altered by different diets. Linear
discriminant analysis effect size (LEfSe) analysis was used to
estimate the impact of each microbiota species on the difference
between the MCD group and the MCD + rifaximin group via linear
discriminant analysis. Kruskal-Wallis test and Dunn's test were
performed to detect the significant differences in abundance, and
the groups with significant differences in abundance were
identified.
Bile acid analysis
Terminal ileum samples (0.1 g) were mixed with NaOH
and acetonitrile and centrifuged (4°C, 5,698 × g, 5 min). After
centrifugation, the supernatants were placed in a chromatographic
bottle to detect bile acid levels (performed by Shanghai Majorbio
Bio-pharm Technology Co., Ltd.). Chlorpropamide was used as an
internal standard for bile acid levels. The bile acid
concentrations of the terminal ileum samples were qualitatively and
quantitatively determined by liquid chromatography-electrospray
ionization-tandem mass spectrometry (LC-ESI-MS/MS) analysis method
[LC-ESI-MS/MS (UHPLC-Qtrap); Waters Corporation]. The mass
spectrometry system consisted of air curtain gas 40, ion spray
voltage of −4,500 V, temperature 550°C, ion source Gas1 50, and ion
source Gas2 50. Chromatographic separation was performed using the
BEH C18 liquid chromatography column (100×2.1 mm, 1.8 µm; Waters
Corporation). The sample size was 5 µl (flow rate, 0.4 ml/min), and
the mobile phases were phase A (0.1% formic acid in water) and
phase B (0.1% formic acid in acetonitrile). The bile acid standard
was used to identify different bile acid metabolites detected by
LC-ESI-MS/MS. Finally, the peak mass spectrum area of the
analytical sample was substituted into a linear equation to
calculate the concentration of bile acid.
Biochemical analysis
Mouse serum was extracted from whole blood via
centrifugation at 3,000 rpm for 15 min at 4°C. Alanine transaminase
(ALT) and aspartate transaminase (AST) levels in mouse serum were
measured using an automated analyser at Shanghai Sipur-Bika
Experimental Animal Co., Ltd.
Statistical analysis
Data are presented as the mean ± SEM. GraphPad Prism
7.0 software (Dotmatics) was used to analyse the experimental data
through one-way ANOVA or Kruskal-Wallis test among multiple groups,
and two-tailed unpaired Student's t-test or Mann-Whitney test
between two groups. The correlation between the gut microbiota and
bile acid levels in the terminal ileum was investigated using a
nonparametric Spearman's test. Gut microbiota clustering analysis
was performed using Pearson's correlation coefficient. P<0.05
was considered to indicate a statistically significant
difference.
Results
Rifaximin ameliorates NASH in MCD
diet-fed mice
After 4 weeks of rifaximin treatment, the livers of
the MCD + rifaximin group were smoother and moister than those of
the MCD group (Fig. 1A).
Furthermore, MCD-fed mice had a significant decrease in body weight
and increase in liver-to-body weight ratio compared with in the
MCS-fed mice, while rifaximin treatment significantly reduced the
liver-to-body weight ratio (Fig.
1B). As expected, the MCD diet caused hepatic steatosis and
inflammation, eventually leading to steatohepatitis in MCD group
mice compared with the MCS group. As shown in Fig. S1B, the body weight of mice in the
MCD + rifaximin group did not significantly differ from that of
mice in the MCD group. However, H&E staining of liver sections
showed decreased lipid deposition, ballooning and interlobular
inflammation in the MCD + rifaximin group compared with that in the
MCD group, revealing a notable decrease in the severity of hepatic
steatosis after rifaximin treatment (Fig. 1C). Additionally, rifaximin
treatment significantly decreased the ballooning score,
interlobular inflammation score and total NAS (Fig. 1D). Oil red O staining also showed
reduced lipid accumulation in the liver after rifaximin treatment
(Fig. 1E). Consistent with these
findings, the mRNA expression levels of Srebp1, Pparγ and CD36,
which are involved in liver lipid synthesis (30), were significantly decreased in the
MCD + rifaximin group compared with in the MCD group (Fig. 1F). Moreover, compared with in the
MCD group, plasma ALT in the MCD + rifaximin group was
significantly lower, and plasma AST exhibited a downward trend,
further indicating improvements in liver injury after rifaximin
treatment (Fig. 1G). These results
indicated that rifaximin treatment ameliorated NASH in MCD diet-fed
mice.
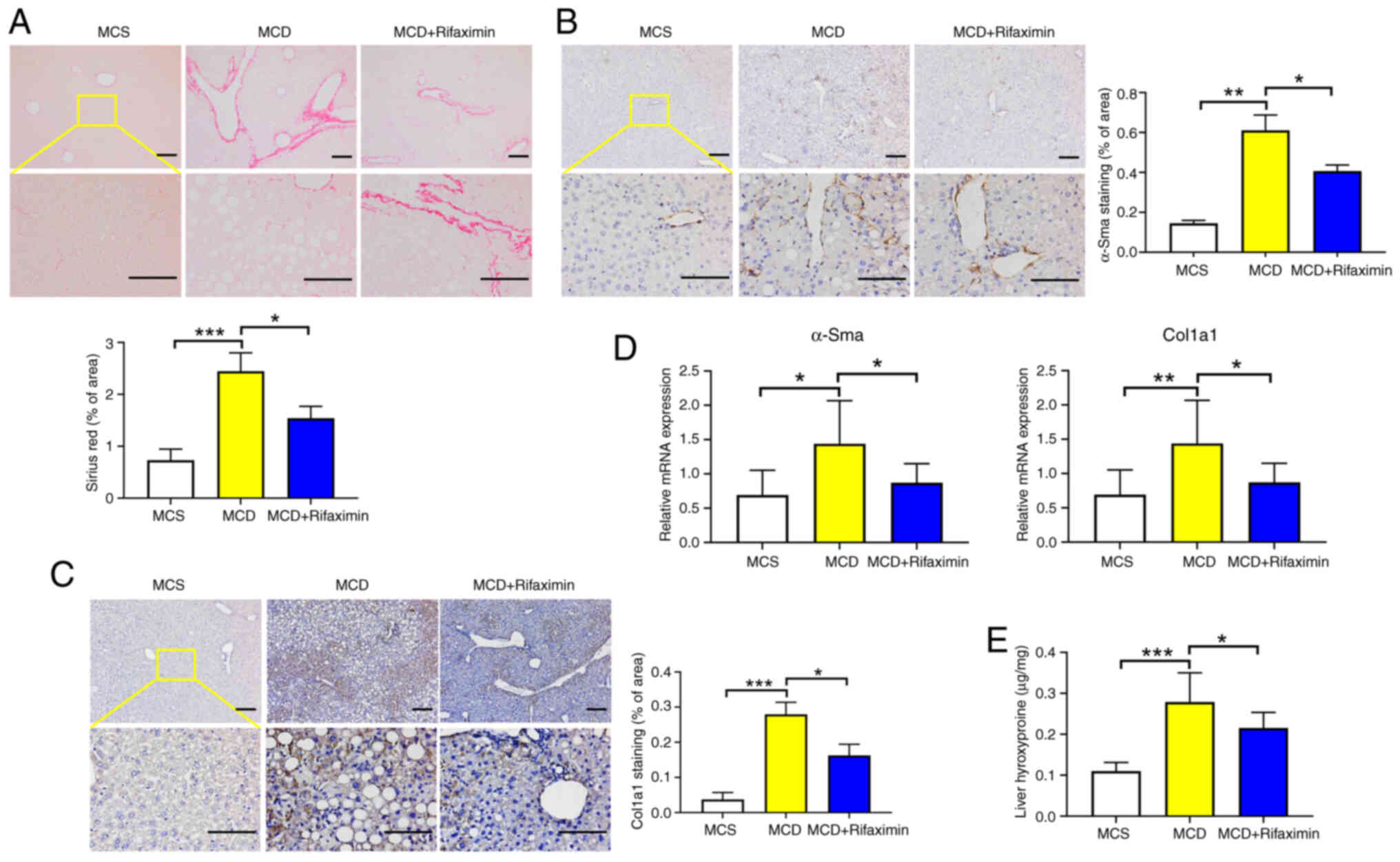
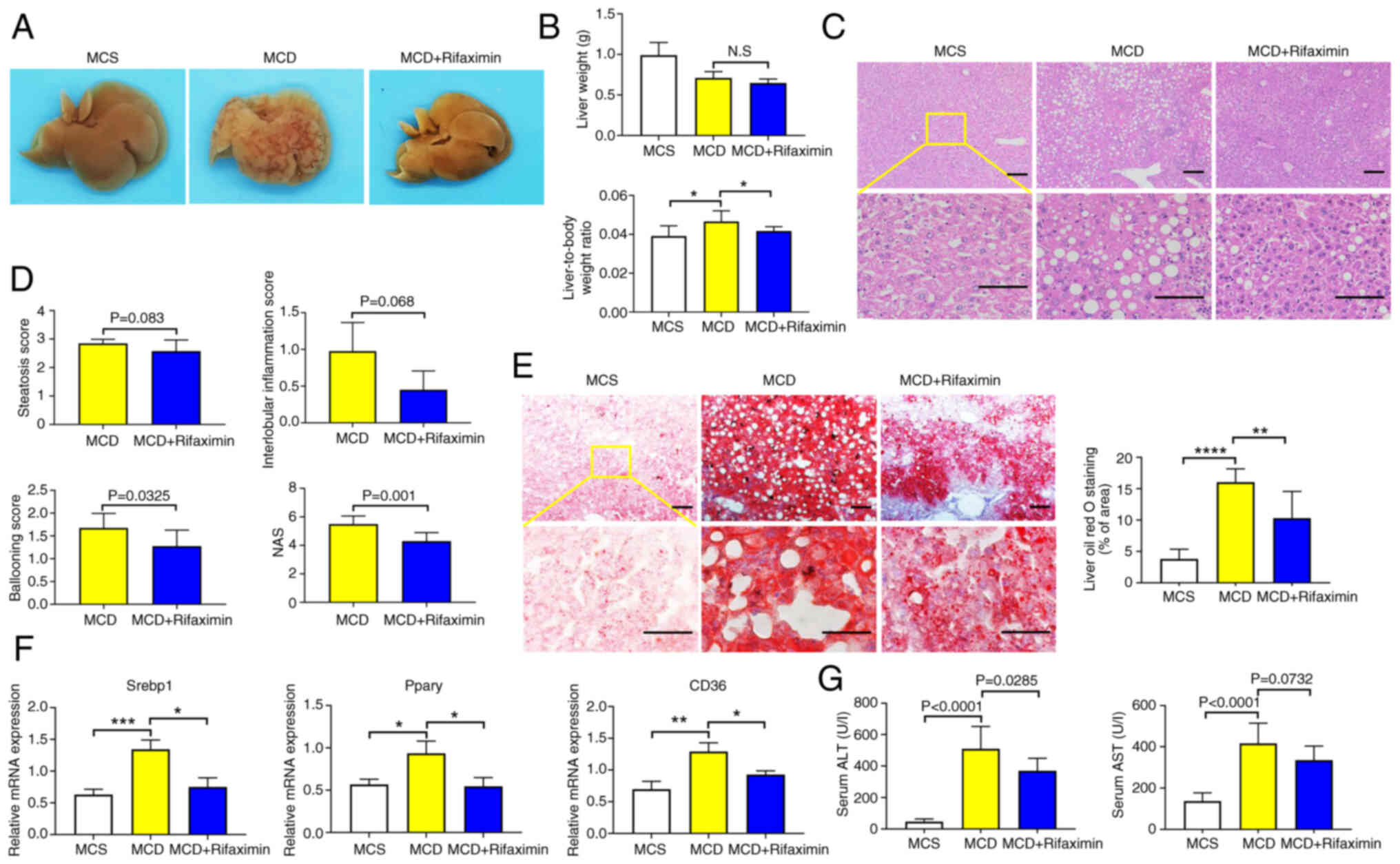 | Figure 1.Rifaximin ameliorates non-alcoholic
steatohepatitis in MCD diet-fed mice. (A) Photographs of livers in
the MCS, MCD and MCD + rifaximin groups. (B) Liver weight, and
liver weight to body weight ratio in each group. (C) Haematoxylin
and eosin staining of livers in the MCS, MCD and MCD + rifaximin
groups. Scale bars, 100 µm. (D) Steatosis score, hepatic ballooning
score, interlobular inflammation score and NAS. (E) Oil red O
staining of livers in the MCS, MCD and MCD + rifaximin groups.
Scale bars, 100 µm. (F) Srebp1, Pparγ and CD36 mRNA expression
levels in mouse livers. (G) Plasma ALT and AST levels in mice. n=8
mice/group. *P<0.05, **P<0.01, ***P<0.001,
****P<0.0001. ALT, alanine transaminase; AST, aspartate
transaminase; MCD, methionine-choline deficient; MCS,
methionine-choline sufficient; NAS, non-alcoholic fatty liver
disease activity score; Srebp1, sterol regulatory element-binding
protein 1. |
Rifaximin alleviates MCD diet-induced
liver fibrosis in mice with NASH
With the development of NASH, hepatic stellate cells
are activated, and hepatic collagen deposition increases, leading
to liver fibrosis (31).
Consistent with the findings of previous studies (32,33),
the present study revealed that collagen deposition was increased
in the livers of mice fed an MCD diet, while rifaximin treatment
significantly reduced collagen deposition (Fig. 2A). Moreover, the α-Sma and Col1a1
mRNA and protein expression levels were significantly decreased
after rifaximin treatment, as confirmed by immunohistochemistry and
RT-qPCR (Fig. 2B-D). Additionally,
compared with that in the MCD group, the liver hydroxyproline
content in the MCD + rifaximin group was lower, which also
indicated that rifaximin treatment alleviated liver fibrosis in
mice with MCD diet-induced NASH (Fig.
2E).
Rifaximin modulates the gut microbiota
of mice with MCD diet-induced NASH
To explore the molecular mechanism by which
rifaximin improves NASH, 16S rRNA pyrosequencing was performed on
the caecal contents of mice with NASH. It was observed that
rifaximin treatment significantly decreased the faecal OTU, Chao1
estimator and Simpson index, but markedly increased the Shannon
index, indicating that rifaximin could markedly reduce the total
intestinal microflora and increase bacterial richness (Fig. 3A). As confirmed by plots from NMDS
analysis, Adonis analysis and PLS-DA, the gut microbiota
composition was substantially reshaped after rifaximin treatment
(Figs. 3B, and S2A and B). Notably, LEfSe bar analysis,
which estimated the magnitude of the influence of the abundance of
each component on the differential effect, indicated that the
phylum Epsilonbacteraeota was the most abundant gut microbiota
associated with group separation between the MCD group and the MCD
+ rifaximin group (Fig. 3C).
Notably, it was observed that, at the phylum level, MCD markedly
increased the abundance of Epsilonbacteraeota, the most
significantly changed bacteria, while rifaximin treatment
significantly decreased the increase in Epsilonbacteraeota
abundance in MCD diet-induced NASH mice (Fig. 3D and E). To further determine
alterations in the gut microbiota, the intestinal microflora was
analysed at the genus level. It was revealed that MCD significantly
increased the abundance of Helicobacter hepaticus, a member
of the Epsilonbacteraeota phylum, while rifaximin treatment
markedly decreased the increase in the abundance of Helicobacter
hepaticus (P=0.0000314; Figs.
3F, 3G and S2C). These results suggested that
rifaximin treatment modulated the gut microbiota community,
especially Helicobacter hepaticus, in the caecal contents of
mice with MCD diet-induced NASH.
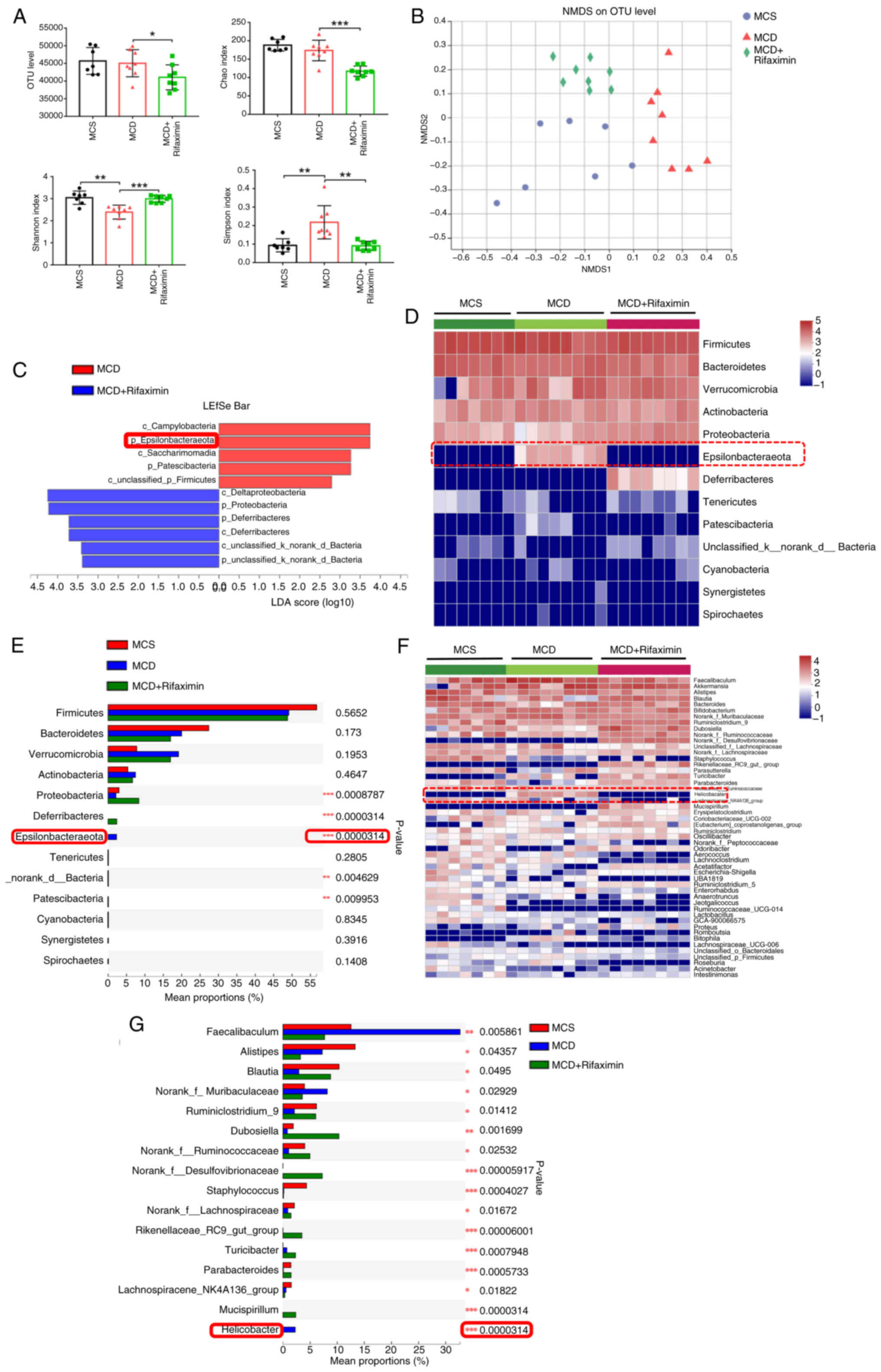 | Figure 3.Rifaximin affects the gut microbiota
in mice with MCD diet-induced non-alcoholic steatohepatitis. (A)
Caecal OTU, Chao index, Simpson index and Shannon index of mice.
(B) Separation of samples by MCS, MCD, and MCD with rifaximin
gavage was observed via NMDS analysis. (C) Relative abundance of
gut microbiota in caecal content identified by LEfSe bar analysis
among MCS, MCD and MCD + rifaximin groups. (D) Heatmap of
separation at the phylum level of mouse faecal microbiota among the
MCS, MCD and MCD + rifaximin groups. (E) Significant difference in
mouse faecal microbiota at the phylum level among the MCS, MCD and
MCD + rifaximin groups. (F) Heatmap of separation at the genus
level of mouse faecal microflora among the MCS, MCD and MCD +
rifaximin groups. (G) Significant difference of mouse faecal
microbiota at the genus level among the MCS, MCD and MCD +
rifaximin groups. Clustering was performed using the Pearson
measurement. *P<0.05, **P<0.01, ***P<0.001. LDA, linear
discriminant analysis; MCD, methionine-choline deficient; MCS,
methionine-choline sufficient; NMDS, non-metric multidimensional
scaling; OTU, operational taxonomic unit. |
Intestinal Helicobacter-DCA-Fxr
signalling pathway is suppressed after rifaximin treatment
A close interaction exists between the intestinal
microflora and intestinal bile acids; in particular, bile acids
directly inhibit the growth of the gut microbiota. In addition,
bile saline hydrolase (BSH), encoded by some intestinal microflora,
can hydrolyse bile acids to resist the antibacterial effect of bile
acids (34). To evaluate the
potential link between rifaximin-induced changes in the intestinal
microflora composition and bile acid levels in the terminal ileum,
Spearman's correlation analysis was performed. As shown in Fig. 4A, the abundance of Helicobacter
hepaticus, which was the most significantly altered bacteria in
mice with MCD diet-induced NASH, was positively correlated with
DCA. Therefore, it was speculated that DCA in the terminal ileum
may decrease with decreasing Helicobacter hepaticus
abundance after rifaximin treatment. As expected, a bile acid assay
of the terminal ileum demonstrated that DCA was significantly
elevated in mice with MCD diet-induced NASH, whereas rifaximin
treatment markedly decreased the increase in DCA (Fig. 4B). In addition, the total bile acid
content, conjugated bile acid/unconjugated bile acid ratio, and
primary bile acid/second bile acid ratio did not significantly
change after rifaximin treatment (Fig.
4C). DCA has been reported to be an intestinal Fxr agonist
involved in activating the intestinal Fxr-fibroblast growth factor
15 (Fgf15) signalling pathway to affect liver cholesterol
metabolism and bile acid synthesis (35). Therefore, the present study further
detected the intestinal Fxr-Fgf15-Cyp7a1 signalling pathway. It was
observed that rifaximin treatment significantly downregulated the
expression of Fxr and Fgf15 in the distal ileum, but markedly
increased Cyp7a1 and Cyp7b1 expression in the liver (Fig. 4D and E), compared with that in the
MCD group. These results indicated that rifaximin may inhibit the
intestinal Helicobacter-DCA-Fxr signalling pathway.
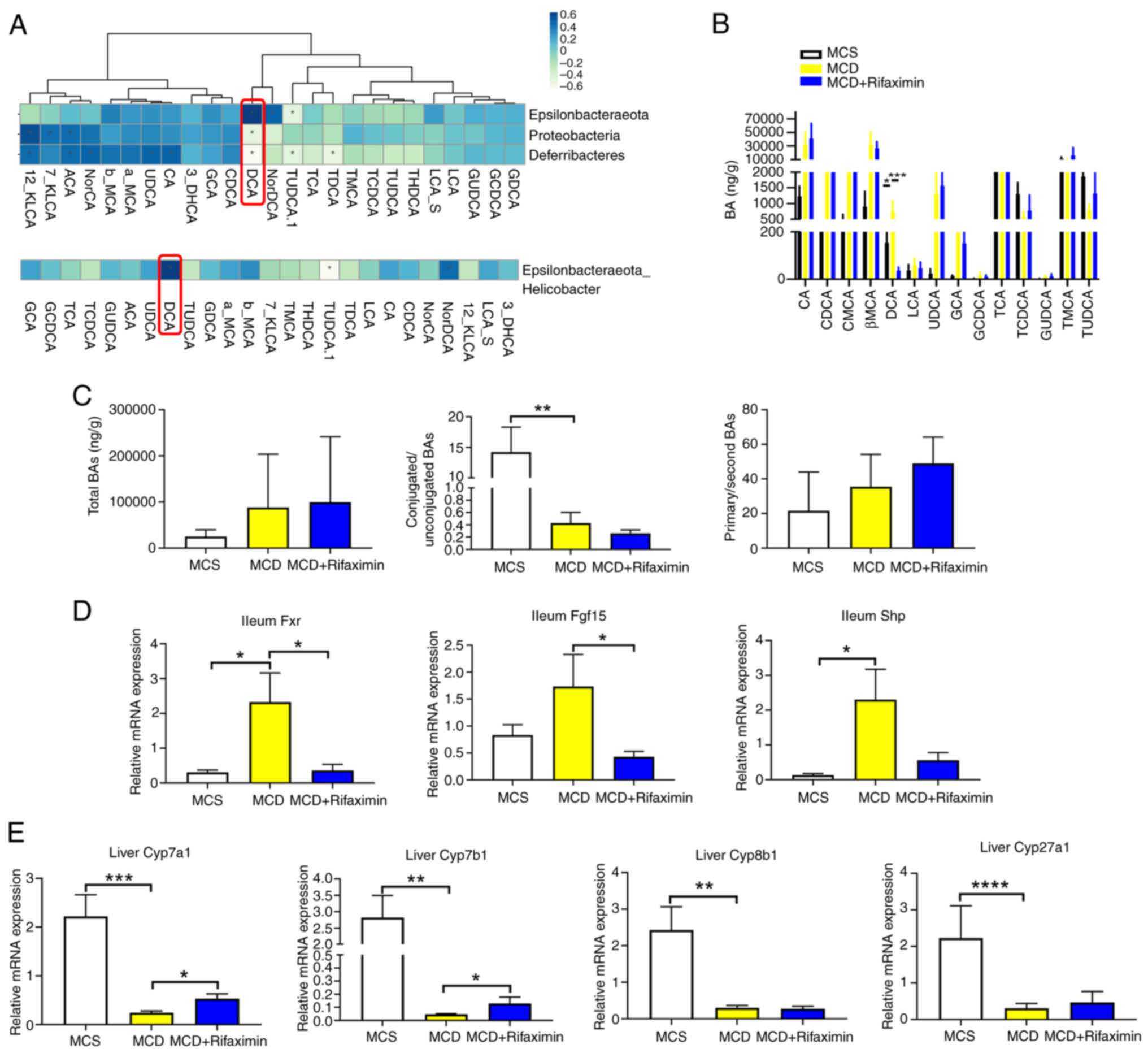 | Figure 4.Rifaximin suppresses the
Helicobacter-DCA-Fxr signalling pathway in MCD diet-fed
mice. (A) Correlation analysis of intestinal microflora and BAs in
the distal ileum was investigated using nonparametric Spearman's
test. (B) BA levels in the distal ileum of mice. (C) Total BAs,
conjugated BAs/unconjugated BAs ratio, and primary BAs/second BAs
ratio in the distal ileum of mice. (D) Expression levels of Fxr,
Fgf15 and Shp in the distal ileum. (E) Expression levels of Cyp7a1,
Cyp7b1, Cyp8b1, and Cyp27a1 mRNA in the liver. n=8 mice/group.
*P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. BA, bile
acid; DCA, deoxycholic acid; Fgf15, fibroblast growth factor 15;
Fxr, farnesoid X receptor; MCD, methionine-choline deficient; MCS,
methionine-choline sufficient. |
Anti-NASH effects of rifaximin are
impaired in mice lacking gut microbiota
To determine whether the anti-NASH effects of
rifaximin are mediated through the gut microbiota, the effects of
rifaximin on hepatic steatosis, inflammation and fibrosis were
assessed in mice with NASH and intestinal decontamination. After 1
week of treatment with broad-spectrum antibiotics (ampicillin 1
g/l, neomycin sulphate 1 g/l, metronidazole 1 g/l and vancomycin
0.5 g/l) in the drinking water, as previously described (23), the mice in the MCD + Abx +
rifaximin group were administered rifaximin 100 mg/kg/day by oral
gavage for 3 weeks, and the mice in the MCD + Abx group were
treated with water by oral gavage according to their body weight
for 3 weeks (Fig. 5A). Notably, no
further improvement was observed in liver steatosis, hepatocyte
ballooning or lobular inflammation after rifaximin treatment in
mice with NASH and intestinal decontamination (Fig. 5B and C). In addition, Oil Red O
staining showed no further reduction in lipid accumulation in the
livers of the MCD + Abx + rifaximin group compared with those of
the MCD + Abx group (Fig. 5D).
Moreover, the protein expression levels of Pparγ and Srebp1 in the
MCD + Abx + rifaximin group were not significantly different from
those in the MCD + Abx group (Fig.
5E). Additionally, α-Sma protein expression was not
significantly reduced after rifaximin treatment in mice with NASH
and intestinal decontamination (Fig.
5F). These findings indicated that intestinal decontamination
could impair the ability of rifaximin to ameliorate hepatic
steatosis, inflammation and fibrosis, suggesting that the
intestinal microbiota is required for the ability of rifaximin to
ameliorate MCD diet-induced NASH in mice.
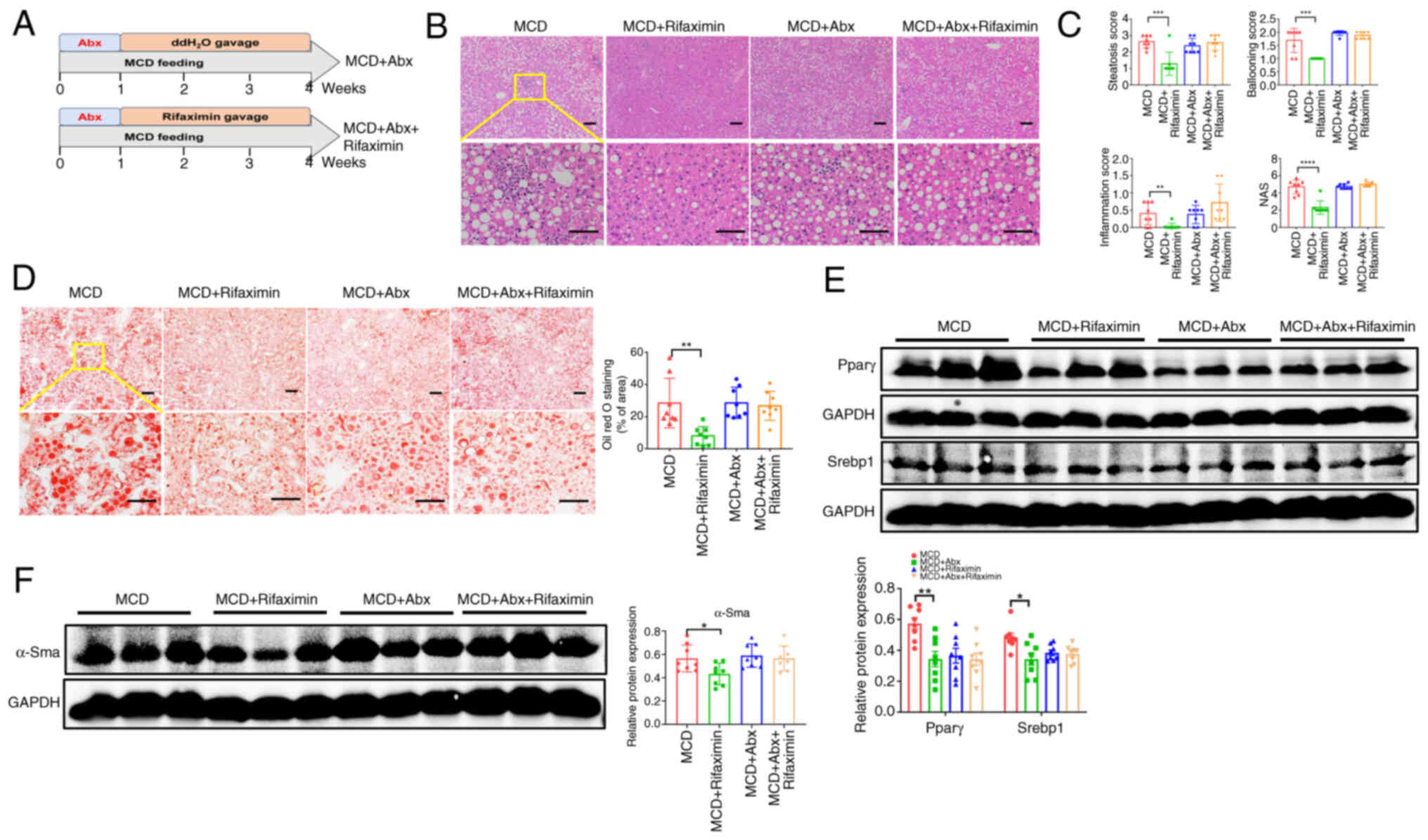 | Figure 5.Anti-NASH effects of rifaximin are
impaired in mice lacking gut microbiota. (A) Schematic illustration
of the experimental design of intestinal decontamination in mice
with NASH. (B) Haematoxylin and eosin staining of livers in the
MCD, MCD + rifaximin, MCD + Abx and MCD + Abx + rifaximin groups.
Scale bars, 100 µm. (C) Steatosis score, hepatic ballooning score,
interlobular inflammation score and NAS. (D) Oil red O staining for
livers of mice in each group. Scale bars, 100 µm. (E) Pparγ and
Srebp1 protein expression levels in mouse livers. (F) α-Sma protein
expression levels in the livers in each group. n=8 mice/group.
*P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. α-Sma,
α-smooth muscle actin; MCD, methionine-choline deficient; MCS,
methionine-choline sufficient; NASH, non-alcoholic steatohepatitis;
NAS, non-alcoholic fatty liver disease activity score; Srebp1,
sterol regulatory element-binding protein 1. |
Activation of hepatic Hnf1α is
required for rifaximin to ameliorate NASH in mice
The present study revealed that rifaximin could
improve bile acid and cholesterol metabolism by regulating the
intestinal Helicobacter-DCA-Fxr pathway. Therefore, the
study aimed to determine how rifaximin specifically affects liver
cell function. Our previous study indicated that
Hnf1αH-KO mice spontaneously develop NASH (24). Notably, the present study revealed
that rifaximin enhanced liver Hnf1α and Fxr mRNA expression
compared with that in the MCD group (Fig. S1C). Therefore, it was hypothesized
that the activation of hepatic Hnf1α may be required for rifaximin
to ameliorate NASH in mice. To confirm this hypothesis,
Hnf1αH-KO mice were generated by crossing mice
homozygous for Hnf1αf/f with Alb-Cre transgenic mice
(Fig. 6A). Male
Hnf1αH-KO mice were fed a normal chow diet for 20 weeks
to induce NASH, then treated with rifaximin by oral gavage for 4
weeks (Fig. 6B). As shown in
Fig. 6C, the protein expression
levels of Hnf1α in the Hnf1αH-KO group and
Hnf1αH-KO + rifaximin groups were lower than those in
Hnf1αf/f group. Notably, it was observed that liver
steatosis, hepatocyte ballooning, lobular inflammation and NAS in
the Hnf1αH-KO + rifaximin group were not significantly
different from those in the Hnf1αH-KO group (Fig. 6D and E). In addition, no further
improvement in lipid accumulation was observed after rifaximin
treatment in mice with Hnf1α knockout (Fig. 6F). Moreover, the protein expression
levels of Pparγ and Srebp1 in the Hnf1αH-KO + rifaximin
group were not significantly different from those in the
Hnf1αH-KO group (Fig.
6G). Additionally, α-Sma protein expression was not
significantly reduced after rifaximin treatment in
Hnf1αH-KO mice with NASH (Fig. 6H). Based on these results, it was
indicated that the anti-NASH biological effects of rifaximin depend
on the activation of liver Hnf1α.
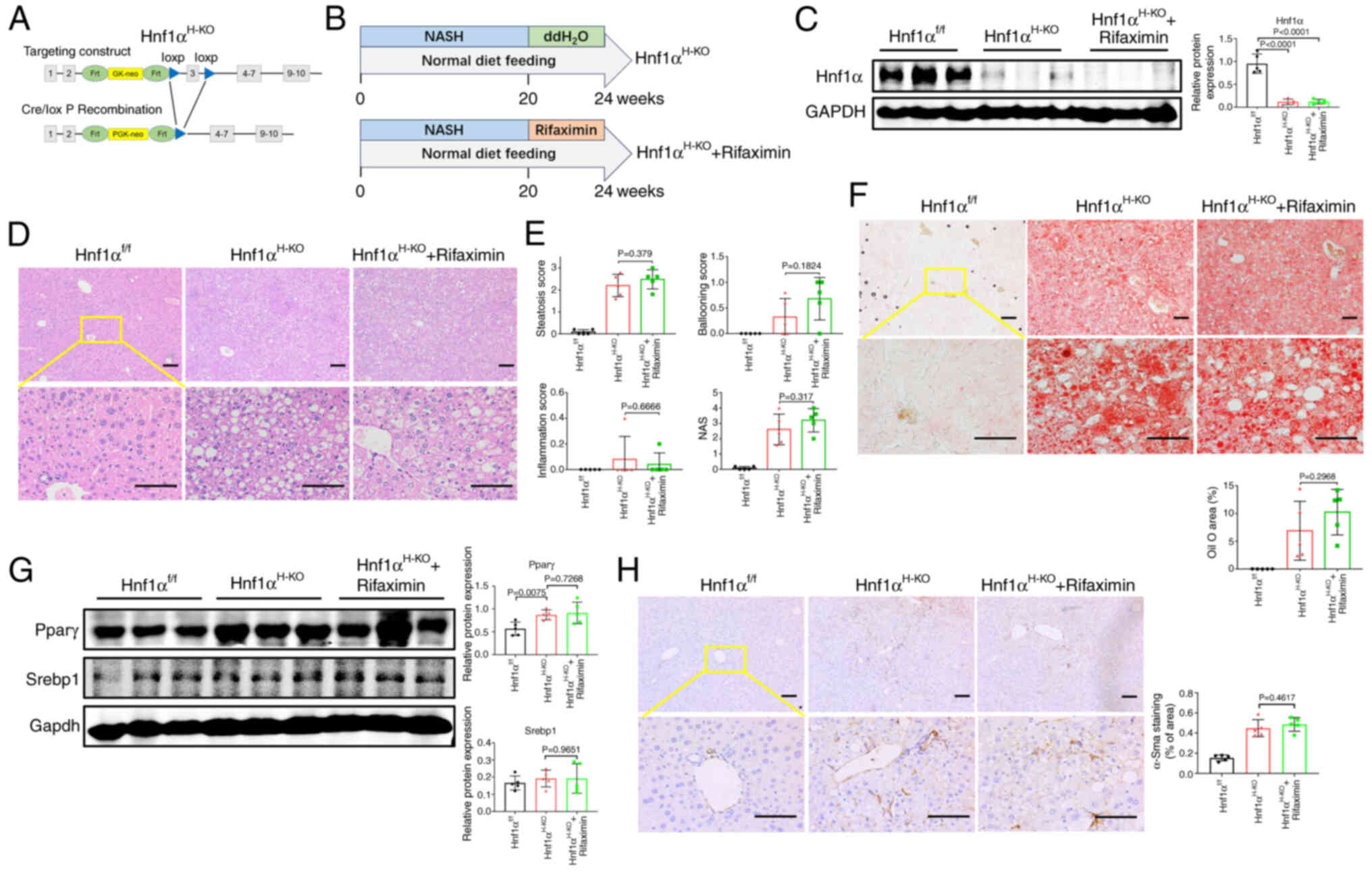 | Figure 6.Activation of hepatic Hnf1α is
required for rifaximin to ameliorate NASH in mice. (A)
Hnf1αH-KO NASH mice were established. (B) Schematic
illustration of the experimental design. (C) Hnf1α protein
expression levels in the mouse livers of each group. (D)
Haematoxylin and eosin staining of livers in each group. Scale
bars, 100 µm. (E) Steatosis score, hepatic ballooning score,
interlobular inflammation score and NAS. (F) Oil red O staining for
livers of mice in each group. Scale bars, 100 µm. (G) Pparγ and
Srebp1 protein expression levels in the livers of each group. (H)
α-Sma protein expression level in the livers in each group. n=5
mice/group. α-Sma, α-smooth muscle actin; Hnf1α, hepatocyte nuclear
factor 1α; Hnf1αf/f, floxed Hnf1α; Hnf1αH-KO,
hepatocyte-specific Hnf1α knockout; MCD, methionine-choline
deficient; MCS, methionine-choline sufficient; NASH, non-alcoholic
steatohepatitis; NAS, non-alcoholic fatty liver disease activity
score; Srebp1, sterol regulatory element-binding protein 1. |
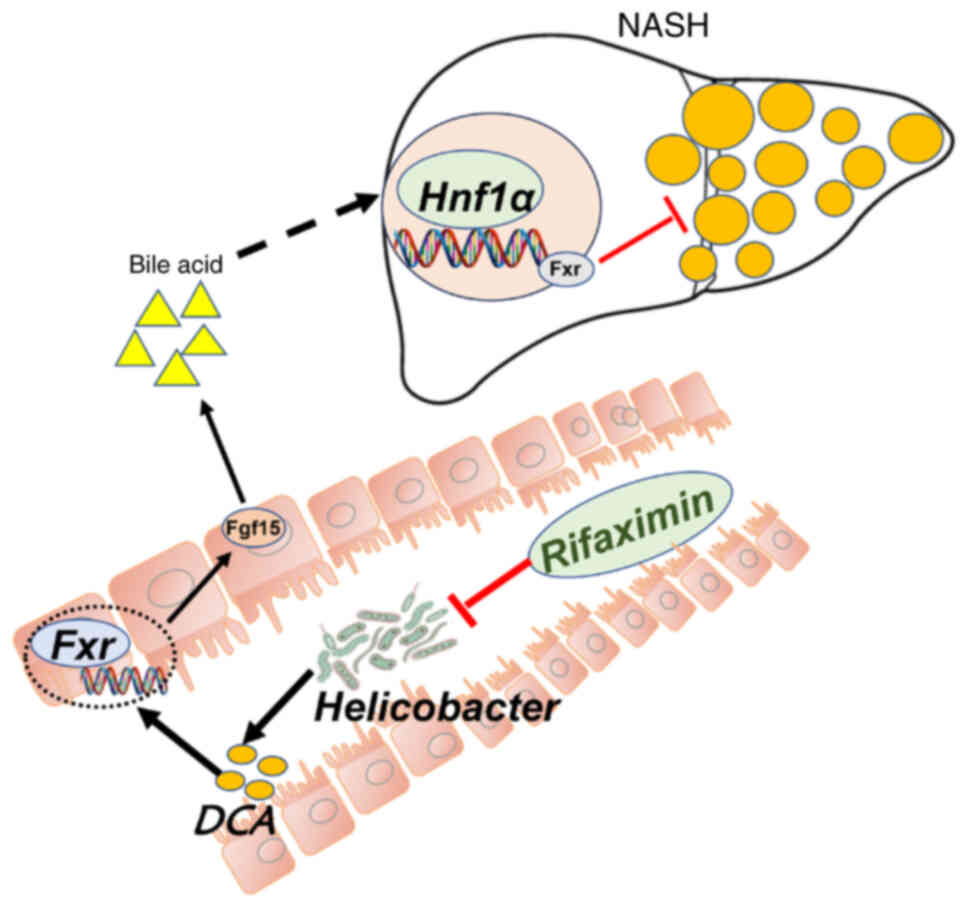
On the basis of these results, it could be concluded
that the anti-NASH biological effects of rifaximin depend on
modulation of the intestinal Helicobacter-DCA-Fxr-Hnf1α
signalling pathway.
Discussion
Although previous clinical studies have indicated
that rifaximin can ameliorate serum endotoxaemia and inhibit
proinflammatory factor expression in patients with NASH (20,21),
the potential molecular biological mechanisms of rifaximin in
treating NASH are currently unknown. The present study showed that
mice in the MCD + rifaximin group had significantly lower
liver-to-body weight ratio than mice in the MCD group. Moreover,
rifaximin treatment significantly improved liver lipid deposition,
hepatocyte ballooning and lobular inflammation. During the
development of NASH, liver inflammation persists, leading to liver
cell damage and increased ALT levels (36). The present results showed that
rifaximin treatment reduced the serum ALT levels in mice with NASH
and improved their liver function. Srebp1 is an important
transcription factor for liver lipid metabolism, which serves a
crucial role in the occurrence and development of NASH (37). Pparγ is a ligand-activated receptor
in the nuclear hormone receptor family that is involved in
regulating cell proliferation, inflammation, and lipid and glucose
homeostasis (38). Consistent with
previous research results, the present study indicated that the
expression levels of Srebp1 and Pparγ were upregulated in mice with
NASH, whereas rifaximin treatment significantly decreased the
increased levels of Srebp1 and Pparγ, and inhibited liver lipid
metabolism.
With the development of NASH, α-Sma expression in
the liver is upregulated, red collagen deposition increases and
liver fibrosis occurs (33). The
present study showed that the expression levels of α-Sma and Col1a1
in the liver of the NASH group were markedly upregulated, and red
collagen deposition and liver hydroxyproline content were
significantly increased. Notably, rifaximin treatment markedly
decreased the increase in α-Sma and Col1a1, and reduced red
collagen deposition and liver hydroxyproline content. These results
indicated that rifaximin treatment may significantly alleviate MCD
diet-induced liver fibrosis in mice with NASH.
The interaction between the intestinal microflora
and host can affect the circadian rhythm of tissues and organs,
which in turn changes metabolic homeostasis (39). It has previously been demonstrated
that gut microbiota dysbiosis is closely related to the occurrence
and development of NAFLD (40).
Rifaximin is an intestinal-specific broad-spectrum antibiotic,
which is not absorbed by the intestine and is not metabolized by
the liver. Most of the time, rifaximin is used as a probiotic that
regulates the gut flora. It has been approved by the U.S. FDA for
the treatment of traveller's diarrhoea and hepatic encephalopathy
due to its ability to improve intestinal microflora imbalance
(16,18). Therefore, in the present study,
mouse caecal content samples were collected for bacterial 16S rRNA
gene sequencing and the potential involvement of the intestinal
microflora in mediating MCD diet-induced NASH was explored. The
results revealed that MCD significantly decreased the faecal OTU,
Chao1 index and Simpson index, but markedly increased the Shannon
index, indicating that rifaximin can markedly reduce the total
intestinal microflora and increase bacterial richness. Furthermore,
it was observed that, compared with that in the MCS group,
Helicobacter hepaticus was the most significantly affected
bacteria in the MCD group, while rifaximin treatment markedly
decreased the increase in Helicobacter hepaticus. In
addition, Epsilonbacteraeota was one of the most significantly
changed bacteria between the MCD group and the MCD + rifaximin
group; however, there was no difference in Epsilonbacteraeota
between the MCS group and the MCD group. A previous study reported
that Helicobacter hepaticus could promote hepatitis
development by regulating the Fxr signalling pathway (41). Therefore, the present study
speculated that rifaximin may improve liver inflammation by
decreasing the abundance of Helicobacter hepaticus.
There is a close interaction between the gut
microbiota and intestinal bile acids. On the one hand, bile acids
directly inhibit the growth of the intestinal microflora due to
their antibacterial effect, whereas on the other hand, BSH, which
is encoded by some intestinal microflora, can hydrolyse bile acids
to resist their antibacterial effect (34). Therefore, Spearman's correlation
analysis was performed between the intestinal microflora and bile
acid levels in the terminal ileum. Notably, it was observed that
Helicobacter hepaticus abundance was significantly
positively correlated with DCA. Therefore, a full-spectrum test of
bile acids was performed in the terminal ileum. As expected, the
results showed that DCA in the terminal ileum was significantly
decreased following rifaximin treatment. There is evidence that DCA
is an intestinal Fxr agonist, which activates the intestinal
Fxr-Fgf15 signalling pathway, and affects liver cholesterol and
bile acid metabolism (35).
Therefore, the present study further detected the intestinal
Fxr-Fgf15-Cyp7a1 signalling pathway. It was observed that rifaximin
significantly downregulated the expression of Fxr and Fgf15 in the
distal ileum, but markedly increased Cyp7a1 and Cyp7b1 expression
in the liver. Based on these results, it could be speculated that
rifaximin may inhibit the growth of Helicobacter hepaticus
and reduce DCA in the distal ileum, thereby inhibiting the
intestinal Fxr-Fgf15 signalling pathway to improve liver
cholesterol and bile acid metabolism in mice with NASH.
To further confirm that rifaximin relies on the
intestinal microbiota to serve a molecular biological role in
preventing NASH, mice with NASH were first treated with
broad-spectrum antibiotics for intestinal decontamination and were
then treated with rifaximin. As expected, no further improvement in
liver steatosis, hepatocyte ballooning or lobular inflammation was
observed in the MCD + Abx + rifaximin group compared with that in
the MCD + Abx group. Moreover, the expression levels of Pparγ,
α-Sma and Col1a1 were not significantly reduced following rifaximin
treatment in mice with NASH and intestinal decontamination. These
findings indicated that intestinal decontamination may impair the
ability of rifaximin to ameliorate hepatic inflammation and
fibrosis, suggesting that the intestinal microbiota is required for
the ability of rifaximin to ameliorate MCD diet-induced NASH in
mice.
Hnf1α is a liver-enriched transcription factor that
modulates liver fatty acid binding protein function to affect liver
fatty acid transport and lipid metabolism (42). A previous study revealed that Hnf1α
could regulate liver Fxr expression, and affect lipid and
cholesterol metabolism by directly binding to the liver Fxr
transcription factor (43). In
vivo experiments in mice also confirmed that activation of the
Hnf1α-Fxr signalling pathway can regulate liver bile acid
metabolism and inhibit the formation of cholesterol stones
(44). Furthermore, our previous
study indicated that Hnf1αH-KO mice develop spontaneous
NASH (24). Notably, the present
study revealed that rifaximin activated liver Hnf1α expression.
Therefore, it was speculated that activation of hepatic Hnf1α was
required for rifaximin to ameliorate NASH in mice. To confirm this
hypothesis, Hnf1αH-KO mice with NASH were treated with
rifaximin. Subsequently, it was observed that liver lipid
deposition, liver inflammation and liver fibrosis were not
significantly improved after rifaximin treatment when Hnf1α was
knocked out. These findings confirmed that the anti-NASH biological
effects of rifaximin depend on the activation of liver Hnf1α.
In conclusion, rifaximin can inhibit the
proliferation of intestinal Helicobacter hepaticus, thereby
decreasing the DCA in the terminal ileum and inhibiting the
intestinal Fxr-Fgf15 signalling pathway. With inhibition of the
intestinal Fxr-Fgf15 signalling pathway, increased Cyp7a1 and
Cyp7b1 in the liver can further activate the liver Hnf1a-Fxr
signalling pathway and improve liver lipid metabolism, ultimately
inhibiting the progression of NASH (Fig. 7). However, humans and mice have
different physiological structures. The present study revealed that
rifaximin can improve lipid and cholesterol metabolism in mice with
NASH; however, it is unclear whether it has the same biological
effect on patients with NASH. In future studies, different doses of
rifaximin will be administered to patients with NASH to observe the
clinical therapeutic effect. The findings of the present study
provide a theoretical basis for the clinical treatment of rifaximin
in patients with NASH.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Professor Mei-Tong
Nie (Department of Gastroenterology, Shanghai East Hospital, Tongji
University School of Medicine) for providing their assistance in
submitting the data to a public curated database.
Funding
This work was supported by the National Natural Science
Foundation of China (grant no. 82360123) and the Natural Science
Foundation of Jiangxi Province (grant nos. 20232BAB206017 and
20242BAB20355).
Availability of data and materials
The sequencing data generated in the present study
may be found in the BioProject database under accession number
PRJNA1172135 or at the following URL: https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1172135.
The other data generated in the present study may be requested from
the corresponding author.
Authors' contributions
JJ conceived and designed the experiments. YPW and
SL wrote the manuscript. YPW, SL, DL and XMH performed the
experiments. JHW analysed data. YPW and JJ revised the manuscript.
YPW and JJ confirm the authenticity of all the raw date. All
authors read and approved the final version of the manuscript.
Ethics approval and consent to
participate
The animal experiments were initially performed at
Naval Medical University and later at Nanchang University. All
animal experiments were performed in accordance with the National
Institute of Health Guide for the Care and Use of Laboratory
Animals, and were approved by the Naval Medical University
(approval no. SYXK2021-0075) and Nanchang University (approval no.
LL-202303280001).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Younossi Z, Tacke F, Arrese M, Chander
Sharma B, Mostafa I, Bugianesi E, Wai-Sun Wong V, Yilmaz Y, George
J, Fan J and Vos MB: Global perspectives on nonalcoholic fatty
liver disease and nonalcoholic Steatohepatitis. Hepatology.
69:2672–2682. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Friedman SL, Neuschwander-Tetri BA,
Rinella M and Sanyal AJ: Mechanisms of NAFLD development and
therapeutic strategies. Nat Med. 24:908–922. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schuster S, Cabrera D, Arrese M and
Feldstein AE: Triggering and resolution of inflammation in NASH.
Nat Rev Gastroenterol Hepatol. 15:349–364. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang X, Coker OO, Chu ES, Fu K, Lau HCH,
Wang YX, Chan AWH, Wei H, Yang X, Sung JJY and Yu J: Dietary
cholesterol drives fatty liver-associated liver cancer by
modulating gut microbiota and metabolites. Gut. 70:761–774. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dr De Sousa SM and Prof Norman RJ :
Metabolic syndrome, diet and exercise. Best Pract Res Clin Obstet
Gynaecol. 37:140–151. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kim D, Touros A and Kim WR: Nonalcoholic
fatty liver disease and metabolic syndrome. Clin Liver Dis.
22:133–140. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sharpton SR, Ajmera V and Loomba R:
Emerging role of the gut microbiome in nonalcoholic fatty liver
disease: From composition to function. Clin Gastroenterol Hepatol.
17:296–306. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ponziani FR, Bhoori S, Castelli C,
Putignani L, Rivoltini L, Del Chierico F, Sanguinetti M, Morelli D,
Paroni Sterbini F, Petito V, et al: Hepatocellular carcinoma is
associated with gut microbiota profile and inflammation in
nonalcoholic fatty liver disease. Hepatology. 69:107–120. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Grabherr F, Grander C, Effenberger M,
Adolph TE and Tilg H: Gut dysfunction and non-alcoholic fatty liver
disease. Front Endocrinol (Lausanne). 10:6112019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sun L, Pang Y, Wang X, Wu Q, Liu H, Liu B,
Liu G, Ye M, Kong W and Jiang C: Ablation of gut microbiota
alleviates obesity-induced hepatic steatosis and glucose
intolerance by modulating bile acid metabolism in hamsters. Acta
Pharm Sin B. 9:702–710. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jasirwan COM, Lesmana CRA, Hasan I,
Sulaiman AS and Gani RA: The role of gut microbiota in
non-alcoholic fatty liver disease: pathways of mechanisms. Biosci
Microbiota Food Health. 38:81–88. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chávez-Talavera O, Tailleux A, Lefebvre P
and Staels B: Bile acid control of metabolism and inflammation in
obesity, type 2 diabetes, dyslipidemia, and nonalcoholic fatty
liver disease. Gastroenterology. 152:1679–1694.e3. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Corbin KD and Zeisel SH: Choline
metabolism provides novel insights into nonalcoholic fatty liver
disease and its progression. Curr Opin Gastroenterol. 28:159–165.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu HY, Walden TB, Cai D, Ahl D,
Bertilsson S, Phillipson M, Nyman M and Holm L: Dietary fiber in
bilberry ameliorates pre-obesity events in rats by regulating lipid
depot, cecal short-chain fatty acid formation and microbiota
composition. Nutrients. 11:13502019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhu L, Baker SS, Gill C, Liu W, Alkhouri
R, Baker RD and Gill SR: Characterization of gut microbiomes in
nonalcoholic steatohepatitis (NASH) patients: A connection between
endogenous alcohol and NASH. Hepatology. 57:601–609. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Coronel-Castillo CE, Contreras-Carmona J,
Frati-Munari AC, Uribe M and Méndez-Sánchez N: Efficacy of
rifaximin in the different clinical scenarios of hepatic
encephalopathy. Rev Gastroenterol Mex (Engl Ed). 85:56–68. 2020.(In
English, Spanish). PubMed/NCBI
|
|
17
|
Chautant F, Guillaume M, Robic MA,
Cadranel JF, Peron JM, Lison H, Cool C, Bureau C and Duhalde V:
Lessons from ‘real life experience’ of rifaximin use in the
management of recurrent hepatic encephalopathy. World J Hepatol.
12:10–20. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fodor AA, Pimentel M, Chey WD, Lembo A,
Golden PL, Israel RJ and Carroll IM: Rifaximin is associated with
modest, transient decreases in multiple taxa in the gut microbiota
of patients with diarrhoea-predominant irritable bowel syndrome.
Gut Microbes. 10:22–33. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shah ED, Saini SD and Chey WD: Value-based
pricing for rifaximin increases access of patients with irritable
bowel syndrome with diarrhea to therapy. Clin Gastroenterol
Hepatol. 17:2687–2695.e11. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Abdel-Razik A, Mousa N, Shabana W, Refaey
M, Elzehery R, Elhelaly R, Zalata K, Abdelsalam M, Eldeeb AA, Awad
M, et al: Rifaximin in nonalcoholic fatty liver disease: Hit
multiple targets with a single shot. Eur J Gastroenterol Hepatol.
30:1237–1246. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gangarapu V, Ince AT, Baysal B, Kayar Y,
Kılıç U, Gök Ö, Uysal Ö and Şenturk H: Efficacy of rifaximin on
circulating endotoxins and cytokines in patients with nonalcoholic
fatty liver disease. Eur J Gastroenterol Hepatol. 27:840–845. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang L, Liu B, Zheng J, Huang J, Zhao Q,
Liu J, Su Z, Wang M, Cui Z, Wang T, et al: Rifaximin alters
intestinal microbiota and prevents progression of ankylosing
spondylitis in mice. Front Cell Infect Microbiol. 9:442019.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhu Y, He C, Li X, Cai Y, Hu J, Liao Y,
Zhao J, Xia L, He W, Liu L, et al: Gut microbiota dysbiosis worsens
the severity of acute pancreatitis in patients and mice. J
Gastroenterol. 54:347–358. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ni Q, Ding K, Wang KQ, He J, Yin C, Shi J,
Zhang X, Xie WF and Shi YQ: Deletion of HNF1α in hepatocytes
results in fatty liver-related hepatocellular carcinoma in mice.
FEBS Lett. 591:1947–1957. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kleiner DE, Brunt EM, Van Natta M, Behling
C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS,
Unalp-Arida A, et al: Design and validation of a histological
scoring system for nonalcoholic fatty liver disease. Hepatology.
41:1313–1321. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yue HY, Yin C, Hou JL, Zeng X, Chen YX,
Zhong W, Hu PF, Deng X, Tan YX, Zhang JP, et al: Hepatocyte nuclear
factor 4alpha attenuates hepatic fibrosis in rats. Gut. 59:236–246.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nie Y, Liu Q, Zhang W, Wan Y, Huang C and
Zhu X: Ursolic acid reverses liver fibrosis by inhibiting
NOX4/NLRP3 inflammasome pathways and bacterial dysbiosis. Gut
Microbes. 13:19727462021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
R Core Team, . R: A language and
environment for statistical computing. R Foundation for Statistical
Computing; Vienna, Austria: 2021
|
|
30
|
Quan T, Zhou F, Chen H, Jian L, Yang Y,
Xia F, Xiang S, Zhou B and Li S: Ficus hirta Vahl. Ameliorates
nonalcoholic fatty liver disease through regulating lipid
metabolism and gut microbiota. Oxid Med Cell Longev.
2022:34747232022. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mukherjee S, Zhelnin L, Sanfiz A, Pan J,
Li Z, Yarde M, McCarty J and Jarai G: Development and validation of
an in vitro 3D model of NASH with severe fibrotic phenotype. Am J
Transl Res. 11:1531–1540. 2019.PubMed/NCBI
|
|
32
|
Kumar S, Duan Q, Wu R, Harris EN and Su Q:
Pathophysiological communication between hepatocytes and
non-parenchymal cells in liver injury from NAFLD to liver fibrosis.
Adv Drug Deliv Rev. 176:1138692021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Huang R, Guo F, Li Y, Liang Y, Li G, Fu P
and Ma L: Activation of AMPK by triptolide alleviates nonalcoholic
fatty liver disease by improving hepatic lipid metabolism,
inflammation and fibrosis. Phytomedicine. 92:1537392021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Begley M, Gahan CGM and Hill C: The
interaction between bacteria and bile. FEMS Microbiol Rev.
29:625–651. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
de Aguiar Vallim TQ, Tarling EJ and
Edwards PA: Pleiotropic roles of bile acids in metabolism. Cell
Metab. 17:657–669. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Honda T, Ishigami M, Luo F, Lingyun M,
Ishizu Y, Kuzuya T, Hayashi K, Nakano I, Ishikawa T, Feng GG, et
al: Branched-chain amino acids alleviate hepatic steatosis and
liver injury in choline-deficient high-fat diet induced NASH mice.
Metabolism. 69:177–187. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang L, Li HX, Pan WS, Khan FU, Qian C,
Qi-Li FR and Xu X: Novel hepatoprotective role of Leonurine
hydrochloride against experimental non-alcoholic steatohepatitis
mediated via AMPK/SREBP1 signallinging pathway. Biomed
Pharmacother. 110:571–581. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cheng S, Qian K, Wang Y, Wang G, Liu X,
Xiao Y and Wang X: PPARγ inhibition regulates the cell cycle,
proliferation and motility of bladder cancer cells. J Cell Mol Med.
23:3724–3736. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Voigt RM, Summa KC, Forsyth CB, Green SJ,
Engen P, Naqib A, Vitaterna MH, Turek FW and Keshavarzian A: The
circadian clock mutation promotes intestinal dysbiosis. Alcohol
Clin Exp Res. 40:335–347. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Canfora EE, Meex RCR, Venema K and Blaak
EE: Gut microbial metabolites in obesity, NAFLD and T2DM. Nat Rev
Endocrinol. 15:261–273. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Swennes AG, Sheh A, Parry NMA, Muthupalani
S, Lertpiriyapong K, García A and Fox JG: Helicobacter
hepaticus infection promotes hepatitis and preneoplastic foci
in farnesoid X receptor (FXR) deficient mice. PLoS One.
9:e1067642014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Qadri I, Hu LJ, Iwahashi M, Al-Zuabi S,
Quattrochi LC and Simon FR: Interaction of hepatocyte nuclear
factors in transcriptional regulation of tissue specific hormonal
expression of human multidrug resistance-associated protein 2
(abcc2). Toxicol Appl Pharmacol. 234:281–292. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Xiong X, Wang X, Lu Y, Wang E, Zhang Z,
Yang J, Zhang H and Li X: Hepatic steatosis exacerbated by
endoplasmic reticulum stress-mediated downregulation of FXR in
aging mice. J Hepatol. 60:847–854. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Purushotham A, Xu Q, Lu J, Foley JF, Yan
X, Kim DH, Kemper JK and Li X: Hepatic deletion of SIRT1 decreases
hepatocyte nuclear factor 1α/farnesoid X receptor signaling and
induces formation of cholesterol gallstones in mice. Mol Cell Biol.
32:1226–1236. 2012. View Article : Google Scholar : PubMed/NCBI
|